
New Therapeutics for Heart Failure: Focusing on cGMP Signaling


Abstract
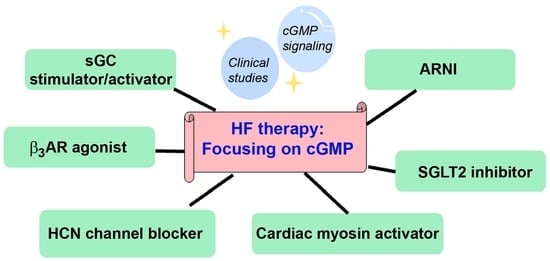
:
1. Introduction
2. Cyclic Guanosine Monophosphate (cGMP) Signaling
2.1. cGMP Production
2.2. cGMP Signaling Effector Molecules
2.2.1. cGMP-Dependent Protein Kinases (PKGs)
2.2.2. Phosphodiesterases (PDEs)
2.2.3. Cyclic Nucleotide-Regulated Cation Channels
2.2.4. Nitric Oxide (NO)
2.3. cGMP Elimination Process
3. New Drugs for the Treatment of HF
3.1. Angiotensin Receptor Blocker/Neprilysin Inhibitor (ARNI)
3.2. Sodium-Glucose Co-Transporter-2 (SGLT2) Inhibitors
3.3. Hyperpolarization-Activated Cyclic Nucleotide-Gated (HCN) Channel Blocker
3.4. Cardiac Myosin Activators
3.5. Soluble Guanylyl Cyclase (sGC) Stimulator/Activator
3.6. SGLT2 Inhibitors and cGMP Signaling
4. β Adrenergic Receptors (βARs) and NO System Signaling
5. Candidate Molecular Therapeutic Targets for HF and Cardiac Remodeling
5.1. Free Fatty Acid Receptors (FFARs)
5.2. Cannabinoids
5.3. Transient Receptor Potential Cation Channel Subfamily V Member 1 (TRPV1) Channel
5.4. Aquaporins
6. Clinical Studies of Drugs and Therapeutic Targets for HF Treatment
6.1. Clinical Studies of ARNI
6.2. Clinical Studies of HCN Channel Blocker
6.3. Clinical Studies of Cardiac Myosin Activators

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Study Population | Treatment | Primary and Secondary Endpoints | Main Findings and Conclusions |
|---|---|---|---|---|
| Danicamtiv [123] |
| Danicamtiv 50, 75, or 100 mg BID or placebo for 7 days | Primary:
|
|
| Omecamtiv (COSMIC-HF trial) [124,125] |
| Omecamtiv 25 mg BID (fixed-dose), 25 mg BID titrated to 50 mg BID or placebo for 20 weeks | Primary:
|
|
|
| |||
| Omecamtiv (METEORIC-HF trial) [127] |
| Omecamtiv 25, 37.5, or 50 mg BID or placebo for 20 weeks | Primary:
|
|
| Omecamtiv (GALACTIC-HF trial) [128,129,130] |
| Omecamtiv 25, 37.5, or 50 mg BID based on target plasma level or placebo for 20 weeks | Primary:
|
|
|
| |||
|
|
6.4. Clinical Studies of SGLT2 Inhibitors
6.5. Clinical Studies of Soluble Guanylyl Cyclase (sGC) Stimulators/Activators
| Drug | Study Population | Treatment | Primary and Secondary Endpoints | Main Findings and Conclusions |
|---|---|---|---|---|
| Praliciguat (CAPACITY-HFpEF) [69] |
| Praliciguat 40 mg OD or placebo for 12 weeks | Primary:
|
|
| Riociguat (LEPHT) [141] |
| Riociguat 0.1, 1, or 2 mg TID or placebo for 16 weeks | Primary:
|
|
| Vericiguat (SOCRATES-REDUCED trial) [138] |
| Vericiguat 1.25, 2.5, 5, or 10 mg OD or placebo for 12 weeks | Primary:
|
|
| Vericiguat (SOCRATES-PRESERVED trial) [139] |
| Vericiguat 1.25–10 mg OD or placebo for 12 weeks | Primary:
|
|
| Vericiguat (VITALITY-HFpEF trial) [68] |
| Vericiguat up-titrated to 10 or 15 mg OD or placebo for 24 weeks | Primary:
|
|
| Vericiguat (VICTORIA trial) [66,140] |
| Vericiguat 10 mg OD or placebo | Primary:
|
|
|
|
6.6. Clinical Studies of β3 Adrenergic Receptor (β3AR) Agonists
7. Conclusions and Further Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: A report of the American College of Cardiology/American Heart Association joint committee on clinical practice guidelines. Circulation 2022, 145, e895–e1032. [Google Scholar] [CrossRef] [PubMed]
- Blanton, R.M. cGMP signaling and modulation in heart failure. J. Cardiovasc. Pharmacol. 2020, 75, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Petraina, A.; Nogales, C.; Krahn, T.; Mucke, H.; Lüscher, T.F.; Fischmeister, R.; Kass, D.A.; Burnett, J.C.; Hobbs, A.J.; Schmidt, H.H.H.W. Cyclic GMP modulating drugs in cardiovascular diseases: Mechanism-based network pharmacology. Cardiovasc. Res. 2022, 118, 2085–2102. [Google Scholar] [CrossRef]
- Bauersachs, J. Heart failure drug treatment: The fantastic four. Eur. Heart J. 2021, 42, 681–683. [Google Scholar] [CrossRef]
- Krishnan, S.M.; Kraehling, J.R.; Eitner, F.; Bénardeau, A.; Sandner, P. The Impact of the nitric oxide (NO)/soluble guanylyl cyclase (sGC) signaling cascade on kidney health and disease: A preclinical perspective. Int. J. Mol. Sci. 2018, 19, 1712. [Google Scholar] [CrossRef]
- Friebe, A.; Sandner, P.; Schmidtko, A. cGMP: A unique 2nd messenger molecule—Recent developments in cGMP research and development. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 393, 287–302. [Google Scholar] [CrossRef] [PubMed]
- Friebe, A.; Kraehling, J.R.; Russwurm, M.; Sandner, P.; Schmidtko, A. The 10th international conference on cGMP 2022: Recent trends in cGMP research and development-meeting report. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2023, 396, 1669–1686. [Google Scholar] [CrossRef]
- Francis, S.H.; Busch, J.L.; Corbin, J.D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef]
- Prysyazhna, O.; Eaton, P. Redox regulation of cGMP-dependent protein kinase Iα in the cardiovascular system. Front. Pharmacol. 2015, 6, 139. [Google Scholar] [CrossRef]
- Ramsey, I.S.; Delling, M.; Clapham, D.E. An introduction to TRP channels. Annu. Rev. Physiol. 2006, 68, 619–647. [Google Scholar] [CrossRef]
- Onohara, N.; Nishida, M.; Inoue, R.; Kobayashi, H.; Sumimoto, H.; Sato, Y.; Mori, Y.; Nagao, T.; Kurose, H. TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J. 2006, 25, 5305–5316. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Sayed, N.; Pyriochou, A.; Roussos, C.; Fulton, D.; Beuve, A.; Papapetropoulos, A. Protein kinase G phosphorylates soluble guanylyl cyclase on serine 64 and inhibits its activity. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1803–1810. [Google Scholar] [CrossRef] [PubMed]
- Thoonen, R.; Giovanni, S.; Govindan, S.; Lee, D.I.; Wang, G.R.; Calamaras, T.D.; Takimoto, E.; Kass, D.A.; Sadayappan, S.; Blanton, R.M. Molecular screen identifies cardiac myosin-binding protein-C as a protein kinase G-Iα substrate. Circ. Heart Fail. 2015, 8, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Blanton, R.M.; Takimoto, E.; Aronovitz, M.; Thoonen, R.; Kass, D.A.; Karas, R.H.; Mendelsohn, M.E. Mutation of the protein kinase I alpha leucine zipper domain produces hypertension and progressive left ventricular hypertrophy: A novel mouse model of age-dependent hypertensive heart disease. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 1351–1355. [Google Scholar] [CrossRef]
- Blanton, R.M.; Takimoto, E.; Lane, A.M.; Aronovitz, M.; Piotrowski, R.; Karas, R.H.; Kass, D.A.; Mendelsohn, M.E. Protein kinase g Iα inhibits pressure overload-induced cardiac remodeling and is required for the cardioprotective effect of sildenafil in vivo. J. Am. Heart. Assoc. 2012, 1, e003731. [Google Scholar] [CrossRef]
- Tokudome, T.; Kishimoto, I.; Horio, T.; Arai, Y.; Schwenke, D.O.; Hino, J.; Okano, I.; Kawano, Y.; Kohno, M.; Miyazato, M.; et al. Regulator of G-protein signaling subtype 4 mediates antihypertrophic effect of locally secreted natriuretic peptides in the heart. Circulation 2008, 117, 2329–2339. [Google Scholar] [CrossRef]
- Riddle, E.L.; Schwartzman, R.A.; Bond, M.; Insel, P.A. Multi-tasking RGS proteins in the heart: The next therapeutic target? Circ. Res. 2005, 96, 401–411. [Google Scholar] [CrossRef]
- Klaiber, M.; Kruse, M.; Völker, K.; Schröter, J.; Feil, R.; Freichel, M.; Gerling, A.; Feil, S.; Dietrich, A.; Londoño, J.E.C.; et al. Novel insights into the mechanisms mediating the local antihypertrophic effects of cardiac atrial natriuretic peptide: Role of cGMP-dependent protein kinase and RGS2. Basic Res. Cardiol. 2010, 105, 583–595. [Google Scholar] [CrossRef]
- Druey, K.M.; Blumer, K.J.; Kang, V.H.; Kehrl, J.H. Inhibition of G-protein-mediated MAP kinase activation by a new mammalian gene family. Nature 1996, 379, 742–746. [Google Scholar] [CrossRef]
- Rainer, P.P.; Kass, D.A. Old dog, new tricks: Novel cardiac targets and stress regulation by protein kinase G. Cardiovasc. Res. 2016, 111, 154–162. [Google Scholar] [CrossRef]
- Lee, D.I.; Vahebi, S.; Tocchetti, C.G.; Barouch, L.A.; Solaro, R.J.; Takimoto, E.; Kass, D.A. PDE5A suppression of acute beta-adrenergic activation requires modulation of myocyte beta-3 signaling coupled to PKG-mediated troponin I phosphorylation. Basic Res. Cardiol. 2010, 105, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.Z.; Jang, J.H.; Kim, H.J.; Wang, Y.; Hwang, I.C.; Sadayappan, S.; Park, B.M.; Kim, S.H.; Jin, Z.H.; Seo, E.Y.; et al. Myofilament Ca2+ desensitization mediates positive lusitropic effect of neuronal nitric oxide synthase in left ventricular myocytes from murine hypertensive heart. J. Mol. Cell. Cardiol. 2013, 60, 107–115. [Google Scholar] [CrossRef]
- Layland, J.; Li, J.M.; Shah, A.M. Role of cyclic GMP-dependent protein kinase in the contractile response to exogenous nitric oxide in rat cardiac myocytes. J. Physiol. 2002, 540, 457–467. [Google Scholar] [CrossRef]
- Nakamura, T.; Ranek, M.J.; Lee, D.I.; Hahn, S.V.; Kim, C.; Eaton, P.; Kass, D.A. Prevention of PKG1alpha oxidation augments cardioprotection in the stressed heart. J. Clin. Investig. 2015, 125, 2468–2472. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, D.; Lucas, J.; Oparil, S.; Xing, D.; Cao, X.; Novak, L.; Renfrow, M.B.; Chen, Y.F. Atrial natriuretic peptide inhibits transforming growth factor beta-induced Smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circ. Res. 2008, 102, 185–192. [Google Scholar] [CrossRef]
- Chen, J.; Shearer, G.C.; Chen, Q.; Healy, C.L.; Beyer, A.J.; Nareddy, V.B.; Gerdes, A.M.; Harris, W.S.; O’Connell, T.D.; Wang, D. Omega-3 fatty acids prevent pressure overload-induced cardiac fibrosis through activation of cyclic GMP/protein kinase G signaling in cardiac fibroblasts. Circulation 2011, 123, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Akashi, S.; Ahmed, K.A.; Sawa, T.; Ono, K.; Tsutsuki, H.; Burgoyne, J.R.; Ida, T.; Horio, E.; Prysyazhna, O.; Oike, Y.; et al. Persistent activation of cGMP-dependent protein kinase by a nitrated cyclic nucleotide via site specific protein S-guanylation. Biochemistry 2016, 55, 751–761. [Google Scholar] [CrossRef]
- Vandecasteele, G.; Verde, I.; Rücker-Martin, C.; Donzeau-Gouge, P.; Fischmeister, R. Cyclic GMP regulation of the L-type Ca2+ channel current in human atrial myocytes. J. Physiol. 2001, 533, 329–340. [Google Scholar] [CrossRef]
- Omori, K.; Kotera, J. Overview of PDEs and their regulation. Circ. Res. 2007, 100, 309–327. [Google Scholar] [CrossRef]
- Brescia, M.; Zaccolo, M. Modulation of compartmentalised cyclic Nucleotide signalling via local inhibition of phosphodiesterase activity. Int. J. Mol. Sci. 2016, 17, 1672. [Google Scholar] [CrossRef]
- Lee, D.I.; Zhu, G.; Sasaki, T.; Cho, G.S.; Hamdani, N.; Holewinski, R.; Jo, S.H.; Danner, T.; Zhang, M.; Rainer, P.P.; et al. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 2015, 519, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Biel, M. Cyclic nucleotide-regulated cation channels. J. Biol. Chem. 2009, 284, 9017–9021. [Google Scholar] [CrossRef] [PubMed]
- Kaupp, U.B.; Seifert, R. Cyclic nucleotide-gated ion channels. Physiol. Rev. 2002, 82, 769–824. [Google Scholar] [CrossRef]
- Craven, K.B.; Zagotta, W.N. CNG and HCN channels: Two peas, one pod. Annu. Rev. Physiol. 2006, 68, 375–401. [Google Scholar] [CrossRef] [PubMed]
- Biel, M.; Zong, X.; Distler, M.; Bosse, E.; Klugbauer, N.; Murakami, M.; Flockerzi, V.; Hofmann, F. Another member of the cyclic nucleotide-gated channel family, expressed in testis, kidney, and heart. Proc. Natl. Acad. Sci. USA 1994, 91, 3505–3509. [Google Scholar] [CrossRef]
- Liao, Z.; Lockhead, D.; Larson, E.D.; Proenza, C. Phosphorylation and modulation of hyperpolarization-activated HCN4 channels by protein kinase A in the mouse sinoatrial node. J. Gen. Physiol. 2010, 136, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Stomberski, C.T.; Hess, D.T.; Stamler, J.S. Protein S-nitrosylation: Determinants of specificity and enzymatic regulation of S-nitrosothiol-based signaling. Antioxid. Redox. Signal. 2019, 30, 1331–1351. [Google Scholar] [CrossRef]
- Lukowski, R.; Feil, R. Recent developments in cGMP research: From mechanisms to medicines and back. Br. J. Pharmacol. 2022, 179, 2321–2327. [Google Scholar] [CrossRef]
- Grange, R.M.H.; Preedy, M.E.J.; Renukanthan, A.; Dignam, J.P.; Lowe, V.J.; Moyes, A.J.; Perez-Ternero, C.; Aubdool, A.A.; Baliga, R.S.; Hobbs, A.J. Multidrug resistance proteins preferentially regulate natriuretic peptide-driven cGMP signalling in the heart and vasculature. Br. J. Pharmacol. 2022, 179, 2443–2459. [Google Scholar] [CrossRef]
- Buggey, J.; Mentz, R.J.; DeVore, A.D.; Velazquez, E.J. Angiotensin receptor neprilysin inhibition in heart failure: Mechanistic action and clinical impact. J. Card. Fail. 2015, 21, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Hubers, S.A.; Brown, N.J. Combined angiotensin receptor antagonism and neprilysin inhibition. Circulation 2016, 133, 1115–1124. [Google Scholar] [CrossRef]
- Bertrand, L.; Auquier, J.; Renguet, E.; Angé, M.; Cumps, J.; Horman, S.; Beauloye, C. Glucose transporters in cardiovascular system in health and disease. Pflügers Arch. 2020, 472, 1385–1399. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Tian, R. Glucose transporters in cardiac metabolism and hypertrophy. Compr. Physiol. 2015, 6, 331–351. [Google Scholar]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Fitchett, D.; Inzucchi, S.E.; Cannon, C.P.; McGuire, D.K.; Scirica, B.M.; Johansen, O.E.; Sambevski, S.; Kaspers, S.; Pfarr, E.; George, J.T.; et al. Empagliflozin reduced mortality and hospitalization for heart failure across the spectrum of cardiovascular risk in the EMPA-REG OUTCOME trial. Circulation 2019, 139, 1384–1395. [Google Scholar] [CrossRef]
- Zannad, F.; Ferreira, J.P.; Pocock, S.J.; Anker, S.D.; Butler, J.; Filippatos, G.; Brueckmann, M.; Ofstad, A.P.; Pfarr, E.; Jamal, W.; et al. SGLT2 inhibitors in patients with heart failure with reduced ejection fraction: A meta-analysis of the EMPEROR-reduced and DAPA-HF trials. Lancet 2020, 396, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Zannad, F. Effects of sodium-glucose cotransporter 2 inhibitors for the treatment of patients with heart failure: Proposal of a novel mechanism of action. JAMA Cardiol. 2017, 2, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Rawat, S.; Ho, K.L.; Wagg, C.S.; Zhang, L.; Teoh, H.; Dyck, J.E.; Uddin, G.M.; Oudit, G.Y.; Mayoux, E.; et al. Empagliflozin increases cardiac energy production in diabetes: Novel translational insights into the heart failure benefits of SGLT2 inhibitors. JACC Basic Transl. Sci. 2018, 3, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Lytvyn, Y.; Bjornstad, P.; Udell, J.A.; Lovshin, J.A.; Cherney, D.Z.I. Sodium glucose cotransporter-2 inhibition in heart failure: Potential mechanisms, clinical applications, and summary of clinical trials. Circulation 2017, 136, 1643–1658. [Google Scholar] [CrossRef]
- Suzuki, M.; Honda, K.; Fukazawa, M.; Ozawa, K.; Hagita, H.; Kawai, T.; Takeda, M.; Yata, T.; Kawai, M.; Fukuzawa, T.; et al. Tofogliflozin, a potent and highly specific sodium/glucose cotransporter 2 inhibitor, improves glycemic control in diabetic rats and mice. J. Pharmacol. Exp. Ther. 2012, 341, 692–701. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.J.V.; Claggett, B.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Felipe Martinez, M.D.; et al. Dapagliflozin in heart failure with mildly reduced or preserved ejection fraction. N. Engl. J. Med. 2022, 387, 1089–1098. [Google Scholar] [CrossRef]
- Bucchi, A.; Tognati, A.; Milanesi, R.; Baruscotti, M.; DiFrancesco, D. Properties of ivabradine-induced block of HCN1 and HCN4 pacemaker channels. J. Physiol. 2006, 572, 335–346. [Google Scholar] [CrossRef] [PubMed]
- DiFrancesco, D.; Camm, J.A. Heart rate lowering by specific and selective If current inhibition with ivabradine: A new therapeutic perspective in cardiovascular disease. Drugs 2004, 64, 1757–1765. [Google Scholar] [CrossRef]
- Baruscotti, M.; Bucchi, A.; DiFrancesco, D. Physiology and pharmacology of the cardiac pacemaker (“funny”) current. Pharmacol. Ther. 2005, 107, 59–79. [Google Scholar] [CrossRef] [PubMed]
- Baruscotti, M.; Bucchi, A.; Viscomi, C.; Mandelli, G.; Consalez, G.; Gnecchi-Rusconi, T.; Montano, N.; Casali, K.R.; Micheloni, S.; Barbuti, A.; et al. Deep bradycardia and heart block caused by inducible cardiac-specific knockout of the pacemaker channel gene Hcn4. Proc. Natl. Acad. Sci. USA 2011, 108, 1705–1710. [Google Scholar] [CrossRef] [PubMed]
- Haechl, N.; Ebner, J.; Hilber, K.; Todt, H.; Koenig, X. Pharmacological profile of the bradycardic agent ivabradine on human cardiac ion channels. Cell. Physiol. Biochem. 2019, 53, 36–48. [Google Scholar]
- Psotka, M.A.; Teerlink, J.R. Direct myosin activation by omecamtiv mecarbil for heart failure with reduced ejection fraction. Handb. Exp. Pharmacol. 2017, 243, 465–490. [Google Scholar]
- Bernier, T.D.; Buckley, L.F. Cardiac myosin activation for the treatment of systolic heart failure. J. Cardiovasc. Pharmacol. 2021, 77, 4. [Google Scholar] [CrossRef]
- Barrick, S.K.; Greenberg, M.J. Cardiac myosin contraction and mechanotransduction in health and disease. J. Biol. Chem. 2021, 297, 101297. [Google Scholar] [CrossRef]
- Planelles-Herrero, V.J.; Hartman, J.J.; Robert-Paganin, J.; Malik, F.I.; Houdusse, A. Mechanistic and structural basis for activation of cardiac myosin force production by omecamtiv mecarbil. Nat. Commun. 2017, 8, 190. [Google Scholar] [CrossRef]
- Kovács, Á.; Alogna, A.; Post, H.; Hamdani, N. Is enhancing cGMP-PKG signalling a promising therapeutic target for heart failure with preserved ejection fraction? Neth. Heart J. 2016, 24, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Sandner, P.; Follmann, M.; Becker-Pelster, E.; Hahn, M.G.; Meier, C.; Freitas, C.; Roessig, L.; Stasch, J.P. Soluble GC stimulators and activators: Past, present and future. Br. J. Pharmacol. 2021, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Lin, M.; Maitz, T.; Egeler, D.J.; Sood, A.; Aronow, W.S.; Rajeswaran, Y.; Ahnert, A.M.; Vyas, A.V.; Frishman, W.H.; et al. A novel soluble guanylate cyclase stimulator for use in patients with heart failure. Cardiol. Rev. 2023, 31, 87–92. [Google Scholar] [CrossRef]
- Kansakar, S.; Guragain, A.; Verma, D.; Sharma, P.; Dhungana, B.; Bhattarai, B.; Yadav, S.; Gautam, N. Soluble guanylate cyclase stimulators in heart failure. Cureus 2021, 13, e17781. [Google Scholar] [CrossRef] [PubMed]
- Schwaerzer, G.K.; Casteel, D.E.; Cividini, F.; Kalyanaraman, H.; Zhuang, S.; Gu, Y.; Dalton, N.D.; Peterson, K.L.; Dillmann, W.H.; Boss, G.R.; et al. Constitutive protein kinase G activation exacerbates stress-induced cardiomyopathy. Br. J. Pharmacol. 2022, 179, 2413–2429. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, P.W.; Pieske, B.; Anstrom, K.J.; Ezekowitz, J.; Hernandez, A.F.; Butler, J.; Lam, C.S.P.; Ponikowski, P.; Voors, A.A.; Jia, G.; et al. Vericiguat in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 2020, 382, 1883–1893. [Google Scholar] [CrossRef]
- De Vecchis, R.; Cesaro, A.; Ariano, C.; Giasi, A.; Carmela Cioppa, C. Phosphodiesterase-5 inhibitors improve clinical outcomes, exercise capacity and pulmonary hemodynamics in patients with heart failure with reduced left ventricular ejection fraction: A meta-analysis. J. Clin. Med. Res. 2017, 9, 488–498. [Google Scholar] [CrossRef]
- Armstrong, P.W.; Lam, C.S.P.; Anstrom, K.J.; Ezekowitz, J.; Hernandez, A.F.; O’Connor, C.M.; Pieske, B.; Ponikowski, P.; Shah, S.J.; Solomon, S.D.; et al. Effect of vericiguat vs placebo on quality of life in patients with heart failure and preserved ejection fraction: The VITALITY-HFpEF randomized clinical trial. J. Am. Med. Assoc. 2020, 324, 1512–1521. [Google Scholar] [CrossRef]
- Udelson, J.E.; Lewis, G.D.; Shah, S.J.; Zile, M.R.; Redfield, M.M.; Burnett, J., Jr.; Parker, J.; Seferovic, J.P.; Wilson, P.; Mittleman, R.S.; et al. Effect of praliciguat on peak rate of oxygen consumption in patients with heart failure with preserved ejection fraction: The CAPACITY HFpEF randomized clinical trial. J. Am. Med. Assoc. 2020, 324, 1522–1531. [Google Scholar] [CrossRef]
- Tam, K.; Richards, D.A.; Aronovitz, M.J.; Martin, G.L.; Pande, S.; Jaffe, I.Z.; Blanton, R.M. Sacubitril/Valsartan improves left ventricular function in chronic pressure overload independent of intact cyclic guanosine monophosphate-dependent protein kinase I alpha signaling. J. Card. Fail. 2020, 26, 769–775. [Google Scholar] [CrossRef]
- Santos-Gallego, C.G.; Requena-Ibanez, J.A.; Antonio, R.S.; Garcia-Ropero, A.; Ishikawa, K.; Watanabe, S.; Picatoste, B.; Vargas-Delgado, A.P.; Flores-Umanzor, E.J.; Sanz, J.; et al. Empagliflozin ameliorates diastolic dysfunction and left ventricular fibrosis/stiffness in nondiabetic heart failure: A multimodality study. J. Am. Coll. Cardiol. Imaging 2021, 14, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Robles-Vera, I.; Toral, M.; de la Visitación, N.; Aguilera-Sánchez, N.; Redondo, J.M.; Duarte, J. Protective effects of short-chain fatty acids on endothelial dysfunction induced by angiotensin II. Front. Physiol. 2020, 11, 277. [Google Scholar] [CrossRef] [PubMed]
- Blad, C.C.; Tang, C.; Offermanns, S. G protein-coupled receptors for energy metabolites as new therapeutic targets. Nat. Rev. Drug Discov. 2012, 11, 603–619. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Suster, M.M.; Borges, J.I. Short-chain fatty acid receptors and cardiovascular function. Int. J. Mol. Sci. 2022, 23, 3303. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Di, W.; Dong, X.; Li, Z.; Zhang, Y.; Xue, X.-D.; Xu, Y.; Zhang, J.; Xiao, X.; Han, J.; et al. Melatonin protects diabetic heart against ischemia-reperfusion injury, role of membrane receptor-dependent cGMP-PKG activation. Biochim. Biophys. Acta-Mol. Basis Dis. 2018, 1864, 563–578. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Liu, J.; Li, X.; Sun, X.; Zhang, J.; Ren, D.; Tong, N.; Li, J. Empagliflozin attenuates ischemia and reperfusion injury through LKB1/AMPK signaling pathway. Mol. Cell. Endocrinol. 2020, 501, 110642. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, Y.; Wang, Z.; Tan, M.; Lin, J.; Qian, X.; Li, H.; Jiang, T. Dapagliflozin alleviates myocardial ischemia/reperfusion injury by reducing ferroptosis via MAPK signaling inhibition. Front. Pharmacol. 2023, 14, 1078205. [Google Scholar] [CrossRef]
- Michel, K.; Herwig, M.; Werner, F.; Spes, K.Š.; Abeßer, M.; Schuh, K.; Dabral, S.; Mügge, A.; Baba, H.A.; Skryabin, B.V.; et al. C-type natriuretic peptide moderates titin-based cardiomyocyte stiffness. JCI Insight 2020, 5, e139910. [Google Scholar] [CrossRef]
- Kolijn, D.; Pabel, S.; Tian, Y.; Lódi, M.; Herwig, M.; Carrizzo, A.; Zhazykbayeva, S.; Kovács, A.; Fülöp, G.A.; Falcão-Pires, I.; et al. Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase Gα oxidation. Cardiovasc. Res. 2021, 117, 495–507. [Google Scholar] [CrossRef]
- Mangmool, S.; Denkaew, T.; Parichatikanond, W.; Kurose, H. β-Adrenergic receptor and insulin resistance in the heart. Biomol. Ther. 2017, 25, 44–56. [Google Scholar] [CrossRef]
- Mangmool, S.; Parichatikanond, W.; Kurose, H. Therapeutic targets for treatment of heart failure: Focus on GRKs and β-arrestins affecting βAR signaling. Front. Pharmacol. 2018, 9, 1336. [Google Scholar] [CrossRef]
- Moore, C.A.C.; Milano, S.K.; Benovic, J.L. Regulation of receptor trafficking by GRKs and arrestins. Annu. Rev. Physiol. 2007, 69, 451–482. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hanada, K.; Status, D.P.; Makara, M.A.; Dahal, G.R.; Chen, Q.; Ahles, A.; Engelhardt, S.; Rockman, H.A. Gαi is required for carvedilol-induced β1 adrenergic receptor β-arrestin biased signaling. Nat. Commun. 2017, 8, 1706. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Lefkowitz, R.J.; Rajagopal, S. Biased signalling: From simple switches to allosteric microprocessors. Nat. Rev. Drug Discov. 2018, 17, 243–260. [Google Scholar] [CrossRef]
- Mangmool, S.; Shukla, A.K.; Rockman, H.A. β-Arrestin-dependent activation of Ca2+/calmodulin kinase II after β1-adrenergic receptor stimulation. J. Cell. Biol. 2010, 189, 573–587. [Google Scholar] [CrossRef]
- Grogan, A.; Lucero, E.Y.; Jiang, H.; Rockman, H.A. Pathophysiology and pharmacology of G protein-coupled receptors in the heart. Cardiovasc. Res. 2023, 119, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Naseem, K.M. The role of nitric oxide in cardiovascular diseases. Mol. Aspects Med. 2005, 26, 33–65. [Google Scholar] [CrossRef]
- Michel, L.Y.M.; Farah, C.; Balligand, J.L. The beta3 adrenergic receptor in healthy and pathological cardiovascular tissues. Cells 2020, 9, 2584. [Google Scholar] [CrossRef]
- Chapple, C.R.; Cardozo, L.; Nitti, V.W.; Siddiqui, E.; Michel, M.C. Mirabegron in overactive bladder: A review of efficacy, safety, and tolerability. Neurourol. Urodyn. 2014, 33, 17–30. [Google Scholar] [CrossRef]
- Bundgaard, H.; Raja, A.A.; Iversen, K.; Valeur, N.; Tønder, N.; Schou, M.; Christensen, A.H.; Bruun, N.E.; Søholm, H.; Ghanizada, M.; et al. Hemodynamic effects of cyclic guanosine monophosphate-dependent signaling through β3 adrenoceptor stimulation in patients with advanced heart failure: A randomized invasive clinical trial. Circ. Heart Fail. 2022, 15, e009120. [Google Scholar] [CrossRef]
- Roy, R.; Koch, W.J. Not all β-receptors appear the same in heart failure: Emergence of β3-agonists as a therapeutic option. Circ. Heart Fail. 2022, 15, e009685. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, Y.; West, T.M.; Liu, Y.; Reddy, G.R.; Barbagallo, F.; Xu, B.; Shi, Q.; Deng, B.; Wei, W.; et al. Carvedilol induces biased β1 adrenergic receptor-nitric oxide synthase 3-cyclic guanylyl monophosphate signalling to promote cardiac contractility. Cardiovasc. Res. 2021, 117, 2237–2251. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Ichimura, A.; Ohue-Kitano, R.; Igarashi, M. Free Fatty Acid Receptors in Health and Disease. Physiol. Rev. 2020, 100, 171–210. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Wu, J.H.Y. Omega-3 fatty acids and cardiovascular disease: Effects on risk factors, molecular pathways, and clinical events. J. Am. Coll. Cardiol. 2011, 58, 2047–2067. [Google Scholar] [CrossRef] [PubMed]
- Eclov, J.A.; Qian, Q.; Redetzke, R.; Chen, Q.; Wu, S.C.; Healy, C.L.; Ortmeier, S.B.; Harmon, E.; Shearer, G.C.; O’Connell, T.D. EPA, not DHA, prevents fibrosis in pressure overload-induced heart failure: Potential role of free fatty acid receptor 4. J. Lipid Res. 2015, 56, 2297–2308. [Google Scholar] [CrossRef]
- Parichatikanond, W.; Luangmonkong, T.; Mangmool, S.; Kurose, H. Therapeutic targets for the treatment of cardiac fibrosis and cancer: Focusing on TGF-β signaling. Front. Cardiovasc. Med. 2020, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Duangrat, R.; Parichatikanond, W.; Mangmool, S. Dual blockade of TGF-β receptor and endothelin receptor synergistically inhibits angiotensin II-induced myofibroblast differentiation: Role of AT1R/Gαq-mediated TGF-β1 and ET-1 signaling. Int. J. Mol. Sci. 2023, 24, 6972. [Google Scholar] [CrossRef] [PubMed]
- Wess, J. Designer GPCRs as novel tools to identify metabolically important signaling pathways. Front. Endocrinol. (Lausanne) 2021, 12, 706957. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, E.; Tian, Q.; Wagner, M.; Barth, M.; Xian, W.; Schröder, L.; Ruppenthal, S.; Kaestner, L.; Boehm, U.; Wartenberg, P.; et al. DREADD technology reveals major impact of Gq signalling on cardiac electrophysiology. Cardiovasc. Res. 2019, 115, 1052–1066. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, Y.; Nishida, M.; Sugimoto, Y.; Tanabe, S.; Turner, J.H.; Kozasa, T.; Wada, T.; Nagao, T.; Kurose, H. Galpha(12/13) mediates alpha(1)-adrenergic receptor-induced cardiac hypertrophy. Circ. Res. 2002, 91, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Duangrat, R.; Parichatikanond, W.; Morales, N.P.; Pinthong, D.; Mangmool, S. Sustained AT1R stimulation induces upregulation of growth factors in human cardiac fibroblasts via Gαq/TGF-β/ERK signaling that influences myocyte hypertrophy. Eur. J. Pharmacol. 2022, 937, 175384. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K.; Nahrendorf, M. Cardioimmunology: The immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Fulmer, M.L.; Thewke, D.P. The Endocannabinoid system and heart disease: The role of cannabinoid receptor type 2. Cardiovasc. Hematol. Disord. Drug Targets 2018, 18, 34–51. [Google Scholar] [CrossRef]
- Howlett, A.C. Cannabinoid receptor signaling. Handb. Exp. Pharmacol. 2005, 168, 53–79. [Google Scholar]
- Montecucco, F.; Lenglet, S.; Braunersreuther, V.; Burger, F.; Pelli, G.; Bertolotto, M.; Mach, F.; Steffens, S. CB(2) cannabinoid receptor activation is cardioprotective in a mouse model of ischemia/reperfusion. J. Mol. Cell. Cardiol. 2009, 46, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ma, S.; Wang, Q.; Hu, W.; Wang, D.; Li, X.; Su, T.; Qin, X.; Zhang, X.; Ma, K.; et al. Effects of cannabinoid receptor type 2 on endogenous myocardial regeneration by activating cardiac progenitor cells in mouse infarcted heart. Sci. China Life Sci. 2014, 57, 201–208. [Google Scholar] [CrossRef]
- Li, X.; Han, D.; Tian, Z.; Gao, B.; Fan, M.; Li, C.; Li, X.; Wang, Y.; Ma, S.; Cao, F. Activation of cannabinoid receptor type II by AM1241 ameliorates myocardial fibrosis via Nrf2-mediated inhibition of TGF-β1/Smad3 pathway in myocardial infarction mice. Cell. Physiol. Biochem. 2016, 39, 1521–1536. [Google Scholar] [CrossRef]
- Peng, G.; Tang, X.; Gui, Y.; Yang, J.; Ye, L.; Wu, L.; Ding, Y.H.; Wang, L. Transient receptor potential vanilloid subtype 1: A potential therapeutic target for fibrotic diseases. Front. Physiol. 2022, 15, 951980. [Google Scholar] [CrossRef]
- Horton, J.S.; Shiraishi, T.; Alfulaij, N.; Small-Howard, A.L.; Turner, H.C.; Tatsuki Kurokawa, T.; Mori, Y.; Stokes, A.J. TRPV1 is a component of the atrial natriuretic signaling complex, and using orally delivered antagonists, presents a valid therapeutic target in the longitudinal reversal and treatment of cardiac hypertrophy and heart failure. Channels (Austin) 2019, 13, 1–16. [Google Scholar] [CrossRef]
- Montiel, V.; Bella, R.; Michel, L.Y.M.; Esfahani, H.; De Mulder, D.; Robinson, E.L.; Deglasse, J.L.; Tiburcy, M.; Chow, P.H.; Jonas, J.C.; et al. Inhibition of aquaporin-1 prevents myocardial remodeling by blocking the transmembrane transport of hydrogen peroxide. Sci. Transl. Med. 2020, 12, eaay2176. [Google Scholar] [CrossRef]
- Nesverova, V.; Törnroth-Horsefield, S. Phosphorylation-dependent regulation of mammalian aquaporins. Cells 2019, 8, 82. [Google Scholar] [CrossRef]
- Solomon, S.D.; Zile, M.; Pieske, B.; Voors, A.; Shah, A.; Kraigher-Krainer, E.; Shi, V.; Bransford, T.; Takeuchi, M.; Gong, J.; et al. The angiotensin receptor neprilysin inhibitor LCZ696 in heart failure with preserved ejection fraction: A phase 2 double-blind randomised controlled trial. Lancet 2012, 380, 1387–1395. [Google Scholar] [CrossRef]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; McMurray, J.J.; Anand, I.S.; Ge, J.; Lam, C.S.; Maggioni, A.P.; Martinez, F.; Packer, M.; Pfeffer, M.A.; Pieske, B.; et al. Angiotensin–neprilysin inhibition in heart failure with preserved ejection fraction. N. Engl. J. Med. 2019, 381, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Scicchitano, P.; Cortese, F.; Ricci, G.; Carbonara, S.; Moncelli, M.; Iacoviello, M.; Cecere, A.; Gesualdo, M.; Zito, A.; Caldarola, P.; et al. Ivabradine, coronary artery disease, and heart failure: Beyond rhythm control. Drug Des. Develop. Ther. 2014, 8, 689–700. [Google Scholar]
- Camm, A.J. How does pure heart rate lowering impact on cardiac tolerability? Eur. Heart J. Suppl. 2006, 8, D9–D15. [Google Scholar] [CrossRef]
- Fox, K.; Ford, I.; Steg, P.G.; Tendera, M.; Ferrari, R. Ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): A randomised, double-blind, placebo-controlled trial. Lancet 2008, 372, 807–816. [Google Scholar] [CrossRef]
- Ceconi, C.; Freedman, S.B.; Tardif, J.C.; Hildebrandt, P.; McDonagh, T.; Gueret, P.; Parrinello, G.; Robertson, M.; Steg, P.G.; Tendera, M.; et al. Effect of heart rate reduction by ivabradine on left ventricular remodeling in the echocardiographic substudy of BEAUTIFUL. Int. J. Cardiol. 2011, 146, 408–414. [Google Scholar] [CrossRef]
- Swedberg, K.; Komajda, M.; Bohm, M.; Borer, J.S.; Ford, I.; Dubost-Braman, A.; Lerebours, G.; Tavazzi, L.; on behalf of the SHIFT Investigators. Ivabradine and outcomes in chronic heart failure (SHIFT): A randomised placebo-controlled study. Lancet 2010, 376, 875–885. [Google Scholar] [CrossRef]
- Tardif, J.C.; O’Meara, E.; Komajda, M.; Bohm, M.; Borer, J.S.; Ford, I.; Tavazzi, L.; Swedberg, K. Effects of selective heart rate reduction with ivabradine on left ventricular remodelling and function: Results from the SHIFT echocardiography substudy. Eur. Heart J. 2011, 32, 2507–2515. [Google Scholar] [CrossRef]
- Biering-Sørensen, T.; Minamisawa, M.; Claggett, B.; Liu, J.; Felker, G.M.; McMurray, J.J.; Malik, F.I.; Abbasi, S.; Kurtz, C.E.; Teerlink, J.R.; et al. Cardiac myosin activator omecamtiv mecarbil improves left ventricular myocardial deformation in chronic heart failure: The COSMIC-HF trial. Circ. Heart Fail. 2020, 13, e008007. [Google Scholar] [CrossRef]
- Shah, S.R.; Ali, A.; Ikram, S. Omecamtiv Mecarbil use in systolic heart failure-Results of the GALACTIC-HF trial. Exp. Rev. Clin. Pharmacol. 2021, 14, 407–409. [Google Scholar] [CrossRef]
- Voors, A.A.; Tamby, J.F.; Cleland, J.G.; Koren, M.; Forgosh, L.B.; Gupta, D.; Lund, L.H.; Camacho, A.; Karra, R.; Swart, H.P.; et al. Effects of danicamtiv, a novel cardiac myosin activator, in heart failure with reduced ejection fraction: Experimental data and clinical results from a phase 2a trial. Eur. J. Heart Fail. 2020, 22, 1649–1658. [Google Scholar] [CrossRef]
- Teerlink, J.; Felker, G.; McMurray, J.; Solomon, S.; Adams, K., Jr.; Cleland, J.; Ezekowitz, J.; Goudev, A.; Macdonald, P.; Metra, M.; et al. Chronic oral study of myosin activation to increase contractility in heart failure (COSMIC-HF): A phase 2, pharmacokinetic, randomised, placebo-controlled trial. Lancet 2016, 388, 2895–2903. [Google Scholar] [CrossRef] [PubMed]
- Felker, G.M.; Solomon, S.D.; McMurray, J.J.V.; Cleland, J.G.F.; Abbasi, S.A.; Malik, F.I.; Zhang, H.; Globe, G.; Teerlink, J.R. Effects of omecamtiv mecarbil on symptoms and health-related quality of life in patients with chronic heart failure: Results from the COSMIC-HF study. Circ. Heart Fail. 2020, 13, 814. [Google Scholar] [CrossRef]
- Clark, A.L. Exercise and heart failure: Assessment and treatment. Heart 2006, 92, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.D.; Voors, A.A.; Cohen-Solal, A.; Metra, M.; Whellan, D.J.; Ezekowitz, J.A.; Böhm, M.; Teerlink, J.R.; Docherty, K.F.; Lopes, R.D.; et al. Effect of omecamtiv mecarbil on exercise capacity in chronic heart failure with reduced ejection fraction: The METEORIC-HF randomized clinical trial. J. Am. Med. Assoc. 2022, 328, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Teerlink, J.R.; Diaz, R.; Felker, G.M.; McMurray, J.J.V.; Metra, M.; Solomon, S.D.; Adams, K.F.; Anand, I.; Arias-Mendoza, A.; Biering-Sorensen, T.; et al. Cardiac myosin activation with omecamtiv mecarbil in systolic heart failure. N. Engl. J. Med. 2021, 384, 105–116. [Google Scholar] [CrossRef]
- Teerlink, J.R.; Diaz, R.; Felker, G.M.; McMurray, J.J.V.; Metra, M.; Solomon, S.D.; Biering-Sorensen, T.; Bohm, M.; Bonderman, D.; Fang, J.C.; et al. Effect of ejection fraction on clinical outcomes in patients treated with omecamtiv mecarbil in GALACTIC-HF. J. Am. Coll. Cardiol. 2021, 78, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Docherty, K.F.; McMurray, J.J.V.; Claggett, B.L.; Miao, Z.M.; Adams, K.F.; Arias-Mendoza, A.; Cleland, J.G.F.; Diaz, R.; Echeverria Correa, L.E.; Felker, M.G.; et al. Efficacy of omecamtiv mecarbil in heart failure with reduced ejection fraction according to N-terminal pro-B-type natriuretic peptide level: Insights from the GALACTIC-HF trial. Eur. J. Heart Fail. 2023, 25, 248–259. [Google Scholar] [CrossRef]
- Vallon, V.; Verma, S. Effects of SGLT2 inhibitors on kidney and cardiovascular function. Annu. Rev. Physiol. 2021, 83, 503–528. [Google Scholar] [CrossRef]
- Salvatore, T.; Galiero, R.; Caturano, A.; Rinaldi, L.; Di Martino, A.; Albanese, G.; Di Salvo, J.; Epifani, R.; Marfella, R.; Docimo, G.; et al. An Overview of the cardiorenal protective mechanisms of SGLT2 inhibitors. Int. J. Mol. Sci. 2022, 23, 3651. [Google Scholar] [CrossRef] [PubMed]
- Spertus, J.A.; Birmingham, M.C.; Nassif, M.; Damaraju, C.V.; Abbate, A.; Butler, J.; Lanfear, D.E.; Lingvay, I.; Kosiborod, M.N.; Januzzi, J.L. The SGLT2 inhibitor canagliflozin in heart failure: The CHIEF-HF remote, patient-centered randomized trial. Nat. Med. 2022, 28, 809–813. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Kober, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Nassif, M.E.; Windsor, S.L.; Tang, F.; Khariton, Y.; Husain, M.; Inzucchi, S.E.; McGuire, D.K.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; et al. Dapagliflozin effects on biomarkers, symptoms, and functional status in patients with heart failure with reduced ejection fra35tion: The DEFINE-HF trial. Circulation 2019, 140, 1463–1476. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Jamal, W. Cardiovascular and renal outcomes with empagliflozin in heart failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner–La Rocca, H.-P.; Choi, D.J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in heart failure with a preserved ejection fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Gheorghiade, M.; Greene, S.J.; Butler, J.; Filippatos, G.; Lam, C.S.P.; Maggioni, A.P.; Ponikowski, P.; Shah, S.J.; Solomon, S.D.; Kraigher-Krainer, E.; et al. Effect of vericiguat, a soluble guanylate cyclase stimulator, on natriuretic peptide levels in patients with worsening chronic heart failure and reduced ejection fraction: The SOCRATES-REDUCED randomized trial. J. Am. Med. Assoc. 2015, 314, 2251–2262. [Google Scholar] [CrossRef]
- Pieske, B.; Maggioni, A.P.; Lam, C.S.P.; Pieske-Kraigher, E.; Filippatos, G.; Butler, J.; Ponikowski, P.; Shah, S.J.; Solomon, S.D.; Scalise, A.V.; et al. Vericiguat in patients with worsening chronic heart failure and preserved ejection fraction: Results of the soluble guanylate cyclase stimulator in heart failure patients with preserved EF (SOCRATES-PRESERVED) study. Eur. Heart J. 2017, 38, 1119–1127. [Google Scholar] [CrossRef]
- Ezekowitz, J.A.; O’Connor, C.M.; Troughton, R.W.; Alemayehu, W.G.; Westerhout, C.M.; Voors, A.A.; Butler, J.; Lam, C.S.P.; Ponikowski, P.; Emdin, M.; et al. N-terminal pro-B-type natriuretic peptide and clinical outcomes: Vericiguat heart failure with reduced ejection fraction study. J. Am. Coll. Cardiol. Heart Fail. 2020, 8, 931–939. [Google Scholar]
- Bonderman, D.; Ghio, S.; Felix, S.B.; Ghofrani, H.A.; Michelakis, E.; Mitrovic, V.; Oudiz, R.J.; Boateng, F.; Scalise, A.-V.; Roessig, L.; et al. Riociguat for patients with pulmonary hypertension caused by systolic left ventricular dysfunction: A phase IIb double-blind, randomized, placebo-controlled, dose-ranging hemodynamic study. Circulation 2013, 128, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Schena, G.; Caplan, M.J. Everything you always wanted to know about β3-AR * (* but were afraid to ask). Cells 2019, 8, 357. [Google Scholar] [CrossRef] [PubMed]
- Bundgaard, H.; Axelsson, A.; Thomsen, J.H.; Sørgaard, M.; Kofoed, K.F.; Hasselbalch, R.; Fry, N.A.S.; Valeur, N.; Boesgaard, S.; Gustafsson, F.; et al. The first-in-man randomized trial of a beta3 adrenoceptor agonist in chronic heart failure: The BEAT-HF trial. Eur. J. Heart Fail. 2017, 19, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.K.; Chi, A.; Melas, I.N.; Alexopoulos, L.G. Phosphoproteomics in drug discovery. Drug Dis. Today 2014, 19, 425–432. [Google Scholar] [CrossRef] [PubMed]



| Type | Molecular Species | Characteristics and Sites of Expression | Ligands and Other Comments |
|---|---|---|---|
| Soluble GC | α1β1 α2β1 | Form heterodimers consisting of two types of α (α1 and α2) and two types of β (β1 and β2). β2 does not form a dimer with α1 or α2. |
|
| Membrane-bound GC | GC-A (NPR-A) | GC-A and GC-B have natriuretic peptide (NP) binding domains in the amino-terminal region, thus GC-A is referred to as NPR-A, and GC-B is referred to as NPR-B. |
|
| GC-B (NPR-B) |
| ||
| NPR-C * | No intracellular GC domain, thus stimulation with NPs does not increase cGMP production. |
| |
| GC-C | Expressed predominantly in the intestines and partly in the kidneys, liver, and brain. |
| |
| GC-D | Pseudogene | - | |
| GC-E | Expressed in the retina | - | |
| GC-F | Expressed in the retina | - | |
| GC-G | Pseudogene | - |
| Effect on sGC Activity | Name of Drug or Compound |
|---|---|
| Stimulators | Riociguat (BAY 63–2521) |
| Vericiguat (BAY 1021189/MK-1242) | |
| Praliciguat (IW-1973) | |
| Zagociguat (CY-6463) | |
| MK-5475 | |
| Activators | Runcaciguat (BAY 1101042) |
| Mosliciguat (BAY 1237592) | |
| BI-685509 | |
| Mosliciguat (BAY 1237592) | |
| BI-685509 |
| PDE Family Members | Selectivity of Cyclic Nucleotides to Hydrolyze | Affinity for cAMP and cGMP | Activity Regulation |
|---|---|---|---|
| PDE1 | cAMP, cGMP | PDE1A: cGMP > cAMP PDE1B: cGMP > cAMP PDE1C: cAMP = cGMP |
|
| PDE2 | cAMP, cGMP | PDE2A: cAMP = cGMP |
|
| PDE3 | cAMP, cGMP | PDE3A, PDE3B: cAMP = cGMP (catalytic rates: cAMP > cGMP) |
|
| PDE4 | cAMP | - | - |
| PDE5 | cGMP | - |
|
| PDE6 | cGMP | - |
|
| PDE7 | cAMP | - | - |
| PDE8 | cAMP | - | - |
| PDE9 | cGMP | - |
|
| PDE10 | cAMP, cGMP | PDE10A: cAMP > cGMP | - |
| PDE11 | cAMP, cGMP | PDE11A: cAMP = cGMP | - |
| Classification | Subfamily | Isoforms | Characteristics |
|---|---|---|---|
| GLUTs | class I | GLUT1 GLUT2 GLUT3 GLUT4 GLUT14 |
|
| Class II | GLUT5 GLUT7 GLUT9 GLUT11 | - | |
| class III | GLUT6 GLUT8 GLUT10 GLUT12 HMIT |
|
| Classification | Isoform | Physiological Functions |
|---|---|---|
| SGLTs | SGLT1 SGLT2 SGLT3 SGLT4 SGLT5 SGLT6 (SMIT2 *) SMIT1 * |
|
| Drug | Study Population | Treatment | Primary and Secondary Endpoints | Main Findings and Conclusions |
|---|---|---|---|---|
| LCZ696 (PARAMOUNT trial) [112] |
| LCZ696 200 mg BID or valsartan 160 mg BID for 36 weeks | Primary:
|
|
| LCZ696 (PARADIGM-HF trial) [113] |
| LCZ696 200 mg BID or enalapril 10 mg BID | Primary:
|
|
| Sacubitril-valsartan (PARAGON-HF trial) [114] |
| Sacubitril-valsartan 200 mg BID or valsartan 160 mg BID | Primary:
|
|
| Drug | Study Population | Treatment | Primary and Secondary Endpoints | Main Findings and Conclusions |
|---|---|---|---|---|
| Ivabradine (BEAUTIFUL trial) [117] |
| Ivabradine 5–7.5 mg BID or placebo | Primary:
|
|
| Ivabradine (Echo substudy of BEAUTIFUL) [118] | Subgroup analysis of the BEAUTIFUL trial (N = 590) | Primary:
|
| |
| Ivabradine (SHIFT trial) [119] |
| Ivabradine titrated to a maximum of 7.5 mg BID or placebo | Primary:
|
|
| Ivabradine (Echo substudy of SHIFT) [120] | Subgroup analysis of the SHIFT trial (N = 411) | Primary:
|
|
| Drug | Study Population | Treatment | Primary and Secondary Endpoints | Main Findings and Conclusions |
|---|---|---|---|---|
| Canagliflozin (CHIEF-HF trial) [133] |
| Canagliflozin 100 mg OD or placebo for 12 weeks. | Primary:
|
|
| Dapagliflozin (DELIVER trial) [51] |
| Dapagliflozin 10 mg OD or placebo | Primary:
|
|
| Dapagliflozin (DAPA-HF trial) [134] |
| Dapagliflozin 10 mg OD or placebo | Primary:
|
|
| Dapagliflozin (DEFINE-HF trial) [135] |
| Dapagliflozin 10 mg OD or placebo for 12 weeks | Primary:
|
|
| Empagliflozin (EMPEROR-Reduced trial) [136] |
| Empagliflozin 10 mg OD or placebo | Primary:
|
|
| Empagliflozin (EMPEROR-Preserved trial) [137] |
| Empagliflozin 10 mg OD or placebo | Primary:
|
|
| Drug | Study Population | Treatment | Primary and Secondary Endpoints | Main Findings and Conclusion |
|---|---|---|---|---|
| Mirabegron (BEAT-HF trial) [143] |
| Mirabegron titrated to 150 mg BID or placebo for 6 months | Primary:
|
|
| Mirabegron (BEAT-HF-II trial) [90] |
| Mirabegron 300 mg daily or placebo for one week |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mangmool, S.; Duangrat, R.; Parichatikanond, W.; Kurose, H. New Therapeutics for Heart Failure: Focusing on cGMP Signaling. Int. J. Mol. Sci. 2023, 24, 12866. https://doi.org/10.3390/ijms241612866
Mangmool S, Duangrat R, Parichatikanond W, Kurose H. New Therapeutics for Heart Failure: Focusing on cGMP Signaling. International Journal of Molecular Sciences. 2023; 24(16):12866. https://doi.org/10.3390/ijms241612866
Chicago/Turabian StyleMangmool, Supachoke, Ratchanee Duangrat, Warisara Parichatikanond, and Hitoshi Kurose. 2023. "New Therapeutics for Heart Failure: Focusing on cGMP Signaling" International Journal of Molecular Sciences 24, no. 16: 12866. https://doi.org/10.3390/ijms241612866
APA StyleMangmool, S., Duangrat, R., Parichatikanond, W., & Kurose, H. (2023). New Therapeutics for Heart Failure: Focusing on cGMP Signaling. International Journal of Molecular Sciences, 24(16), 12866. https://doi.org/10.3390/ijms241612866