

Osteoarthritis: Role of Peroxisome Proliferator-Activated Receptors

 ,
,
Abstract
:1. Introduction
2. Overview of PPAR
2.1. Family of PPAR
2.2. PPAR Ligands
2.3. Function of PPAR
3. The Role of PPARα/γ in OA Chondrocytes/Cartilage
3.1. Inhibition of Matrix Metalloproteinases (MMPs) in OA Chondrocytes
3.2. Inhibition of Inflammatory-Related Factors in OA Cartilage
3.3. Inhibition of Chondrocyte Apoptosis
3.4. Regulating the Autophagic Activity of Chondrocytes
4. Effect of PPARs on OA-Related Signaling Pathways
4.1. NF-κB Signaling Pathway
4.2. Akt/mTOR Signaling Pathway
4.3. MAPK Signaling Pathway
5. Active Small Molecule Drugs Targeting Activation of PPARγ
5.1. Abietic Acid
5.2. Astragalin
5.3. Betulinic Acid
5.4. Mangiferin
5.5. Oridonin
5.6. Losartan
6. Adjusting PPARγ Promoter Methylation in Chondrocytes to Alleviate OA
6.1. 5-Aza-2′-deoxycytidine (5Aza)
6.2. Diacerein
6.3. The Active Constituents of Dabushen Decoction
7. Association between PPARα/γ and OA In Vivo
8. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zheng, S.; Tu, L.; Cicuttini, F.; Zhu, Z.; Han, W.; Antony, B.; Wluka, A.E.; Winzenberg, T.; Aitken, D.; Blizzard, L.; et al. Depression in patients with knee osteoarthritis: Risk factors and associations with joint symptoms. BMC Musculoskelet. Disord. 2021, 22, 40. [Google Scholar] [CrossRef]
- Hunter, D.J.; March, L.; Chew, M. Osteoarthritis in 2020 and beyond: A Lancet Commission. Lancet 2020, 396, 1711–1712. [Google Scholar] [CrossRef]
- Wang, M.; Shen, J.; Jin, H.; Im, H.J.; Sandy, J.; Chen, D. Recent progress in understanding molecular mechanisms of cartilage degeneration during osteoarthritis. Ann. N. Y. Acad. Sci. 2011, 1240, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Martel-Pelletier, J. Pathophysiology of osteoarthritis. Osteoarthr. Cartil. 1999, 7, 371–373. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, F.E.; Faich, G.; Goldstein, J.L.; Simon, L.S.; Pincus, T.; Whelton, A.; Makuch, R.; Eisen, G.; Agrawal, N.M.; Stenson, W.F.; et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: The CLASS study: A randomized controlled trial. Celecoxib long-term arthritis safety study. JAMA 2000, 284, 1247–1255. [Google Scholar] [CrossRef]
- Sun, M.M.; Beier, F.; Ratneswaran, A. Nuclear receptors as potential drug targets in osteoarthritis. Curr. Opin. Pharmacol. 2018, 40, 81–86. [Google Scholar] [CrossRef]
- Ratneswaran, A.; Sun, M.M.; Dupuis, H.; Sawyez, C.; Borradaile, N.; Beier, F. Nuclear receptors regulate lipid metabolism and oxidative stress markers in chondrocytes. J. Mol. Med. 2017, 95, 431–444. [Google Scholar] [CrossRef]
- Guan, Y. Peroxisome proliferator-activated receptor family and its relationship to renal complications of the metabolic syndrome. J. Am. Soc. Nephrol. 2004, 15, 2801–2815. [Google Scholar] [CrossRef]
- Dubois, V.; Eeckhoute, J.; Lefebvre, P.; Staels, B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J. Clin. Investig. 2017, 127, 1202–1214. [Google Scholar] [CrossRef]
- Genolet, R.; Wahli, W.; Michalik, L. PPARs as drug targets to modulate inflammatory responses? Curr. Drug Targets Inflamm. Allergy 2004, 3, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Ting, A.T.; Seed, B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 1998, 391, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef]
- Blanquart, C.; Barbier, O.; Fruchart, J.C.; Staels, B.; Glineur, C. Peroxisome proliferator-activated receptors: Regulation of transcriptional activities and roles in inflammation. J. Steroid Biochem. Mol. Biol. 2003, 85, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Delerive, P.; De Bosscher, K.; Besnard, S.; Vanden Berghe, W.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.C.; Tedgui, A.; Haegeman, G.; Staels, B. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J. Biol. Chem. 1999, 274, 32048–32054. [Google Scholar] [CrossRef]
- Hartley, A.; Ahmad, I. The role of PPARγ in prostate cancer development and progression. Br. J. Cancer 2023, 128, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef]
- Greene, M.E.; Blumberg, B.; McBride, O.W.; Yi, H.F.; Kronquist, K.; Kwan, K.; Hsieh, L.; Greene, G.; Nimer, S.D. Isolation of the human peroxisome proliferator activated receptor gamma cDNA: Expression in hematopoietic cells and chromosomal mapping. Gene Expr. 1995, 4, 281–299. [Google Scholar]
- Moreno, M.; Lombardi, A.; Silvestri, E.; Senese, R.; Cioffi, F.; Goglia, F.; Lanni, A.; de Lange, P. PPARs: Nuclear receptors controlled by, and controlling, nutrient handling through nuclear and cytosolic signaling. PPAR Res. 2010, 2010, 435689. [Google Scholar] [CrossRef]
- Kersten, S.; Desvergne, B.; Wahli, W. Roles of PPARs in health and disease. Nature 2000, 405, 421–424. [Google Scholar] [CrossRef]
- Glass, C.K.; Ogawa, S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 44–55. [Google Scholar] [CrossRef]
- Keller, H.; Dreyer, C.; Medin, J.; Mahfoudi, A.; Ozato, K.; Wahli, W. Fatty acids and retinoids control lipid metabolism through activation of peroxisome proliferator-activated receptor-retinoid X receptor heterodimers. Proc. Natl. Acad. Sci. USA 1993, 90, 2160–2164. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Umesono, K.; Noonan, D.J.; Heyman, R.A.; Evans, R.M. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature 1992, 358, 771–774. [Google Scholar] [CrossRef]
- Tan, C.K.; Zhuang, Y.; Wahli, W. Synthetic and natural Peroxisome Proliferator-Activated Receptor (PPAR) agonists as candidates for the therapy of the metabolic syndrome. Expert Opin. Ther. Targets 2017, 21, 333–348. [Google Scholar] [CrossRef]
- Bordji, K.; Grillasca, J.P.; Gouze, J.N.; Magdalou, J.; Schohn, H.; Keller, J.M.; Bianchi, A.; Dauça, M.; Netter, P.; Terlain, B. Evidence for the presence of peroxisome proliferator-activated receptor (PPAR) alpha and gamma and retinoid Z receptor in cartilage. PPARgamma activation modulates the effects of interleukin-1beta on rat chondrocytes. J. Biol. Chem. 2000, 275, 12243–12250. [Google Scholar] [CrossRef]
- Delerive, P.; Martin-Nizard, F.; Chinetti, G.; Trottein, F.; Fruchart, J.C.; Najib, J.; Duriez, P.; Staels, B. Peroxisome proliferator-activated receptor activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway. Circ. Res. 1999, 85, 394–402. [Google Scholar] [CrossRef]
- Stienstra, R.; Mandard, S.; Patsouris, D.; Maass, C.; Kersten, S.; Müller, M. Peroxisome proliferator-activated receptor alpha protects against obesity-induced hepatic inflammation. Endocrinology 2007, 148, 2753–2763. [Google Scholar] [CrossRef] [PubMed]
- Mandard, S.; Müller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha target genes. Cell. Mol. Life Sci. 2004, 61, 393–416. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, C.; Krey, G.; Keller, H.; Givel, F.; Helftenbein, G.; Wahli, W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell 1992, 68, 879–887. [Google Scholar] [CrossRef]
- Chawla, A.; Schwarz, E.J.; Dimaculangan, D.D.; Lazar, M.A. Peroxisome proliferator-activated receptor (PPAR) gamma: Adipose-predominant expression and induction early in adipocyte differentiation. Endocrinology 1994, 135, 798–800. [Google Scholar] [CrossRef]
- Tontonoz, P.; Hu, E.; Graves, R.A.; Budavari, A.I.; Spiegelman, B.M. mPPAR gamma 2: Tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994, 8, 1224–1234. [Google Scholar] [CrossRef]
- Tontonoz, P.; Graves, R.A.; Budavari, A.I.; Erdjument-Bromage, H.; Lui, M.; Hu, E.; Tempst, P.; Spiegelman, B.M. Adipocyte-specific transcription factor ARF6 is a heterodimeric complex of two nuclear hormone receptors, PPAR gamma and RXR alpha. Nucleic Acids Res. 1994, 22, 5628–5634. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Qi, C.; Korenberg, J.R.; Chen, X.N.; Noya, D.; Rao, M.S.; Reddy, J.K. Structural organization of mouse peroxisome proliferator-activated receptor gamma (mPPAR gamma) gene: Alternative promoter use and different splicing yield two mPPAR gamma isoforms. Proc. Natl. Acad. Sci. USA 1995, 92, 7921–7925. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Yamamoto, J.; Iwasaki, S.; Asaba, H.; Hamura, H.; Ikeda, Y.; Watanabe, M.; Magoori, K.; Ioka, R.X.; Tachibana, K.; et al. Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929. [Google Scholar] [CrossRef]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- Christofides, A.; Konstantinidou, E.; Jani, C.; Boussiotis, V.A. The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metabolism 2021, 114, 154338. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Fruchart, J.C. Therapeutic roles of peroxisome proliferator-activated receptor agonists. Diabetes 2005, 54, 2460–2470. [Google Scholar] [CrossRef]
- Park, S.; Baek, I.J.; Ryu, J.H.; Chun, C.H.; Jin, E.J. PPARα-ACOT12 axis is responsible for maintaining cartilage homeostasis through modulating de novo lipogenesis. Nat. Commun. 2022, 13, 3. [Google Scholar] [CrossRef]
- Vasheghani, F.; Zhang, Y.; Li, Y.H.; Blati, M.; Fahmi, H.; Lussier, B.; Roughley, P.; Lagares, D.; Endisha, H.; Saffar, B.; et al. PPAR gamma deficiency results in severe, accelerated osteoarthritis associated with aberrant mTOR signalling in the articular cartilage. Ann. Rheum. Dis. 2015, 74, 569–578. [Google Scholar] [CrossRef]
- Clockaerts, S.; Bastiaansen-Jenniskens, Y.M.; Feijt, C.; Verhaar, J.A.N.; Somville, J.; De Clerck, L.S.; Van Osch, G.J.V.M. Peroxisome proliferator activated receptor alpha activation decreases inflammatory and destructive responses in osteoarthritic cartilage. Osteoarthr. Cartil. 2011, 19, 895–902. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, X.L.; Qu, N.; Zhang, B.; Xia, C. Chondroprotection of PPAR alpha activation by WY14643 via autophagy involving Akt and ERK in LPS-treated mouse chondrocytes and osteoarthritis model. J. Cell. Mol. Med. 2019, 23, 2782–2793. [Google Scholar] [CrossRef]
- Nogueira-Recalde, U.; Lorenzo-Gómez, I.; Blanco, F.J.; Loza, M.I.; Grassi, D.; Shirinsky, V.; Shirinsky, I.; Lotz, M.; Robbins, P.D.; Domínguez, E.; et al. Fibrates as drugs with senolytic and autophagic activity for osteoarthritis therapy. EBioMedicine 2019, 45, 588–605. [Google Scholar] [CrossRef]
- Sabatini, M.; Bardiot, A.; Lesur, C.; Moulharat, N.; Thomas, M.; Richard, I.; Fradin, A. Effects of agonists of peroxisome proliferator-activated receptor gamma on proteoglycan degradation and matrix metalloproteinase production in rat cartilage in vitro. Osteoarthr. Cartil. 2002, 10, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ma, C.; Zhang, Y.; Zeng, Y.; Li, Y.; Wang, W. Pioglitazone inhibits advanced glycation end product-induced TNF-α and MMP-13 expression via the antagonism of NF-κB activation in chondrocytes. Pharmacology 2014, 94, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Chan, D.C.; Lan, K.C.; Wang, C.C.; Chen, C.M.; Chao, S.C.; Tsai, K.S.; Yang, R.S.; Liu, S.H. PPARγ is involved in the hyperglycemia-induced inflammatory responses and collagen degradation in human chondrocytes and diabetic mouse cartilages. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2015, 33, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-B.; Zhang, Y.; Chen, C.; Li, Y.-Q.; Ma, C.; Wang, Z.-J. Pioglitazone inhibits advanced glycation end product-induced matrix metalloproteinases and apoptosis by suppressing the activation of MAPK and NF-κB. Apoptosis Int. J. Program. Cell Death 2016, 21, 1082–1093. [Google Scholar] [CrossRef]
- Ma, C.; Zhang, Y.; Li, Y.-Q.; Chen, C.; Cai, W.; Zeng, Y.L. The role of PPARγ in advanced glycation end products-induced inflammatory response in human chondrocytes. PLoS ONE 2015, 10, e0125776. [Google Scholar] [CrossRef]
- Wang, Z.-J.; Zhang, H.B.; Chen, C.; Huang, H.; Liang, J.X. Effect of PPARG on AGEs-induced AKT/MTOR signaling-associated human chondrocytes autophagy. Cell Biol. Int. 2018, 42, 841–848. [Google Scholar] [CrossRef]
- Ni, S.; Li, D.; Wei, H.; Miao, K.S.; Zhuang, C. PPARγ attenuates interleukin-1β -Induced cell apoptosis by inhibiting NOX2/ROS/p38MAPK activation in osteoarthritis chondrocytes. Oxidative Med. Cell Longev. 2021, 2021, 5551338. [Google Scholar] [CrossRef]
- Burrage, P.S.; Mix, K.S.; Brinckerhoff, C.E. Matrix metalloproteinases: Role in arthritis. Front. Biosci. 2006, 11, 529–543. [Google Scholar] [CrossRef]
- Konttinen, Y.T.; Ainola, M.; Valleala, H.; Ma, J.; Ida, H.; Mandelin, J.; Kinne, R.W.; Santavirta, S.; Sorsa, T.; López-Otín, C.; et al. Analysis of 16 different matrix metalloproteinases (MMP-1 to MMP-20) in the synovial membrane: Different profiles in trauma and rheumatoid arthritis. Ann. Rheum. Dis. 1999, 58, 691–697. [Google Scholar] [CrossRef]
- Yoshihara, Y.; Nakamura, H.; Obata, K.; Yamada, H.; Hayakawa, T.; Fujikawa, K.; Okada, Y. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in synovial fluids from patients with rheumatoid arthritis or osteoarthritis. Ann. Rheum. Dis. 2000, 59, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Mehana, E.E.; Khafaga, A.F.; El-Blehi, S.S. The role of matrix metalloproteinases in osteoarthritis pathogenesis: An updated review. Life Sci. 2019, 234, 116786. [Google Scholar] [CrossRef]
- Hu, Q.C.; Ecker, M. Overview of MMP-13 as a promising target for the treatment of osteoarthritis. Int. J. Mol. Sci. 2021, 22, 1742. [Google Scholar] [CrossRef] [PubMed]
- Abramson, S.B. Nitric oxide in inflammation and pain associated with osteoarthritis. Arthritis Res. Ther. 2008, 10 (Suppl. S2), S2. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Levy, B. Success of prostaglandin E2 in structure-function is a challenge for structure-based therapeutics. Proc. Natl. Acad. Sci. USA 2003, 100, 8609–8611. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Pillinger, M.H.; Abramson, S.B. Prostaglandin E2 synthesis and secretion: The role of PGE2 synthases. Clin. Immunol. 2006, 119, 229–240. [Google Scholar] [CrossRef]
- Lamark, T.; Johansen, T. Aggrephagy: Selective disposal of protein aggregates by macroautophagy. Int. J. Cell Biol. 2012, 2012, 736905. [Google Scholar] [CrossRef]
- Caramés, B.; Olmer, M.; Kiosses, W.B.; Lotz, M.K. The relationship of autophagy defects to cartilage damage during joint aging in a mouse model. Arthritis Rheumatol. 2015, 67, 1568–1576. [Google Scholar] [CrossRef]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a promoter of longevity: Insights from model organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef]
- Bouderlique, T.; Vuppalapati, K.K.; Newton, P.T.; Li, L.; Barenius, B.; Chagin, A.S. Targeted deletion of Atg5 in chondrocytes promotes age-related osteoarthritis. Ann. Rheum. Dis. 2016, 75, 627–631. [Google Scholar] [CrossRef]
- Caramés, B.; Taniguchi, N.; Seino, D.; Blanco, F.J.; D’Lima, D.; Lotz, M. Mechanical injury suppresses autophagy regulators and pharmacologic activation of autophagy results in chondroprotection. Arthritis Rheum. 2012, 64, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Vinatier, C.; Domínguez, E.; Guicheux, J.; Caramés, B. Role of the inflammation-autophagy-senescence integrative network in osteoarthritis. Front. Physiol. 2018, 9, 706. [Google Scholar] [CrossRef]
- Abshirini, M.; Ilesanmi-Oyelere, B.L.; Kruger, M.C. Potential modulatory mechanisms of action by long-chain polyunsaturated fatty acids on bone cell and chondrocyte metabolism. Prog. Lipid. Res. 2021, 83, 101113. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Zhao, Q.; Liu, H.; Guo, Y.; Xu, H. PPAR-α Agonist WY-14643 Inhibits LPS-Induced Inflammation in Synovial Fibroblasts via NF-kB Pathway. J. Mol. Neurosci. 2016, 59, 544–553. [Google Scholar] [CrossRef]
- Wang, J.S.; Xiao, W.W.; Zhong, Y.S.; Li, X.D.; Du, S.X.; Xie, P.; Zheng, G.Z.; Han, J.M. Galectin-3 deficiency protects lipopolysaccharide-induced chondrocytes injury via regulation of TLR4 and PPAR-γ-mediated NF-κB signaling pathway. J. Cell Biochem. 2019, 120, 10195–10204. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guo, C.; Wei, Z.; He, X.; Kou, J.; Zhou, E.; Yang, Z.; Fu, Y. Morin suppresses inflammatory cytokine expression by downregulation of nuclear factor-κB and mitogen-activated protein kinase (MAPK) signaling pathways in lipopolysaccharide-stimulated primary bovine mammary epithelial cells. J. Dairy Sci. 2016, 99, 3016–3022. [Google Scholar] [CrossRef]
- Roman-Blas, J.A.; Jimenez, S.A. NF-kappaB as a potential therapeutic target in osteoarthritis and rheumatoid arthritis. Osteoarthr. Cartil. 2006, 14, 839–848. [Google Scholar] [CrossRef]
- Lipinski, M.M.; Hoffman, G.; Ng, A.; Zhou, W.; Py, B.F.; Hsu, E.; Liu, X.; Eisenberg, J.; Liu, J.; Blenis, J.; et al. A genome-wide siRNA screen reveals multiple mTORC1 independent signaling pathways regulating autophagy under normal nutritional conditions. Dev. Cell 2010, 18, 1041–1052. [Google Scholar] [CrossRef]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Crawford, R.; Xiao, Y. Vertical inhibition of the PI3K/Akt/mTOR pathway for the treatment of osteoarthritis. J. Cell Biochem. 2013, 114, 245–249. [Google Scholar] [CrossRef]
- Xue, J.F.; Shi, Z.M.; Zou, J.; Li, X.L. Inhibition of PI3K/AKT/mTOR signaling pathway promotes autophagy of articular chondrocytes and attenuates inflammatory response in rats with osteoarthritis. Biomed. Pharmacother. 2017, 89, 1252–1261. [Google Scholar] [CrossRef]
- Tang, Y.; Li, Y.; Xin, D.; Chen, L.; Xiong, Z.; Yu, X. Icariin alleviates osteoarthritis by regulating autophagy of chondrocytes by mediating PI3K/AKT/mTOR signaling. Bioengineered 2021, 12, 2984–2999. [Google Scholar] [CrossRef]
- Pan, X.; Shan, H.; Bai, J.; Gao, T.; Chen, B.; Shen, Z.; Zhou, H.; Lu, H.; Sheng, L.; Zhou, X. Four-octyl itaconate improves osteoarthritis by enhancing autophagy in chondrocytes via PI3K/AKT/mTOR signalling pathway inhibition. Commun. Biol. 2022, 5, 641. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.P.; Xu, W.C.; Zhao, X.; Zhang, C.; Lin, X.W.; Gong, M.X.; Fu, Z.J. Ozone induces autophagy by activating PPAR gamma/mTOR in rat chondrocytes treated with IL-1 beta. J. Orthop. Surg. Res. 2022, 17, 351. [Google Scholar] [CrossRef] [PubMed]
- Burgermeister, E.; Seger, R. MAPK kinases as nucleo-cytoplasmic shuttles for PPARgamma. Cell Cycle 2007, 6, 1539–1548. [Google Scholar] [CrossRef]
- Gardner, O.S.; Dewar, B.J.; Graves, L.M. Activation of mitogen-activated protein kinases by peroxisome proliferator-activated receptor ligands: An example of nongenomic signaling. Mol. Pharmacol. 2005, 68, 933–941. [Google Scholar] [CrossRef]
- Kong, D.; Zheng, T.; Zhang, M.; Wang, D.; Du, S.; Li, X.; Fang, J.; Cao, X. Static mechanical stress induces apoptosis in rat endplate chondrocytes through MAPK and mitochondria-dependent caspase activation signaling pathways. PLoS ONE 2013, 8, e69403. [Google Scholar] [CrossRef]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta 2007, 1773, 1213–1226. [Google Scholar] [CrossRef]
- Li, X.; Feng, K.; Li, J.; Yu, D.; Fan, Q.; Tang, T.; Yao, X.; Wang, X. Curcumin inhibits apoptosis of chondrocytes through activation ERK1/2 signaling pathways induced autophagy. Nutrients 2017, 9, 414. [Google Scholar] [CrossRef]
- Xu, C.; Ni, S.; Zhuang, C.; Li, C.; Zhao, G.; Jiang, S.; Wang, L.; Zhu, R.; van Wijnen, A.J.; Wang, Y. Polysaccharide from Angelica sinensis attenuates SNP-induced apoptosis in osteoarthritis chondrocytes by inducing autophagy via the ERK1/2 pathway. Arthritis Res. Ther. 2021, 23, 47. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Zhang, J.; Yuan, Y. Abietic acid attenuates IL-1β-induced inflammation in human osteoarthritis chondrocytes. Int. Immunopharmacol. 2018, 64, 110–115. [Google Scholar] [CrossRef]
- Ma, Z.; Piao, T.; Wang, Y.; Liu, J. Astragalin inhibits IL-1β-induced inflammatory mediators production in human osteoarthritis chondrocyte by inhibiting NF-κB and MAPK activation. Int. Immunopharmacol. 2015, 25, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.B.; Chen, A.M.; Wu, Q.; Li, X.; Li, H.N. Betulinic acid inhibits IL-1 beta-induced inflammation by activating PPAR-gamma in human osteoarthritis chondrocytes. Int. Immunopharmacol. 2015, 29, 687–692. [Google Scholar]
- Qu, Y.L.; Zhou, L.; Wang, C.L. Mangiferin Inhibits IL-1 beta-Induced Inflammatory Response by Activating PPAR-gamma in Human Osteoarthritis Chondrocytes. Inflammation 2017, 40, 52–57. [Google Scholar] [CrossRef]
- Jia, T.; Cai, M.M.; Ma, X.; Li, M.; Qiao, J.T.; Chen, T.X. Oridonin inhibits IL-1 beta-induced inflammation in human osteoarthritis chondrocytes by activating PPAR-gamma. Int. Immunopharmacol. 2019, 69, 382–388. [Google Scholar] [CrossRef]
- Deng, Z.; Chen, F.; Liu, Y.; Wang, J.; Lu, W.; Jiang, W.; Zhu, W. Losartan protects against osteoarthritis by repressing the TGF-β1 signaling pathway via upregulation of PPARγ. J. Orthop. Transl. 2021, 29, 30–41. [Google Scholar] [CrossRef]
- Kang, X.; Yang, Z.; Sheng, J.; Liu, J.B.; Xie, Q.Y.; Zheng, W.; Chen, K. Oleanolic acid prevents cartilage degeneration in diabetic mice via PPARγ associated mitochondrial stabilization. Biochem. Biophys. Res. Commun. 2017, 490, 834–840. [Google Scholar] [CrossRef]
- Qiu, L.; Zhang, M.; Li, C.H.; Hou, Y.H.; Liu, H.; Lin, J.; Yao, J.; Duan, D.Z.; Zhang, Y.X.; Li, M.; et al. Deciphering the active constituents of Dabushen decoction of ameliorating osteoarthritis via PPAR gamma preservation by targeting DNMT1. Front. Pharmacol. 2022, 13, 993498. [Google Scholar] [CrossRef]
- Justino, G.C.; Correia, C.F.; Mira, L.; Borges Dos Santos, R.M.; Martinho Simões, J.A.; Silva, A.M.; Santos, C.; Gigante, B. Antioxidant activity of a catechol derived from abietic acid. J. Agric. Food Chem. 2006, 54, 342–348. [Google Scholar] [CrossRef]
- Cho, I.H.; Gong, J.H.; Kang, M.K.; Lee, E.J.; Park, J.H.; Park, S.J.; Kang, Y.H. Astragalin inhibits airway eotaxin-1 induction and epithelial apoptosis through modulating oxidative stress-responsive MAPK signaling. BMC Pulm. Med. 2014, 14, 122. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kim, S.H. Inhibitory effect of astragalin on expression of lipopolysaccharide-induced inflammatory mediators through NF-κB in macrophages. Arch. Pharm. Res. 2011, 34, 2101–2107. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.K.; Tseng, C.K.; Chen, K.H.; Wu, S.H.; Liaw, C.C.; Lee, J.C. Betulinic acid exerts anti-hepatitis C virus activity via the suppression of NF-κB- and MAPK-ERK1/2-mediated COX-2 expression. Br. J. Pharmacol. 2015, 172, 4481–4492. [Google Scholar] [CrossRef]
- Cui, H.W.; He, Y.; Wang, J.; Gao, W.; Liu, T.; Qin, M.; Wang, X.; Gao, C.; Wang, Y.; Liu, M.Y.; et al. Synthesis of heterocycle-modified betulinic acid derivatives as antitumor agents. Eur. J. Med. Chem. 2015, 95, 240–248. [Google Scholar] [CrossRef]
- Xu, Y.; Xue, Y.; Wang, Y.; Feng, D.; Lin, S.; Xu, L. Multiple-modulation effects of oridonin on the production of proinflammatory cytokines and neurotrophic factors in LPS-activated microglia. Int. Immunopharmacol. 2009, 9, 360–365. [Google Scholar] [CrossRef]
- Thomas, M.; Fronk, Z.; Gross, A.; Willmore, D.; Arango, A.; Higham, C.; Nguyen, V.; Lim, H.; Kale, V.; McMillan, G.; et al. Losartan attenuates progression of osteoarthritis in the synovial temporomandibular and knee joints of a chondrodysplasia mouse model through inhibition of TGF-β1 signaling pathway. Osteoarthr. Cartil. 2019, 27, 676–686. [Google Scholar] [CrossRef]
- Lev Maor, G.; Yearim, A.; Ast, G. The alternative role of DNA methylation in splicing regulation. Trends Genet. 2015, 31, 274–280. [Google Scholar] [CrossRef]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; D’Souza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y.; et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253–257. [Google Scholar] [CrossRef]
- Zhu, X.; Chen, F.; Lu, K.; Wei, A.; Jiang, Q.; Cao, W. PPARγ preservation via promoter demethylation alleviates osteoarthritis in mice. Ann. Rheum. Dis. 2019, 78, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Pavelka, K.; Bruyère, O.; Cooper, C.; Kanis, J.A.; Leeb, B.F.; Maheu, E.; Martel-Pelletier, J.; Monfort, J.; Pelletier, J.P.; Rizzoli, R.; et al. Diacerein: Benefits, risks and place in the management of osteoarthritis. An opinion-based report from the ESCEO. Drugs Aging 2017, 34, 413. [Google Scholar] [CrossRef]
- Chen, X.R.; Zhu, X.B.; Dong, J.; Chen, F.; Gao, Q.; Zhang, L.J.; Cai, D.W.; Dong, H.; Ruan, B.J.; Wang, Y.X.; et al. Reversal of epigenetic peroxisome proliferator-activated receptor-γ suppression by diacerein alleviates oxidative stress and osteoarthritis in mice. Antioxid. Redox Signal. 2022, 37, 40–53. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Pelletier, J.P. Effects of diacerein at the molecular level in the osteoarthritis disease process. Ther. Adv. Musculoskelet. Dis. 2010, 2, 95–104. [Google Scholar] [CrossRef]
- Pelletier, J.P.; Raynauld, J.P.; Dorais, M.; Bessette, L.; Dokoupilova, E.; Morin, F.; Pavelka, K.; Paiement, P.; Martel-Pelletier, J. An international, multicentre, double-blind, randomized study (DISSCO): Effect of diacerein vs celecoxib on symptoms in knee osteoarthritis. Rheumatology 2020, 59, 3858–3868. [Google Scholar] [CrossRef]
- Kobayashi, T.; Notoya, K.; Naito, T.; Unno, S.; Nakamura, A.; Martel-Pelletier, J.; Pelletier, J.P. Pioglitazone, a peroxisome proliferator-activated receptor γ agonist, reduces the progression of experimental osteoarthritis in guinea pigs. Arthritis Rheum. 2005, 52, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Boileau, C.; Martel-Pelletier, J.; Fahmi, H.; Mineau, F.; Boily, M.; Pelletier, J.-P. The peroxisome proliferator-activated receptor gamma agonist pioglitazone reduces the development of cartilage lesions in an experimental dog model of osteoarthritis: In vivo protective effects mediated through the inhibition of key signaling and catabolic pathways. Arthritis Rheum. 2007, 56, 2288–2298. [Google Scholar] [PubMed]
- Li, Y.; Zhang, Y.; Chen, C.; Zhang, H.; Ma, C.; Xia, Y. Establishment of a rabbit model to study the influence of advanced glycation end products accumulation on osteoarthritis and the protective effect of pioglitazone. Osteoarthr. Cartil. 2016, 24, 307–314. [Google Scholar] [CrossRef] [PubMed]
- DeGroot, J.; Verzijl, N.; Wenting-van Wijk, M.J.; Jacobs, K.M.; Van El, B.; Van Roermund, P.M.; Bank, R.A.; Bijlsma, J.W.; TeKoppele, J.M.; Lafeber, F.P. Accumulation of advanced glycation end products as a molecular mechanism for aging as a risk factor in osteoarthritis. Arthritis Rheum. 2004, 50, 1207–1215. [Google Scholar] [CrossRef]
- Leong, D.J.; Sun, H.B. Events in articular chondrocytes with aging. Curr. Osteoporos. Rep. 2011, 9, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Hart, D.J.; Doyle, D.V.; Spector, T.D. Association between metabolic factors and knee osteoarthritis in women: The Chingford Study. J. Rheumatol. 1995, 22, 1118–1123. [Google Scholar]
- Sarkar, P.; Pain, S.; Sarkar, R.N.; Ghosal, R.; Mandal, S.K.; Banerjee, R. Rheumatological manifestations in diabetes mellitus. J. Indian Med. Assoc. 2008, 106, 593–594. [Google Scholar] [PubMed]
- Waine, H.; Nevinny, D.; Rosenthal, J.; Joffe, I.B. Association of osteoarthritis and diabetes mellitus. Tufts Folia Med. 1961, 7, 13–19. [Google Scholar]
- Schett, G.; Kleyer, A.; Perricone, C.; Sahinbegovic, E.; Iagnocco, A.; Zwerina, J.; Lorenzini, R.; Aschenbrenner, F.; Berenbaum, F.; D’Agostino, M.A.; et al. Diabetes is an independent predictor for severe osteoarthritis: Results from a longitudinal cohort study. Diabetes Care 2013, 36, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Nieves-Plaza, M.; Castro-Santana, L.E.; Font, Y.M.; Mayor, A.M.; Vilá, L.M. Association of hand or knee osteoarthritis with diabetes mellitus in a population of Hispanics from Puerto Rico. J. Clin. Rheumatol. 2013, 19, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Huedo, M.A.; Villanueva, M.; de Andres, A.L.; Hernandez-Barrera, V.; Carrasco-Garrido, P.; Gil, A.; Martinez, D.; Jiménez-Garcia, R. Trends 2001 to 2008 in incidence and immediate postoperative outcomes for major joint replacement among Spanish adults suffering diabetes. Eur. J. Orthop. Surg. Traumatol. 2013, 23, 53–59. [Google Scholar] [CrossRef]
- King, K.B.; Findley, T.W.; Williams, A.E.; Bucknell, A.L. Veterans with diabetes receive arthroplasty more frequently and at a younger age. Clin. Orthop. Relat. Res. 2013, 471, 3049–3054. [Google Scholar] [CrossRef]




{kind=link}
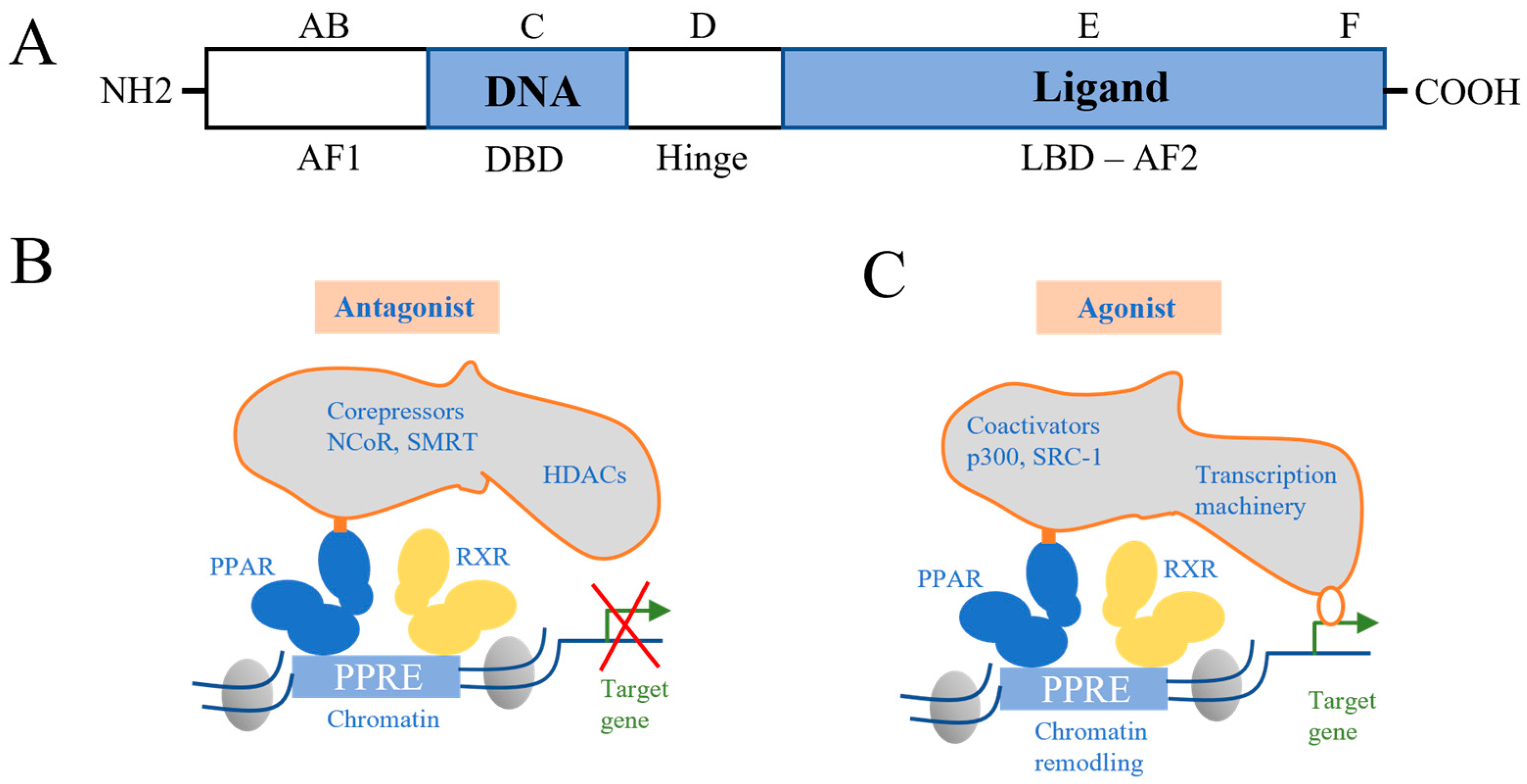{kind=link}
{kind=link}
| Subtype | Chromosome | Site of Expression | Natural Ligands | Synthetic Ligands | Biological Effects |
|---|---|---|---|---|---|
| PPARα | 22 | Liver, heart, skeletal muscles, BAT, intestine, kidneys, cartilage | Palmitic acid Palmitoleic acid Oleic acid Linoleic acid Stearic acid Pristanic acid Arachidonic acid Eicosatetraenoic acid Leukotriene B4 | Fenofibrate Bezafibrate Gemfibrozil Wy-14643 | fatty acid uptake and oxidation inflammation vascular function |
| PPARγ | 3 | WAT, liver, skeletal muscles, brain, stomach intestine, immune cells | Linoleic acid Arachidonic acid Eicosatetraenoic acid PGJ2, Linoleic acid 9-HODE 13-HODE | Troglitazone Pioglitazone Rosiglitazone BRL49653 GW1929 | fatty acid uptake and storage inflammation glucose homeostasis |
| PPARβ/δ | 6 | Ubiquitous | FA Retinoic acid Carbaprostacyclin | GW501516 GW0742 GW501516 | fatty acid metabolism inflammation macrophage lipid homeostasis |
| Isotype of PPAR | Therapeutic Agent | Author (Year) | Subjects | Findings |
|---|---|---|---|---|
| PPARα | Wy-14643 (PPARα agonist) | Clockaerts et al., 2011 [39] | Human OA cartilage explants | Inhibited the inflammatory and destructive responses |
| PPARα | Wy-14643 (PPARα agonist) | Zhou et al., 2018 [40] | Mouse chondrocytes | Promoted proteoglycan synthesis via autophagy enhancement in OA chondrocytes concomiant with the elevation of Akt and ERK phosphorylation |
| PPARα | Fenofibrate (PPARα agonist) | Nogueira-Recalde et al., 2019 [41] | Ageing human and OA Chondrocytes | Protect against cartilage degeneration seen with ageing and OA targeting lipid metabolism |
| PPARγ | 15d-PGJ2 and GI262570 (PPARγ agonist) | Sabatini et al., 2002 [42] | Rat cartilage | Inhibit cytokine-induced proteoglycan degradation mediated by both aggrecanase and MMPs |
| PPARγ | Pioglitazone (PPARγ agonist) | Chen et al., 2014 [43] | Rabbit chondrocytes | Modulates TNF-α and MMP-13 expression in cultured rabbit chondrocytes via NF-κB signaling |
| PPARγ | Pioglitazone (PPARγ agonist) | Chen et al., 2015 [44] | Human chondrocytes | Ameliorate hyperglyce mia-induced inflammatory responses and collagen degradation |
| PPARγ | Pioglitazone (PPARγ agonist) | Zhang et al., 2016 [45] | Human chondrocytes | Inhibits AGEs-induced MMPs and apoptosis by suppressing the activation of MAPK and NF-κB |
| PPARγ | Pioglitazone (PPARγ agonist) | Ma et al., 2015 [46] | Human chondrocytes | Inhibit the effects of AGEs-induced inflammatory response |
| PPARγ | Pioglitazone (PPARγ agonist) | Wang et al., 2018 [47] | Human chondrocytes | Maintains cell viability by activating the Akt/mTOR signaling pathway as well as inducing chondrocyte autophagy |
| PPARγ | GW1929 (PPARγ agonist) | Ni et al., 2021 [48] | Rat chondrocyte | Attenuates IL-1β-induced cell apoptosis by inhibiting NOX2/ ROS/p38MAPK activation |
| Isotype of PPAR | Therapeutic Agent | Author (Year) | Subjects | Findings |
|---|---|---|---|---|
| PPARγ | Ozone | Sun et al., 2022 [76] | Rat chondrocytes | Improved autophagy via activating PPARγ/ mTOR signaling and suppressing inflammation in chondrocytes treated with IL-1β |
| PPARγ | Abietic acid | Kang et al., 2018 [83] | Human OA chondrocytes | Suppressed IL-1β-induced inflammation in human OA chondrocytes |
| PPARγ | Astragalin | Ma et al., 2015 [84] | Human OA chondrocytes | Suppressed IL-1β-induced inflammatory mediators; inhibited IL-1β-induced NF-κB and MAPK activation |
| PPARγ | Betulinic acid | Wang et al., 2015 [85] | Human OA chondrocytes | Inhibited IL-1β-induced inflammation |
| PPARγ | Mangiferin | Qu et al., 2017 [86] | Human OA chondrocytes | Inhibits IL-1β-induced inflammatory response |
| PPARγ | Oridonin | Jia et al., 2019 [87] | Human OA chondrocytes | Inhibits IL-1β-induced inflammatory response |
| PPARγ | Losartan | Deng et al., 2021 [88] | Human OA chondrocytes | Arrest the progression of OA via upregulating PPARγ expression and inactivating the TGF-β1 signaling pathway |
| PPARγ | Oleanolic acid | Kang et al., 2017 [89] | Primary mouse articular chondrocytes | Prevented the high glucose-induced cell injury |
| PPARγ | Dabushen decoction | Qiu et al., 2022 [90] | Rat chondrocytes | Ameliorate IL-1β-induced downregulation of COL II and the production of MMP-13 |
| Isotype of PPAR | Therapeutic Agent | Author (Year) | Subjects | Findings |
|---|---|---|---|---|
| PPARα | Wy-14643 (PPARα agonist) | Zhou et al., 2018 [40] | DMM OA mice | Promoted proteoglycan synthesis via autophagy enhancement in OA chondrocytes concomitant with the elevation of Akt and ERK phosphorylation |
| PPARγ | Pioglitazone (PPARγ agonist) | Chen et al., 2015 [44] | Diabetic Mice | Show chondroprotection on mouse cartilage damage in diabetic mice |
| PPARγ | Pioglitazone (PPARγ agonist) | Zhang et al., 2016 [45] | AGEs-induced OA mice | Inhibiting the apoptosis and cartilage degradation |
| PPARγ | Losartan | Deng et al., 2021 [88] | DMM OA mice | Alleviates OA in DMM mice via PPARγ -mediated inactivation of the TGF-β1/Smad 2/3 signaling pathway |
| PPARγ | The active constituents dabushen decoction | Qiu et al., 2022 [90] | Papain with L-cysteine-induced OA rats | Ameliorate OA via PPARγ preservation by targeting DNMT1 |
| PPARγ | DNA demethyla- ting agent 5Aza (5-Aza-2′-deoxyc ytidine) | Zhu et al., 2019 [100] | Wild type C57BL/6 mice; PPARγ knockout mice | PPARγ presservation via promoter demethylation alleviates OA in mice |
| PPARγ | Diacerein | Chen et al., 2022 [102] | DMM OA mice | Alleviates oxidative stress and OA in mice by reversing epigenetic PPARγ suppression |
| PPARγ | Pioglitazone (PPARγ agonist) | Kobayashi et al., 2005 [105] | Guinea pigs | Reduce the progression of experimental OA in guinea pigs |
| PPARγ | Pioglitazone (PPARγ agonist) | Boileau et al., 2017 [106] | ACLT dogs | Inhibit major signaling pathways of inflammation and reduce the synthesis of cartilage catabolic factors responsible for articular cartilage degradation |
| PPARγ | Pioglitazone (PPARγ agonist) | Li et al., 2016 [107] | Rabbit OA model | Reduce the severity of the AGEs-induced OA in a rabbit model |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheng, W.; Wang, Q.; Qin, H.; Cao, S.; Wei, Y.; Weng, J.; Yu, F.; Zeng, H. Osteoarthritis: Role of Peroxisome Proliferator-Activated Receptors. Int. J. Mol. Sci. 2023, 24, 13137. https://doi.org/10.3390/ijms241713137
Sheng W, Wang Q, Qin H, Cao S, Wei Y, Weng J, Yu F, Zeng H. Osteoarthritis: Role of Peroxisome Proliferator-Activated Receptors. International Journal of Molecular Sciences. 2023; 24(17):13137. https://doi.org/10.3390/ijms241713137
Chicago/Turabian StyleSheng, Weibei, Qichang Wang, Haotian Qin, Siyang Cao, Yihao Wei, Jian Weng, Fei Yu, and Hui Zeng. 2023. "Osteoarthritis: Role of Peroxisome Proliferator-Activated Receptors" International Journal of Molecular Sciences 24, no. 17: 13137. https://doi.org/10.3390/ijms241713137
APA StyleSheng, W., Wang, Q., Qin, H., Cao, S., Wei, Y., Weng, J., Yu, F., & Zeng, H. (2023). Osteoarthritis: Role of Peroxisome Proliferator-Activated Receptors. International Journal of Molecular Sciences, 24(17), 13137. https://doi.org/10.3390/ijms241713137