
Diabetes Mellitus Secondary to Endocrine Diseases: An Update of Diagnostic and Treatment Particularities
 , and
, and 
Abstract
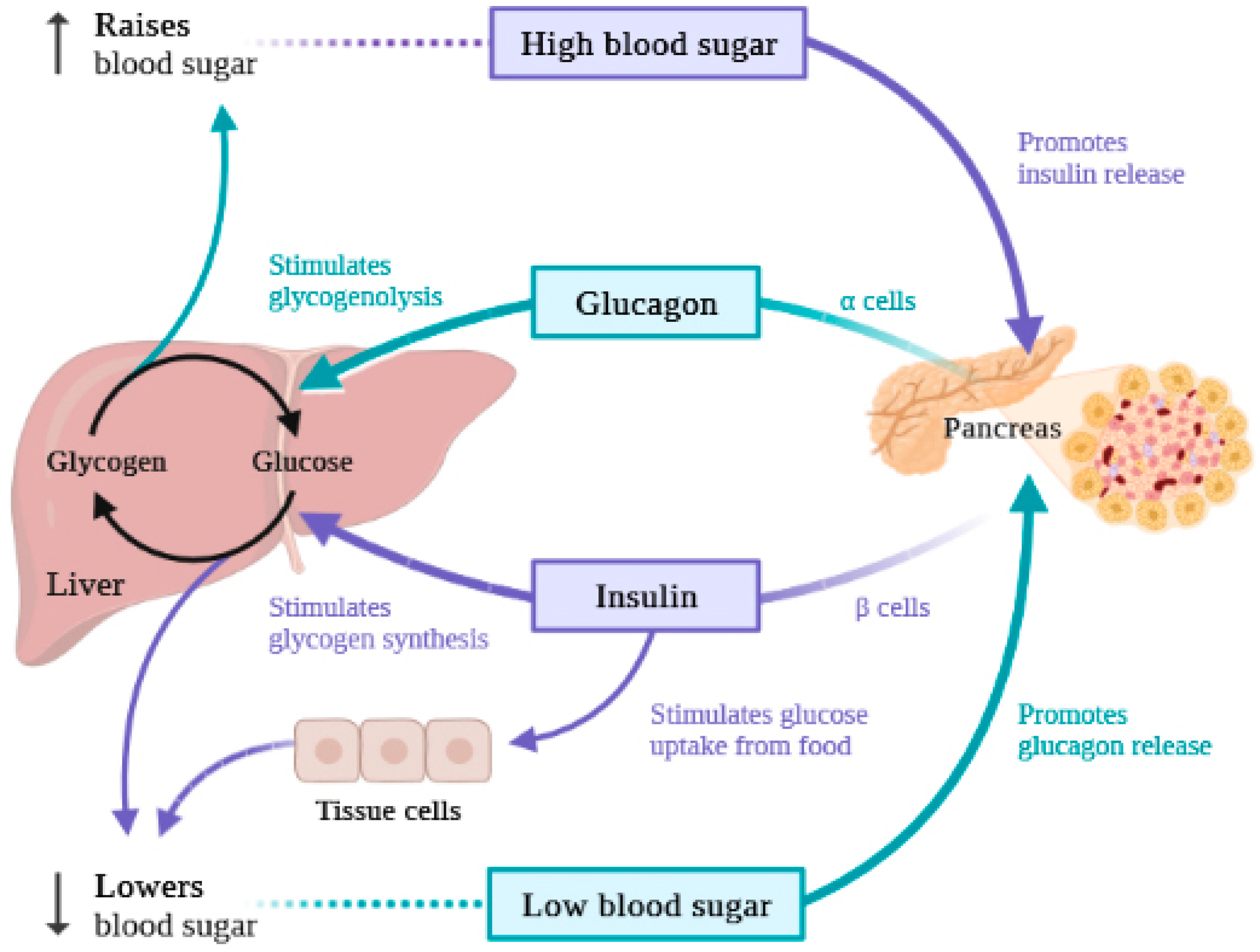
:1. Introduction
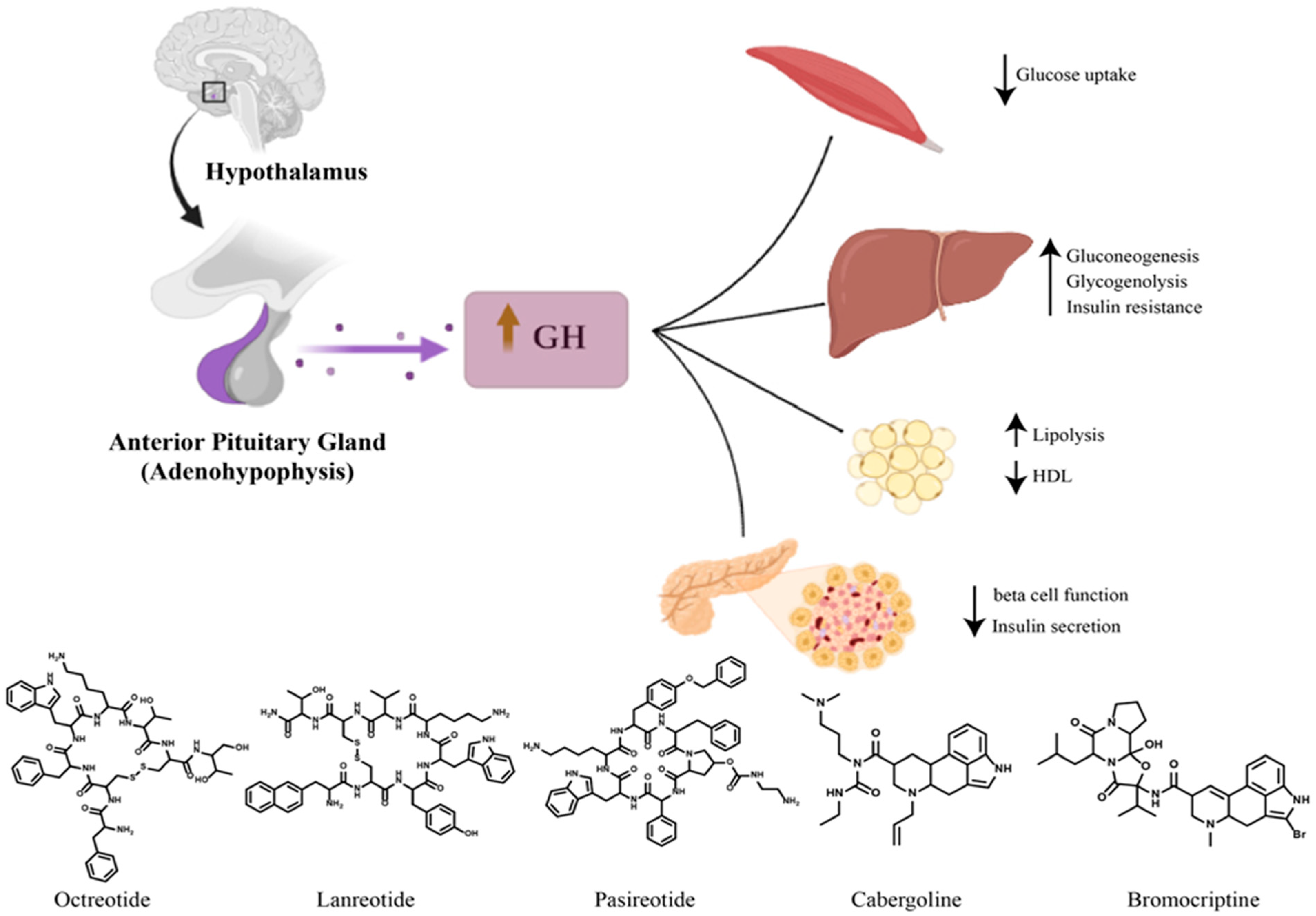
2. Diabetes Mellitus Secondary to Acromegaly
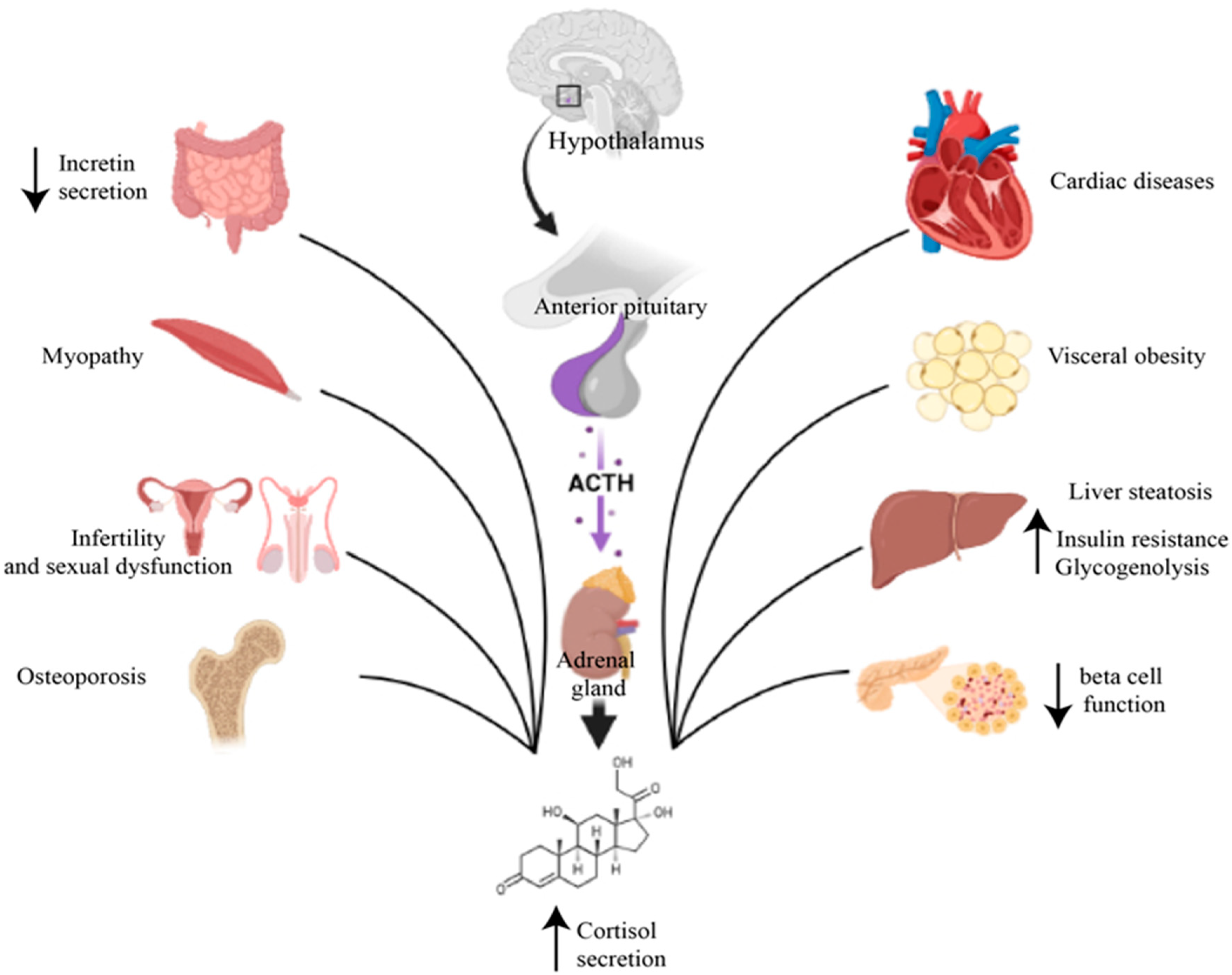
3. Cushing’s Syndrome and Diabetes
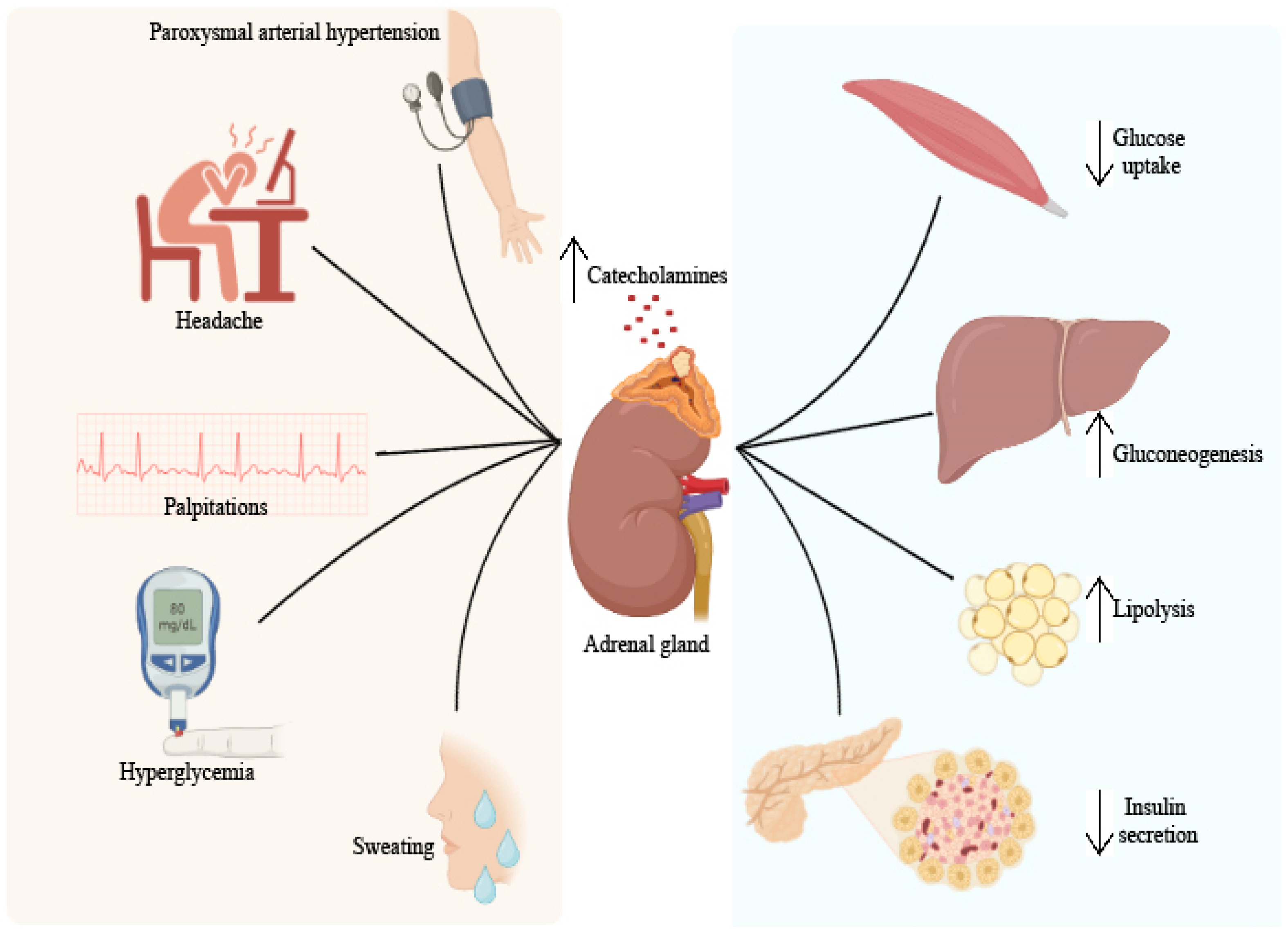
4. Pheochromocytoma and Diabetes
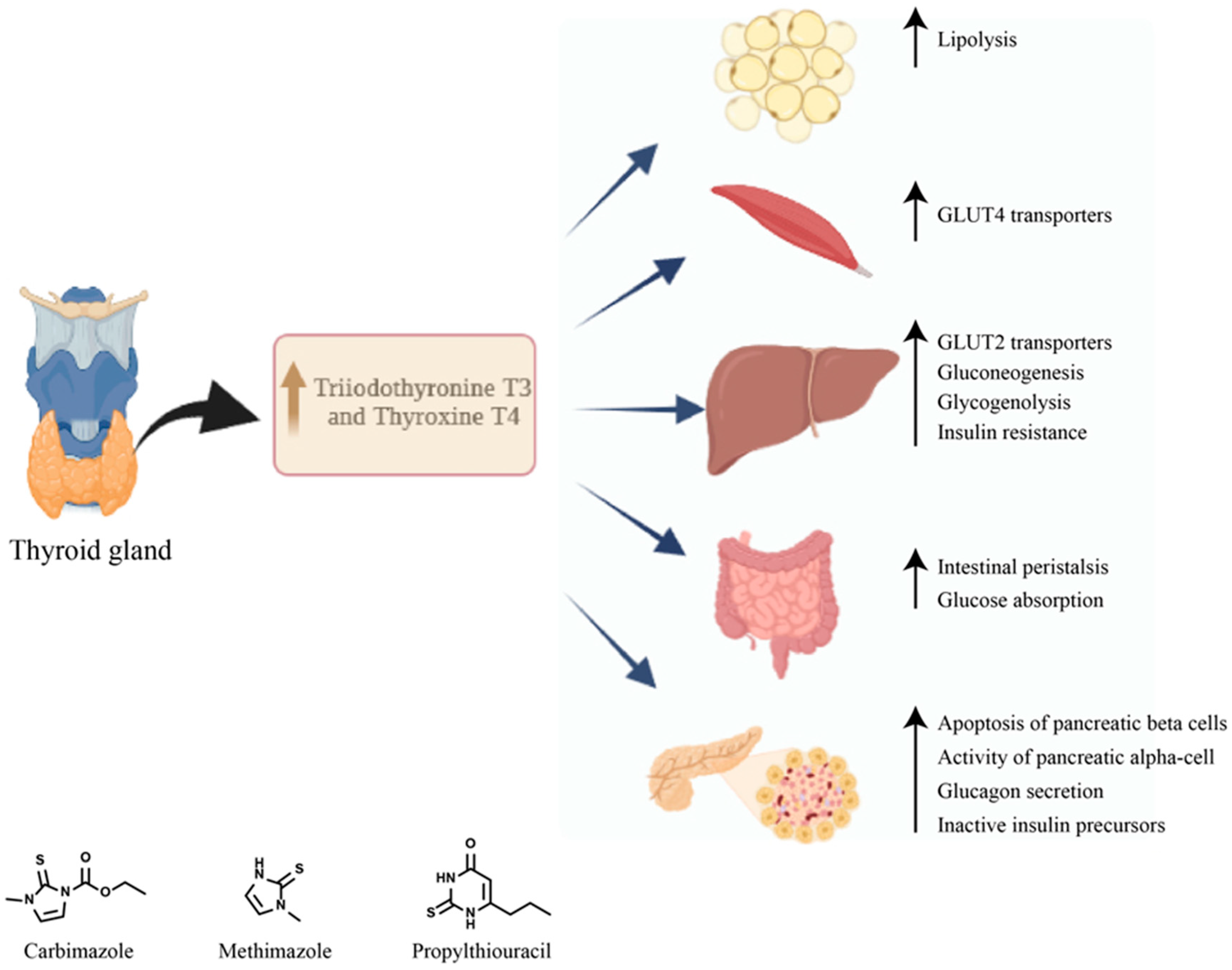
5. Diabetes Mellitus Secondary to Graves’ Disease
6. Diabetes Mellitus Secondary to Primary Aldosteronism
7. Diabetes Mellitus and Somatostatinoma
8. Diabetes Mellitus Secondary to Glucagonoma
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef]
- Resmini, E.; Minuto, F.; Colao, A.; Ferone, D. Secondary diabetes associated with principal endocrinopathies: The impact of new treatment modalities. Acta Diabetol. 2009, 46, 85–95. [Google Scholar] [CrossRef]
- American Diabetes Association. Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 2014, 37 (Suppl. S1), S81–S90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Expert Committee. International Expert Committee report on the role of the A1C assay in the diagnosis of diabetes. Diabetes Care 2009, 32, 1327–1334. [Google Scholar] [CrossRef] [Green Version]
- ElSayed, N.A.; Aleppo, G.; Aroda, V.R.; Bannuru, R.R.; Brown, F.M.; Bruemmer, D.; Collins, B.S.; Gaglia, J.L.; Hilliard, M.E.; Isaacs, D.; et al. 2. Classification and Diagnosis of Diabetes: Standards of Care in Diabetes—2023. Diabetes Care 2022, 46, S19–S40. [Google Scholar] [CrossRef] [PubMed]
- International Diabetes Federation. IDF Diabetes Atlas, 10th ed.; International Diabetes Federation: Brussels, Belgium, 2021; Available online: https://www.diabetesatlas.org (accessed on 7 August 2023).
- Sherlock, M.; Ayuk, J.; Tomlinson, J.W.; Toogood, A.A.; Aragon-Alonso, A.; Sheppard, M.C.; Bates, A.S.; Stewart, P.M. Mortality in Patients with Pituitary Disease. Endocr. Rev. 2010, 31, 301–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ershadinia, N.; Tritos, N.A. Diagnosis and Treatment of Acromegaly: An Update. Mayo Clin. Proc. 2022, 97, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Giustina, A.; Veldhuis, J.D. Pathophysiology of the Neuroregulation of Growth Hormone Secretion in Experimental Animals and the Human. Endocr. Rev. 1998, 19, 717–797. [Google Scholar] [CrossRef]
- Clemmons, D.R. Consensus Statement on the Standardization and Evaluation of Growth Hormone and Insulin-like Growth Factor Assays. Clin. Chem. 2011, 57, 555–559. [Google Scholar] [CrossRef] [Green Version]
- Caron, P.; Brue, T.; Raverot, G.; Tabarin, A.; Cailleux, A.; Delemer, B.; Renoult, P.P.; Houchard, A.; Elaraki, F.; Chanson, P. Correction to: Signs and symptoms of acromegaly at diagnosis: The physician’s and the patient’s perspectives in the ACRO-POLIS study. Endocrine 2018, 63, 130. [Google Scholar] [CrossRef] [Green Version]
- Johannsson, G.; Bidlingmaier, M.; Biller, B.M.K.; Boguszewski, M.; Casanueva, F.F.; Chanson, P.; E Clayton, P.; Choong, C.S.; Clemmons, D.; Dattani, M.; et al. Growth Hormone Research Society perspective on biomarkers of GH action in children and adults. Endocr. Connect. 2018, 7, R126–R134. [Google Scholar] [CrossRef] [Green Version]
- Katznelson, L.; Laws, E.R.; Melmed, S.; Molitch, M.E.; Murad, M.H.; Utz, A.; Wass, J.A.H. Acromegaly: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2014, 99, 3933–3951. [Google Scholar] [CrossRef] [PubMed]
- Pivonello, R.; Auriemma, R.S.; Grasso, L.F.S.; Pivonello, C.; Simeoli, C.; Patalano, R.; Galdiero, M.; Colao, A. Complications of acromegaly: Cardiovascular, respiratory and metabolic comorbidities. Pituitary 2017, 20, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, O.; Bex, M.; Kamenicky, P.; Mvoula, A.B.; Chanson, P.; Maiter, D. Prevalence and risk factors of impaired glucose tolerance and diabetes mellitus at diagnosis of acromegaly: A study in 148 patients. Pituitary 2013, 17, 81–89. [Google Scholar] [CrossRef]
- Ciresi, A.; Amato, M.C.; Pivonello, R.; Nazzari, E.; Grasso, L.F.; Minuto, F.; Ferone, D.; Colao, A.; Giordano, C. The Metabolic Profile in Active Acromegaly is Gender-Specific. J. Clin. Endocrinol. Metab. 2013, 98, E51–E59. [Google Scholar] [CrossRef] [Green Version]
- Dal, J.; List, E.O.; Jørgensen, J.O.L.; Berryman, D.E. Glucose and Fat Metabolism in Acromegaly: From Mice Models to Patient Care. Neuroendocrinology 2015, 103, 96–105. [Google Scholar] [CrossRef]
- del Rincon, J.-P.; Iida, K.; Gaylinn, B.D.; McCurdy, C.E.; Leitner, J.W.; Barbour, L.A.; Kopchick, J.J.; Friedman, J.E.; Draznin, B.; Thorner, M.O. Growth Hormone Regulation of p85α Expression and Phosphoinositide 3-Kinase Activity in Adipose Tissue. Diabetes 2007, 56, 1638–1646. [Google Scholar] [CrossRef] [Green Version]
- Kasayama, S.; Otsuki, M.; Takagi, M.; Saito, H.; Sumitani, S.; Kouhara, H.; Koga, M.; Saitoh, Y.; Ohnishi, T.; Arita, N. Impaired β-cell function in the presence of reduced insulin sensitivity determines glucose tolerance status in acromegalic patients. Clin. Endocrinol. 2000, 52, 549–555. [Google Scholar] [CrossRef]
- Kinoshita, Y.; Fujii, H.; Takeshita, A.; Taguchi, M.; Miyakawa, M.; Oyama, K.; Yamada, S.; Takeuchi, Y. Impaired glucose metabolism in Japanese patients with acromegaly is restored after successful pituitary surgery if pancreatic β-cell function is preserved. Eur. J. Endocrinol. 2011, 164, 467–473. [Google Scholar] [CrossRef] [Green Version]
- Frara, S.; Maffezzoni, F.; Mazziotti, G.; Giustina, A. Current and Emerging Aspects of Diabetes Mellitus in Acromegaly. Trends Endocrinol. Metab. 2016, 27, 470–483. [Google Scholar] [CrossRef]
- Melmed, S. Pituitary-Tumor Endocrinopathies. N. Engl. J. Med. 2020, 382, 937–950. [Google Scholar] [CrossRef] [PubMed]
- Colao, A.; Auriemma, R.S.; Galdiero, M.; Lombardi, G.; Pivonello, R. Effects of Initial Therapy for Five Years with Somatostatin Analogs for Acromegaly on Growth Hormone and Insulin-Like Growth Factor-I Levels, Tumor Shrinkage, and Cardiovascular Disease: A Prospective Study. J. Clin. Endocrinol. Metab. 2009, 94, 3746–3756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraù, F.; Albani, A.; Ciresi, A.; Giordano, C.; Cannavò, S. Diabetes Secondary to Acromegaly: Physiopathology, Clinical Features and Effects of Treatment. Front. Endocrinol. 2018, 9, 358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadelha, M.R.; Bronstein, M.D.; Brue, T.; Coculescu, M.; Fleseriu, M.; Guitelman, M.; Pronin, V.; Raverot, G.; Shimon, I.; Lievre, K.K.; et al. Pasireotide versus continued treatment with octreotide or lanreotide in patients with inadequately controlled acromegaly (PAOLA): A randomised, phase 3 trial. Lancet Diabetes Endocrinol. 2014, 2, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Hannon, A.M.; Thompson, C.J.; Sherlock, M. Diabetes in Patients with Acromegaly. Curr. Diabetes Rep. 2017, 17, 8. [Google Scholar] [CrossRef]
- Giustina, A.; Ambrosio, M.R.; Peccoz, P.B.; Bogazzi, F.; Cannavo’, S.; De Marinis, L.; De Menis, E.; Grottoli, S.; Pivonello, R. Use of Pegvisomant in acromegaly. An Italian Society of Endocrinology guideline. J. Endocrinol. Investig. 2014, 37, 1017–1030. [Google Scholar] [CrossRef] [Green Version]
- Pijl, H.; Ohashi, S.; Matsuda, M.; Miyazaki, Y.; Mahankali, A.; Kumar, V.; Pipek, R.; Iozzo, P.; Lancaster, J.L.; Cincotta, A.H.; et al. Bromocriptine: A novel approach to the treatment of type 2 diabetes. Diabetes Care 2000, 23, 1154–1161. [Google Scholar] [CrossRef] [Green Version]
- Giustina, A.; Barkan, A.; Beckers, A.; Biermasz, N.; Biller, B.M.K.; Boguszewski, C.; Bolanowski, M.; Bonert, V.; Bronstein, M.D.; Casanueva, F.F.; et al. A Consensus on the Diagnosis and Treatment of Acromegaly Comorbidities: An Update. J. Clin. Endocrinol. Metab. 2019, 105, e937–e946. [Google Scholar] [CrossRef]
- Zaina, A.; Prencipe, N.; Golden, E.; Berton, A.M.; Arad, E.; Abid, A.; Shehadeh, J.; Kassem, S.; Ghigo, E. How to position sodium-glucose co-transporter 2 inhibitors in the management of diabetes in acromegaly patients. Endocrine 2023, 80, 491–499. [Google Scholar] [CrossRef]
- Youssef, M.E.; Yahya, G.; Popoviciu, M.S.; Cavalu, S.; Abd-Eldayem, M.A.; Saber, S. Unlocking the Full Potential of SGLT2 Inhibitors: Expanding Applications beyond Glycemic Control. Int. J. Mol. Sci. 2023, 24, 6039. [Google Scholar] [CrossRef]
- Chaudhry, H.S.; Singh, G. Cushing Syndrome. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Raff, H.; Carroll, T. Cushing’s syndrome: From physiological principles to diagnosis and clinical care. J. Physiol. 2015, 593, 493–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiga, F.; Walker, J.J.; Terry, J.R.; Lightman, S.L. HPA Axis-Rhythms. Compr. Physiol. 2014, 4, 1273–1298. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, L. Hypothalamic–Pituitary–Adrenocortical Axis Regulation. Endocrinol. Metab. Clin. N. Am. 2005, 34, 271–292. [Google Scholar] [CrossRef] [PubMed]
- Nieman, L.K.; Biller, B.M.K.; Findling, J.W.; Newell-Price, J.; Savage, M.O.; Stewart, P.M.; Montori, V.M. The Diagnosis of Cushing’s Syndrome: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2008, 93, 1526–1540. [Google Scholar] [CrossRef] [PubMed]
- Badrick, E.; Kirschbaum, C.; Kumari, M. The Relationship between Smoking Status and Cortisol Secretion. J. Clin. Endocrinol. Metab. 2007, 92, 819–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, P.J.; Barth, J.H.; Freedman, D.B.; Perry, L.; Sheridan, B. Evidence for the Low Dose Dexamethasone Suppression Test to Screen for Cushing’s Syndrome—Recommendations for a Protocol for Biochemistry Laboratories. Ann. Clin. Biochem. Int. J. Biochem. Lab. Med. 1997, 34, 222–229. [Google Scholar] [CrossRef] [Green Version]
- Lacroix, A.; Feelders, R.A.; Stratakis, C.A.; Nieman, L.K. Cushing’s syndrome. Lancet 2015, 386, 913–927. [Google Scholar] [CrossRef]
- Tsagarakis, S.; Tsigos, C.; Vasiliou, V.; Tsiotra, P.; Kaskarelis, J.; Sotiropoulou, C.; Raptis, S.A.; Thalassinos, N. The Desmopressin and Combined CRH-Desmopressin Tests in the Differential Diagnosis of ACTH-Dependent Cushing’s Syndrome: Constraints Imposed by the Expression of V2 Vasopressin Receptors in Tumors with Ectopic ACTH Secretion. J. Clin. Endocrinol. Metab. 2002, 87, 1646–1653. [Google Scholar] [CrossRef] [Green Version]
- Fleseriu, M.; Auchus, R.; Bancos, I.; Ben-Shlomo, A.; Bertherat, J.; Biermasz, N.R.; Boguszewski, C.L.; Bronstein, M.D.; Buchfelder, M.; Carmichael, J.D.; et al. Consensus on diagnosis and management of Cushing’s disease: A guideline update. Lancet Diabetes Endocrinol. 2021, 9, 847–875. [Google Scholar] [CrossRef]
- Li, D.; El Kawkgi, O.M.; Henriquez, A.F.; Bancos, I. Cardiovascular risk and mortality in patients with active and treated hypercortisolism. Gland. Surg. 2020, 9, 43–58. [Google Scholar] [CrossRef]
- Scaroni, C.; Zilio, M.; Foti, M.; Boscaro, M. Glucose Metabolism Abnormalities in Cushing Syndrome: From Molecular Basis to Clinical Management. Endocr. Rev. 2017, 38, 189–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbot, M.; Ceccato, F.; Scaroni, C. Diabetes Mellitus Secondary to Cushing’s Disease. Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Seckl, J.R. Glucocorticoids and 11beta-Hydroxysteroid Dehydrogenase in Adipose Tissue. Recent Prog. Horm. Res. 2004, 59, 359–393. [Google Scholar] [CrossRef]
- Pivonello, R.; De Leo, M.; Vitale, P.; Cozzolino, A.; Simeoli, C.; De Martino, M.C.; Lombardi, G.; Colao, A. Pathophysiology of Diabetes Mellitus in Cushing’s Syndrome. Neuroendocrinology 2010, 92, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Brennan-Speranza, T.C.; Henneicke, H.; Gasparini, S.J.; Blankenstein, K.I.; Heinevetter, U.; Cogger, V.C.; Svistounov, D.; Zhang, Y.; Cooney, G.J.; Buttgereit, F.; et al. Osteoblasts mediate the adverse effects of glucocorticoids on fuel metabolism. J. Clin. Investig. 2012, 122, 4172–4189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieman, L.K.; Biller, B.M.K.; Findling, J.W.; Murad, M.H.; Newell-Price, J.; Savage, M.O.; Tabarin, A. Treatment of Cushing’s Syndrome: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2015, 100, 2807–2831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcuff, J.-B.; Young, J.; Masquefa-Giraud, P.; Chanson, P.; Baudin, E.; Tabarin, A. Rapid control of severe neoplastic hypercortisolism with metyrapone and ketoconazole. Eur. J. Endocrinol. 2015, 172, 473–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herndon, J.; Kaur, R.J.; Romportl, M.; Smith, E.; Koenigs, A.; Partlow, B.; Arteaga, L.; Bancos, I. The Effect of Curative Treatment on Hyperglycemia in Patients with Cushing Syndrome. J. Endocr. Soc. 2021, 6, bvab169. [Google Scholar] [CrossRef]
- Suh, S.; Park, M.K. Glucocorticoid-Induced Diabetes Mellitus: An Important but Overlooked Problem. Endocrinol. Metab. 2017, 32, 180–189. [Google Scholar] [CrossRef]
- Popoviciu, M.-S.; Păduraru, L.; Yahya, G.; Metwally, K.; Cavalu, S. Emerging Role of GLP-1 Agonists in Obesity: A Comprehensive Review of Randomised Controlled Trials. Int. J. Mol. Sci. 2023, 24, 10449. [Google Scholar] [CrossRef]
- Clore, J.N.; Thurby-Hay, L. Glucocorticoid-Induced Hyperglycemia. Endocr. Pract. 2009, 15, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Lansang, M.C.; Hustak, L.K. Glucocorticoid-induced diabetes and adrenal suppression: How to detect and manage them. Clevel. Clin. J. Med. 2011, 78, 748–756. [Google Scholar] [CrossRef]
- Dahia, P.L. Pheochromocytomas and Paragangliomas, Genetically Diverse and Minimalist, All at Once! Cancer Cell 2017, 31, 159–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrugia, F.-A.; Charalampopoulos, A. Pheochromocytoma. Endocr. Regul. 2019, 53, 191–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tevosian, S.G.; Ghayee, H.K. Pheochromocytomas and Paragangliomas. Endocrinol. Metab. Clin. N. Am. 2019, 48, 727–750. [Google Scholar] [CrossRef]
- Zuber, S.M.; Kantorovich, V.; Pacak, K. Hypertension in Pheochromocytoma: Characteristics and Treatment. Endocrinol. Metab. Clin. N. Am. 2011, 40, 295–311. [Google Scholar] [CrossRef] [Green Version]
- Manger, W.M. The Protean Manifestations of Pheochromocytoma. Horm. Metab. Res. 2009, 41, 658–663. [Google Scholar] [CrossRef]
- Jain, A.; Baracco, R.; Kapur, G. Pheochromocytoma and paraganglioma—An update on diagnosis, evaluation, and management. Pediatr. Nephrol. 2019, 35, 581–594. [Google Scholar] [CrossRef]
- Lenders, J.W.M.; Duh, Q.-Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.-P.; Grebe, S.K.G.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr. Pheochromocytoma and Paraganglioma: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef] [Green Version]
- La Batide-Alanore, A.; Chatellier, G.; Plouin, P.-F. Diabetes as a marker of pheochromocytoma in hypertensive patients. J. Hypertens. 2003, 21, 1703–1707. [Google Scholar] [CrossRef]
- Beninato, T.; Kluijfhout, W.P.; Drake, F.T.; Lim, J.; Kwon, J.S.; Xiong, M.; Shen, W.T.; Gosnell, J.E.; Liu, C.; Suh, I.; et al. Resection of Pheochromocytoma Improves Diabetes Mellitus in the Majority of Patients. Ann. Surg. Oncol. 2016, 24, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hodin, R.A.; Pandolfi, C.; Ruan, D.T.; McKenzie, T.J. Hypoglycemia after resection of pheochromocytoma. Surgery 2014, 156, 1404–1409. [Google Scholar] [CrossRef] [PubMed]
- Sherwin, R.S.; Sacca, L. Effect of epinephrine on glucose metabolism in humans: Contribution of the liver. Am. J. Physiol. Metab. 1984, 247, E157–E165. [Google Scholar] [CrossRef]
- Smith, T.J.; Hegedüs, L. Graves’ Disease. N. Engl. J. Med. 2016, 375, 1552–1565. [Google Scholar] [CrossRef] [Green Version]
- McLeod, D.S.A.; Cooper, D.S. The incidence and prevalence of thyroid autoimmunity. Endocrine 2012, 42, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Davies, T.F.; Andersen, S.; Latif, R.; Nagayama, Y.; Barbesino, G.; Brito, M.; Eckstein, A.K.; Stagnaro-Green, A.; Kahaly, G.J. Graves’ disease. Nat. Rev. Dis. Prim. 2020, 6, 1–23. [Google Scholar] [CrossRef]
- Bahn, R.S. Graves’ Ophthalmopathy. N. Engl. J. Med. 2010, 362, 726–738. [Google Scholar] [CrossRef] [Green Version]
- Wémeau, J.-L.; Klein, M.; Sadoul, J.-L.; Briet, C.; Vélayoudom-Céphise, F.-L. Graves’ disease: Introduction, epidemiology, endogenous and environmental pathogenic factors. Ann. d’Endocrinologie 2018, 79, 599–607. [Google Scholar] [CrossRef]
- Boelaert, K.; Torlinska, B.; Holder, R.L.; Franklyn, J.A. Older Subjects with Hyperthyroidism Present with a Paucity of Symptoms and Signs: A Large Cross-Sectional Study. J. Clin. Endocrinol. Metab. 2010, 95, 2715–2726. [Google Scholar] [CrossRef] [Green Version]
- Burch, H.B.; Cooper, D.S. Management of Graves Disease. JAMA 2015, 314, 2544–2554. [Google Scholar] [CrossRef]
- Díez, J.J. Hyperthyroidism in Patients Older than 55 Years: An Analysis of the Etiology and Management. Gerontology 2003, 49, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Rundle, F.F.; Wilson, C.W. Development and course of exophthalmos and ophthalmoplegia in Graves’ disease with special reference to the effect of thyroidectomy. Clin. Sci. 1945, 5. [Google Scholar]
- Antonelli, A.; Fallahi, P.; Elia, G.; Ragusa, F.; Paparo, S.R.; Ruffilli, I.; Patrizio, A.; Gonnella, D.; Giusti, C.; Virili, C.; et al. Graves’ disease: Clinical manifestations, immune pathogenesis (cytokines and chemokines) and therapy. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101388. [Google Scholar] [CrossRef]
- Drexhage, H.A. Are There More than Antibodies to the Thyroid-Stimulating Hormone Receptor that Meet the Eye in Graves’ Disease? Endocrinology 2006, 147, 9–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappelli, C.; Pirola, I.; De Martino, E.; Agosti, B.; Delbarba, A.; Castellano, M.; Rosei, E.A. The role of imaging in Graves’ disease: A cost-effectiveness analysis. Eur. J. Radiol. 2008, 65, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Kahaly, G.J.; Bartalena, L.; Hegedüs, L.; Leenhardt, L.; Poppe, K.; Pearce, S.H. 2018 European Thyroid Association Guideline for the Management of Graves’ Hyperthyroidism. Eur. Thyroid. J. 2018, 7, 167–186. [Google Scholar] [CrossRef]
- Subekti, I.; Pramono, L.A. Current Diagnosis and Management of Graves’ Disease. Acta Medica Indones. 2018, 50, 177–182. [Google Scholar]
- Corvilain, B.; Hamy, A.; Brunaud, L.; Borson-Chazot, F.; Orgiazzi, J.; Hachmi, L.B.; Semrouni, M.; Rodien, P.; Lussey-Lepoutre, C. Treatment of adult Graves’ disease. Ann. d’Endocrinologie 2018, 79, 618–635. [Google Scholar] [CrossRef] [Green Version]
- Kahaly, G.J. Management of Graves Thyroidal and Extrathyroidal Disease: An Update. J. Clin. Endocrinol. Metab. 2020, 105, 3704–3720. [Google Scholar] [CrossRef]
- Bartalena, L.; Baldeschi, L.; Boboridis, K.; Eckstein, A.; Kahaly, G.J.; Marcocci, C.; Perros, P.; Salvi, M.; Wiersinga, W.M.; on behalf of the European Group on Graves’ Orbitopathy (EUGOGO). The 2016 European Thyroid Association/European Group on Graves’ Orbitopathy Guidelines for the Management of Graves’ Orbitopathy. Eur. Thyroid. J. 2016, 5, 9–26. [Google Scholar] [CrossRef] [Green Version]
- Song, E.; Koo, M.J.; Noh, E.; Hwang, S.Y.; Park, M.J.; A Kim, J.; Roh, E.; Choi, K.M.; Baik, S.H.; Cho, G.J.; et al. Risk of Diabetes in Patients with Long-Standing Graves’ Disease: A Longitudinal Study. Endocrinol. Metab. 2021, 36, 1277–1286. [Google Scholar] [CrossRef]
- O’Meara, N.M.; Blackman, J.D.; Sturis, J.; Polonsky, K.S. Alterations in the kinetics of C-peptide and insulin secretion in hyperthyroidism. J. Clin. Endocrinol. Metab. 1993, 76, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Bech, K.; Damsbo, P.; Eldrup, E.; Beck-Nielsen, H.; Røder, M.E.; Hartling, S.G.; Vølund, A.; Madsbad, S. β-Cell function and glucose and lipid oxidation in Graves’ disease. Clin. Endocrinol. 1996, 44, 59–66. [Google Scholar] [CrossRef]
- Nishi, M. Diabetes mellitus and thyroid diseases. Diabetol. Int. 2018, 9, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Duntas, L.H.; Orgiazzi, J.; Brabant, G. The interface between thyroid and diabetes mellitus. Clin. Endocrinol. 2011, 75, 1–9. [Google Scholar] [CrossRef]
- Hage, M.; Zantout, M.S.; Azar, S.T. Thyroid Disorders and Diabetes Mellitus. J. Thyroid. Res. 2011, 2011, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Maxon, H.R.; Kreines, K.W.; Goldsmith, R.E.; Knowles, H.C. Long-Term Observations of Glucose Tolerance in Thyrotoxic Patients. Arch. Intern. Med. 1975, 135, 1477–1480. [Google Scholar] [CrossRef]
- Perumal, N.L.; Selvi, J.; Sridharan, K.; Sahoo, J.; Kamalanathan, S. Insulin Sensitivity and Beta-Cell Function in Graves’ Disease and Their Changes with the Carbimazole-Induced Euthyroid State. Eur. Thyroid. J. 2019, 8, 59–63. [Google Scholar] [CrossRef]
- Biondi, B.; Kahaly, G.J.; Robertson, R.P. Thyroid Dysfunction and Diabetes Mellitus: Two Closely Associated Disorders. Endocr. Rev. 2019, 40, 789–824. [Google Scholar] [CrossRef] [Green Version]
- Anil, C.; Kut, A.; Atesagaoglu, B.; Nar, A.; Tutuncu, N.B.; Gursoy, A. Metformin Decreases Thyroid Volume and Nodule Size in Subjects with Insulin Resistance: A Preliminary Study. Med. Princ. Pract. 2016, 25, 233–236. [Google Scholar] [CrossRef]
- Funder, J.W.; Carey, R.M.; Mantero, F.; Murad, M.H.; Reincke, M.; Shibata, H.; Stowasser, M.; Young, W.F. The Management of Primary Aldosteronism: Case Detection, Diagnosis, and Treatment: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2016, 101, 1889–1916. [Google Scholar] [CrossRef] [PubMed]
- Calhoun, D.A.; Nishizaka, M.K.; Zaman, M.A.; Thakkar, R.B.; Weissmann, P. Hyperaldosteronism among Black and White Subjects With Resistant Hypertension. Hypertension 2002, 40, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Carey, R.M.; Siragy, H.M. Newly Recognized Components of the Renin-Angiotensin System: Potential Roles in Cardiovascular and Renal Regulation. Endocr. Rev. 2003, 24, 261–271. [Google Scholar] [CrossRef] [Green Version]
- Carey, R.M. Aldosterone and cardiovascular disease. Curr. Opin. Endocrinol. Diabetes 2010, 17, 194–198. [Google Scholar] [CrossRef]
- Carey, R.M. Primary aldosteronism. J. Surg. Oncol. 2012, 106, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Mulatero, P.; Stowasser, M.; Loh, K.-C.; Fardella, C.E.; Gordon, R.D.; Mosso, L.; Gomez-Sanchez, C.E.; Veglio, F.; Young, W.F. Increased Diagnosis of Primary Aldosteronism, Including Surgically Correctable Forms, in Centers from Five Continents. J. Clin. Endocrinol. Metab. 2004, 89, 1045–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calhoun, D.A.; Jones, D.; Textor, S.; Goff, D.C.; Murphy, T.P.; Toto, R.D.; White, A.; Cushman, W.C.; White, W.; Sica, D.; et al. Resistant Hypertension: Diagnosis, Evaluation, and Treatment. Hypertension 2008, 51, 1403–1419. [Google Scholar] [CrossRef] [Green Version]
- Rossi, G.-P.; Sechi, L.A.; Giacchetti, G.; Ronconi, V.; Strazzullo, P.; Funder, J.W. Primary aldosteronism: Cardiovascular, renal and metabolic implications. Trends Endocrinol. Metab. 2008, 19, 88–90. [Google Scholar] [CrossRef]
- Monticone, S.; D’Ascenzo, F.; Moretti, C.; Williams, T.A.; Veglio, F.; Gaita, F.; Mulatero, P. Cardiovascular events and target organ damage in primary aldosteronism compared with essential hypertension: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2018, 6, 41–50. [Google Scholar] [CrossRef]
- Sowers, J.R.; Whaley-Connell, D.A.; Epstein, M. Narrative Review: The Emerging Clinical Implications of the Role of Aldosterone in the Metabolic Syndrome and Resistant Hypertension. Ann. Intern. Med. 2009, 150, 776–783. [Google Scholar] [CrossRef]
- Tuck, M.L.; Sowers, J.; Dornfeld, L.; Kledzik, G.; Maxwell, M. The Effect of Weight Reduction on Blood Pressure, Plasma Renin Activity, and Plasma Aldosterone Levels in Obese Patients. N. Engl. J. Med. 1981, 304, 930–933. [Google Scholar] [CrossRef]
- Gregoire, J.R. Adjustment of the Osmostat in Primary Aldosteronism. Mayo Clin. Proc. 1994, 69, 1108–1110. [Google Scholar] [CrossRef] [PubMed]
- Tomaschitz, A.; Pilz, S. Aldosterone to Renin Ratio—A Reliable Screening Tool for Primary Aldosteronism? Horm. Metab. Res. 2010, 42, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.T.; Elaraj, D. Evaluation and Management of Primary Hyperaldosteronism. Surg. Clin. N. Am. 2019, 99, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Mulatero, P.; Monticone, S.; Bertello, C.; Mengozzi, G.; Tizzani, D.; Iannaccone, A.; Veglio, F. Confirmatory Tests in the Diagnosis of Primary Aldosteronism. Horm. Metab. Res. 2010, 42, 406–410. [Google Scholar] [CrossRef]
- Conn, J.W.; Knopf, R.F.; Nesbit, R.M. Clinical characteristics of primary aldosteronism from an analysis of 145 cases. Am. J. Surg. 1964, 107, 159–172. [Google Scholar] [CrossRef]
- Luther, J.M. Effects of aldosterone on insulin sensitivity and secretion. Steroids 2014, 91, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Stavropoulos, K.; Imprialos, K.; Papademetriou, V.; Faselis, C.; Tsioufis, K.; Dimitriadis, K.; Doumas, M. Primary Aldosteronism: Novel Insights. Curr. Hypertens. Rev. 2020, 16, 19–23. [Google Scholar] [CrossRef]
- Young, W.F.; Stanson, A.W.; Thompson, G.B.; Grant, C.S.; Farley, D.R.; van Heerden, J.A. Role for adrenal venous sampling in primary aldosteronism. Surgery 2004, 136, 1227–1235. [Google Scholar] [CrossRef]
- Mulatero, P.; Bertello, C.; Rossato, D.; Mengozzi, G.; Milan, A.; Garrone, C.; Giraudo, G.; Passarino, G.; Garabello, D.; Verhovez, A.; et al. Roles of Clinical Criteria, Computed Tomography Scan, and Adrenal Vein Sampling in Differential Diagnosis of Primary Aldosteronism Subtypes. J. Clin. Endocrinol. Metab. 2008, 93, 1366–1371. [Google Scholar] [CrossRef]
- Zennaro, M.-C.; Boulkroun, S.; Fernandes-Rosa, F.L. Pathogenesis and treatment of primary aldosteronism. Nat. Rev. Endocrinol. 2020, 16, 578–589. [Google Scholar] [CrossRef]
- Steichen, O.; Zinzindohoué, F.; Plouin, P.-F.; Amar, L. Outcomes of Adrenalectomy in Patients with Unilateral Primary Aldosteronism: A Review. Horm. Metab. Res. 2012, 44, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Rossi, G.P.; Cesari, M.; Cuspidi, C.; Maiolino, G.; Cicala, M.V.; Bisogni, V.; Mantero, F.; Pessina, A.C. Long-Term Control of Arterial Hypertension and Regression of Left Ventricular Hypertrophy with Treatment of Primary Aldosteronism. Hypertension 2013, 62, 62–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, V.-C.; Wang, S.-M.; Chang, C.-H.; Hu, Y.-H.; Lin, L.-Y.; Lin, Y.-H.; Chueh, S.-C.J.; Chen, L.; Wu, K.-D. Long term outcome of Aldosteronism after target treatments. Sci. Rep. 2016, 6, 32103. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-H.; Lin, L.-Y.; Chen, A.; Wu, X.-M.; Lee, J.-K.; Su, T.-C.; Wu, V.-C.; Chueh, S.-C.; Lin, W.-C.; Lo, M.-T.; et al. Adrenalectomy improves increased carotid intima-media thickness and arterial stiffness in patients with aldosterone producing adenoma. Atherosclerosis 2011, 221, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Komada, H.; Hirota, Y.; So, A.; Nakamura, T.; Okuno, Y.; Fukuoka, H.; Iguchi, G.; Takahashi, Y.; Sakaguchi, K.; Ogawa, W. Insulin secretion and sensitivity before and after surgical treatment for aldosterone-producing adenoma. Diabetes Metab. 2019, 46, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Catena, C.; Lapenna, R.; Baroselli, S.; Nadalini, E.; Colussi, G.; Novello, M.; Favret, G.; Melis, A.; Cavarape, A.; Sechi, L.A. Insulin Sensitivity in Patients with Primary Aldosteronism: A Follow-Up Study. J. Clin. Endocrinol. Metab. 2006, 91, 3457–3463. [Google Scholar] [CrossRef]
- de Herder, W.W.; Zandee, W.T.; Hofland, J. Somatostatinoma. In Endotext [Internet]; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Hofland, J., Dungan, K., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- de Herder, W.W.; Rehfeld, J.F.; Kidd, M.; Modlin, I.M. A short history of neuroendocrine tumours and their peptide hormones. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 30, 3–17. [Google Scholar] [CrossRef]
- Rorsman, P.; Huising, M.O. The somatostatin-secreting pancreatic δ-cell in health and disease. Nat. Rev. Endocrinol. 2018, 14, 404–414. [Google Scholar] [CrossRef]
- Kazanjian, K.K.; Reber, H.A.; Hines, O.J. Resection of Pancreatic Neuroendocrine Tumors. Arch. Surg. 2006, 141, 765–770. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.-P.; Liu, Y.-Y.; Xiao, H.-P.; Li, Y.-B.; Wang, L.-T.; Xiao, P. Pancreatic somatostatinoma characterized by extreme hypoglycemia. Chin. Med. J. 2009, 122, 1709–1712. [Google Scholar]
- Wright, J.; Abolfathi, A.; Penman, E.; Marks, V. Pancreatic Somatostatinoma Presenting with Hypoglycaemia. Clin. Endocrinol. 1980, 12, 603–608. [Google Scholar] [CrossRef] [PubMed]
- O’Grady, H.; Conlon, K. Pancreatic neuroendocrine tumours. Eur. J. Surg. Oncol. (EJSO) 2008, 34, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.L.; Rehfeld, J.F. Gastric Peptides-Gastrin and Somatostatin. Buy Physiol. 2019, 10, 197–228. [Google Scholar] [CrossRef]
- Anderson, C.W.; Bennett, J.J. Clinical Presentation and Diagnosis of Pancreatic Neuroendocrine Tumors. Surg. Oncol. Clin. N. Am. 2016, 25, 363–374. [Google Scholar] [CrossRef]
- Elangovan, A.; Zulfiqar, H. Somatostatinoma. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Mansour, J.C.; Chen, H. Pancreatic endocrine tumors. J. Surg. Res. 2004, 120, 139–161. [Google Scholar] [CrossRef]
- O’Brien, T.D.; Chejfec, G.; Prinz, A.R. Clinical features of duodenal somatostatinomas. Surgery 1993, 114. [Google Scholar]
- Tanaka, S.; Yamasaki, S.; Matsushita, H.; Ozawa, Y.; Kurosaki, A.; Takeuchi, K.; Hoshihara, Y.; Doi, T.; Watanabe, G.; Kawaminami, K. Duodenal somatostatinoma: A case report and review of 31 cases with special reference to the relationship between tumor size and metastasis. Pathol. Int. 2000, 50, 146–152. [Google Scholar] [CrossRef]
- Gallo, M.; Ruggeri, R.M.; Muscogiuri, G.; Pizza, G.; Faggiano, A.; Colao, A. Diabetes and pancreatic neuroendocrine tumours: Which interplays, if any? Cancer Treat. Rev. 2018, 67, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mozell, E.; Woltering, E.A.; Stenzel, P.; Rösch, J.; O’Dorisio, T.M. Functional endocrine tumors of the pancreas: Clinical presentation, diagnosis, and treatment. Curr. Probl. Surg. 1990, 27, 309–386. [Google Scholar] [CrossRef]
- Ito, T.; Igarashi, H.; Jensen, R.T. Pancreatic neuroendocrine tumors: Clinical features, diagnosis and medical treatment: Advances. Best Pract. Res. Clin. Gastroenterol. 2012, 26, 737–753. [Google Scholar] [CrossRef] [Green Version]
- Ramage, J.K.; Ahmed, A.; Ardill, J.; Bax, N.; Breen, D.J.; E Caplin, M.; Corrie, P.; Davar, J.; Davies, A.H.; Lewington, V.; et al. Guidelines for the management of gastroenteropancreatic neuroendocrine (including carcinoid) tumours (NETs). Gut 2011, 61, 6–32. [Google Scholar] [CrossRef] [PubMed]
- Kunz, P.L.; Reidy-Lagunes, D.; Anthony, L.B.; Bertino, E.M.; Brendtro, K.B.; Chan, J.A.; Chen, H.; Jensen, R.T.; Kim, M.K.M.; Klimstra, D.S.; et al. Consensus Guidelines for the Management and Treatment of Neuroendocrine Tumors. Pancreas 2013, 42, 557–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, J.; Thorn, C.; Spalding, D.; Williamson, R. Pancreatic and peripancreatic somatostatinomas. Ind. Mark. Manag. 2011, 93, 356–360. [Google Scholar] [CrossRef]
- Öberg, K.; Knigge, U.; Kwekkeboom, D.; Perren, A.; on behalf of the ESMO Guidelines Working Group. Neuroendocrine gastro-entero-pancreatic tumors: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2012, 23, vii124–vii130. [Google Scholar] [CrossRef]
- Angeletti, S.; Corleto, V.D.; Schillaci, O.; Marignani, M.; Annibale, B.; Moretti, A.; Silecchia, G.; Scopinaro, F.; Basso, N.; Bordi, C.; et al. Use of the somatostatin analogue octreotide to localise and manage somatostatin-producing tumours. Gut 1998, 42, 792–794. [Google Scholar] [CrossRef] [Green Version]
- Caplin, M.E.; Pavel, M.; Ćwikła, J.B.; Phan, A.T.; Raderer, M.; Sedláčková, E.; Cadiot, G.; Wolin, E.M.; Capdevila, J.; Wall, L.; et al. Lanreotide in Metastatic Enteropancreatic Neuroendocrine Tumors. N. Engl. J. Med. 2014, 371, 224–233. [Google Scholar] [CrossRef]
- Al-Toubah, T.; Schell, M.J.; Cives, M.; Zhou, J.-M.; Soares, H.P.; Strosberg, J.R. A Phase II Study of Ibrutinib in Advanced Neuroendocrine Neoplasms. Neuroendocrinology 2019, 110, 377–383. [Google Scholar] [CrossRef]
- Ellison, T.A.; Edil, B.H. The Current Management of Pancreatic Neuroendocrine Tumors. Adv. Surg. 2012, 46, 283–296. [Google Scholar] [CrossRef]
- Adeva-Andany, M.M.; Rañal-Muíño, E.; Vila-Altesor, M.; Fernández-Fernández, C.; Funcasta-Calderón, R.; Castro-Quintela, E. Dietary habits contribute to define the risk of type 2 diabetes in humans. Clin. Nutr. ESPEN 2019, 34, 8–17. [Google Scholar] [CrossRef]
- Sandru, F.; Carsote, M.; Albu, S.E.; Valea, A.; Petca, A.; Dumitrascu, M.C. Glucagonoma: From skin lesions to the neuroendocrine component (Review). Exp. Ther. Med. 2020, 20, 3389–3393. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H. Glucagon and the Insulin:Glucagon Ratio in Diabetes and Other Catabolic Illnesses. Diabetes 1971, 20, 834–838. [Google Scholar] [CrossRef]
- Reaven, G.M.; Chen, Y.-D.I.; Golay, A.; Swislocki, A.L.M.; Jaspan, J.B. Documentation of Hyperglucagonemia throughout the Day in Nonobese and Obese Patients with Noninsulin-Dependent Diabetes Mellitus*. J. Clin. Endocrinol. Metab. 1987, 64, 106–110. [Google Scholar] [CrossRef]
- Knop, F.K.; Aaboe, K.; Vilsbøll, T.; Vølund, A.; Holst, J.J.; Krarup, T.; Madsbad, S. Impaired incretin effect and fasting hyperglucagonaemia characterizing type 2 diabetic subjects are early signs of dysmetabolism in obesity. Diabetes Obes. Metab. 2011, 14, 500–510. [Google Scholar] [CrossRef]
- Adeva-Andany, M.M.; Funcasta-Calderón, R.; Fernández-Fernández, C.; Castro-Quintela, E.; Carneiro-Freire, N. Metabolic effects of glucagon in humans. J. Clin. Transl. Endocrinol. 2018, 15, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Öberg, K. Management of functional neuroendocrine tumors of the pancreas. Gland. Surg. 2018, 7, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavalu, S.; Popa, A.; Bratu, I.; Borodi, G.; Maghiar, A. New Evidences of Key Factors Involved in “Silent Stones” Etiopathogenesis and Trace Elements: Microscopic, Spectroscopic, and Biochemical Approach. Biol. Trace Element Res. 2015, 168, 311–320. [Google Scholar] [CrossRef]
- Sandhu, S.; Jialal, I. Glucagonoma Syndrome. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- McGavran, M.H.; Unger, R.H.; Recant, L.; Polk, H.C.; Kilo, C.; Levin, M.E. A Glucagon-Secreting Alpha-Cell Carcinoma of the Pancreas. N. Engl. J. Med. 1966, 274, 1408–1413. [Google Scholar] [CrossRef]
- Kindmark, H.; Sundin, A.; Granberg, D.; Dunder, K.; Skogseid, B.; Janson, E.T.; Welin, S.; Öberg, K.; Eriksson, B. Endocrine pancreatic tumors with glucagon hypersecretion: A retrospective study of 23 cases during 20 years. Med. Oncol. 2007, 24, 330–337. [Google Scholar] [CrossRef]
- Yao, J.C.; Eisner, M.P.; Leary, C.; Dagohoy, C.; Phan, A.; Rashid, A.; Hassan, M.; Evans, D.B. Population-Based Study of Islet Cell Carcinoma. Ann. Surg. Oncol. 2007, 14, 3492–3500. [Google Scholar] [CrossRef] [Green Version]
- Filho, F.d.A.C.; Feitosa, R.G.F.; Fechine, C.O.C.; de Matos, C.M.M.; Cardoso, A.L.; Cardoso, D.L. Glucagonoma syndrome associated with necrolytic migratory erythema. Rev. Assoc. Méd. Bras. 2015, 61, 203–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wermers, R.A.; Fatourechi, V.; Wynne, A.G.; Kvols, L.K.; Lloyd, R.V. The Glucagonoma Syndrome Clinical and Pathologic Features in 21 Patients. Medicine 1996, 75, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zheng, S.; Yang, G.; Xiong, G.; Cao, Z.; Feng, M.; Zhang, T.; Zhao, Y. Glucagonoma and the glucagonoma syndrome (Review). Oncol. Lett. 2017, 15, 2749–2755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobo, I.; Carvalho, A.; Amaral, C.; Machado, S.; Carvalho, R. Glucagonoma syndrome and necrolytic migratory erythema. Int. J. Dermatol. 2010, 49, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Toberer, F.; Hartschuh, W.; Wiedemeyer, K. Glucagonoma-Associated Necrolytic Migratory Erythema: The Broad Spectrum of the Clinical and Histopathological Findings and Clues to the Diagnosis. Am. J. Dermatopathol. 2019, 41, e29–e32. [Google Scholar] [CrossRef]
- Soga, J.; Yakuwa, Y. Glucagonomas/diabetico-dermatogenic syndrome (DDS): A statistical evaluation of 407 reported cases. J. Hepato-Biliary-Pancreatic Surg. 1998, 5, 312–319. [Google Scholar] [CrossRef]
- Stacpoole, P.W. The Glucagonoma Syndrome: Clinical Features, Diagnosis, and Treatment. Endocr. Rev. 1981, 2, 347–361. [Google Scholar] [CrossRef]
- Wu, S.-L.; Bai, J.-G.; Xu, J.; Ma, Q.-Y.; Wu, Z. Necrolytic migratory erythema as the first manifestation of pancreatic neuroendocrine tumor. World J. Surg. Oncol. 2014, 12, 220. [Google Scholar] [CrossRef] [Green Version]
- Lv, W.-F.; Han, J.-K.; Liu, X.; Wang, S.-C.; Pan, B.; Xu, A. Imaging features of glucagonoma syndrome: A case report and review of the literature. Oncol. Lett. 2015, 9, 1579–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanakis, G.; Kaltsas, G. Biochemical markers for gastroenteropancreatic neuroendocrine tumours (GEP-NETs). Best Pract. Res. Clin. Gastroenterol. 2012, 26, 791–802. [Google Scholar] [CrossRef]
- Partelli, S.; Bartsch, D.K.; Capdevila, J.; Chen, J.; Knigge, U.; Niederle, B.; van Dijkum, E.J.N.; Pape, U.-F.; Pascher, A.; Ramage, J.; et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumours: Surgery for Small Intestinal and Pancreatic Neuroendocrine Tumours. Neuroendocrinology 2017, 105, 255–265. [Google Scholar] [CrossRef]
- Kimura, W.; Tezuka, K.; Hirai, I. Surgical management of pancreatic neuroendocrine tumors. Surg. Today 2011, 41, 1332–1343. [Google Scholar] [CrossRef]
- Ferrarese, A.; Borello, A.; Gentile, V.; Bindi, M.; Ferrara, Y.; Solej, M.; Martino, V.; Nano, M. Meso-pancreatectomy for pancreatic neuroendocrine tumor. Int. J. Surg. 2014, 12, S123–S125. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.C.; Hassan, M.; Phan, A.; Dagohoy, C.; Leary, C.; Mares, J.E.; Abdalla, E.K.; Fleming, J.B.; Vauthey, J.N.; Rashid, A.; et al. One hundred years after “carcinoid”: Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J. Clin. Oncol. 2008, 26, 3063–3072. [Google Scholar] [PubMed] [Green Version]
- Yalcin, S.; Oyan, B.; Bayraktar, Y. Current medical treatment of pancreatic neuroendocrine tumors. Hepato-Gastroenterology 2007, 54. [Google Scholar]
- Kimbara, S.; Fujiwara, Y.; Toyoda, M.; Chayahara, N.; Imamura, Y.; Kiyota, N.; Mukohara, T.; Fukunaga, A.; Oka, M.; Nishigori, C.; et al. Rapid improvement of glucagonoma-related necrolytic migratory erythema with octreotide. Clin. J. Gastroenterol. 2014, 7, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Eldor, R.; Glaser, B.; Fraenkel, M.; Doviner, V.; Salmon, A.; Gross, D.J. Glucagonoma and the glucagonoma syndrome—Cumulative experience with an elusive endocrine tumour. Clin. Endocrinol. 2011, 74, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, D.; Kuang, T.; Rong, Y.; Lou, W. Glucagonoma syndrome: Report of one case. Transl. Gastroenterol. Hepatol. 2016, 1, 70. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Treatment | References |
|---|---|---|
| Acromegaly |
| [21] |
| [8,22,23] | |
| [20] | |
| [8] | |
| [29] | |
| Cushing’s Syndrome |
| [48] |
| [48] | |
| [49] | |
| Pheochromocytoma |
| [61] |
| [65] | |
| Graves’ disease |
| [78,79] |
| [81] | |
| [78] | |
| [80] | |
| [82] | |
| Primary aldosteronism |
| [112] |
| [113] | |
| Somatostatinoma |
| [137,138] |
| [139,142] | |
| Glucagonoma |
| [169] |
| [172] | |
| [173] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popoviciu, M.S.; Paduraru, L.; Nutas, R.M.; Ujoc, A.M.; Yahya, G.; Metwally, K.; Cavalu, S. Diabetes Mellitus Secondary to Endocrine Diseases: An Update of Diagnostic and Treatment Particularities. Int. J. Mol. Sci. 2023, 24, 12676. https://doi.org/10.3390/ijms241612676
Popoviciu MS, Paduraru L, Nutas RM, Ujoc AM, Yahya G, Metwally K, Cavalu S. Diabetes Mellitus Secondary to Endocrine Diseases: An Update of Diagnostic and Treatment Particularities. International Journal of Molecular Sciences. 2023; 24(16):12676. https://doi.org/10.3390/ijms241612676
Chicago/Turabian StylePopoviciu, Mihaela Simona, Lorena Paduraru, Raluca Marinela Nutas, Alexandra Maria Ujoc, Galal Yahya, Kamel Metwally, and Simona Cavalu. 2023. "Diabetes Mellitus Secondary to Endocrine Diseases: An Update of Diagnostic and Treatment Particularities" International Journal of Molecular Sciences 24, no. 16: 12676. https://doi.org/10.3390/ijms241612676
APA StylePopoviciu, M. S., Paduraru, L., Nutas, R. M., Ujoc, A. M., Yahya, G., Metwally, K., & Cavalu, S. (2023). Diabetes Mellitus Secondary to Endocrine Diseases: An Update of Diagnostic and Treatment Particularities. International Journal of Molecular Sciences, 24(16), 12676. https://doi.org/10.3390/ijms241612676