
Hormonal Regulation and Stimulation Response of Jatropha curcas L. Homolog Overexpression on Tobacco Leaf Growth by Transcriptome Analysis


Abstract
:1. Introduction
2. Results
2.1. Effect of JcFTOE on Leaf Size and Sampling
2.2. Overview of RNA-Seq Data and Biological Processes Affected by JcFT Overexpression
2.3. Regulation of Cytokinin and Auxin Levels
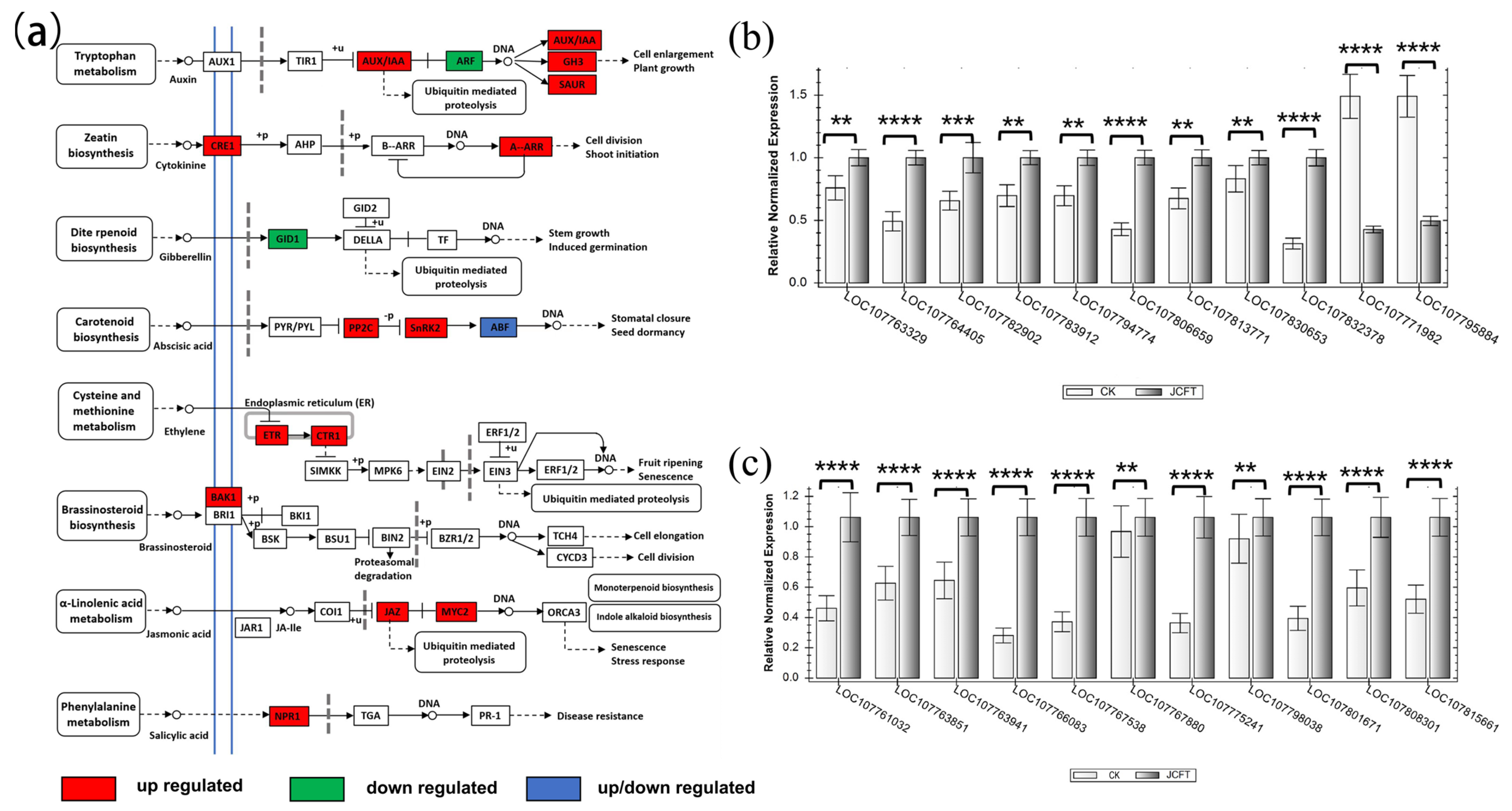
2.4. Regulation of Plant Hormone Signal Transduction
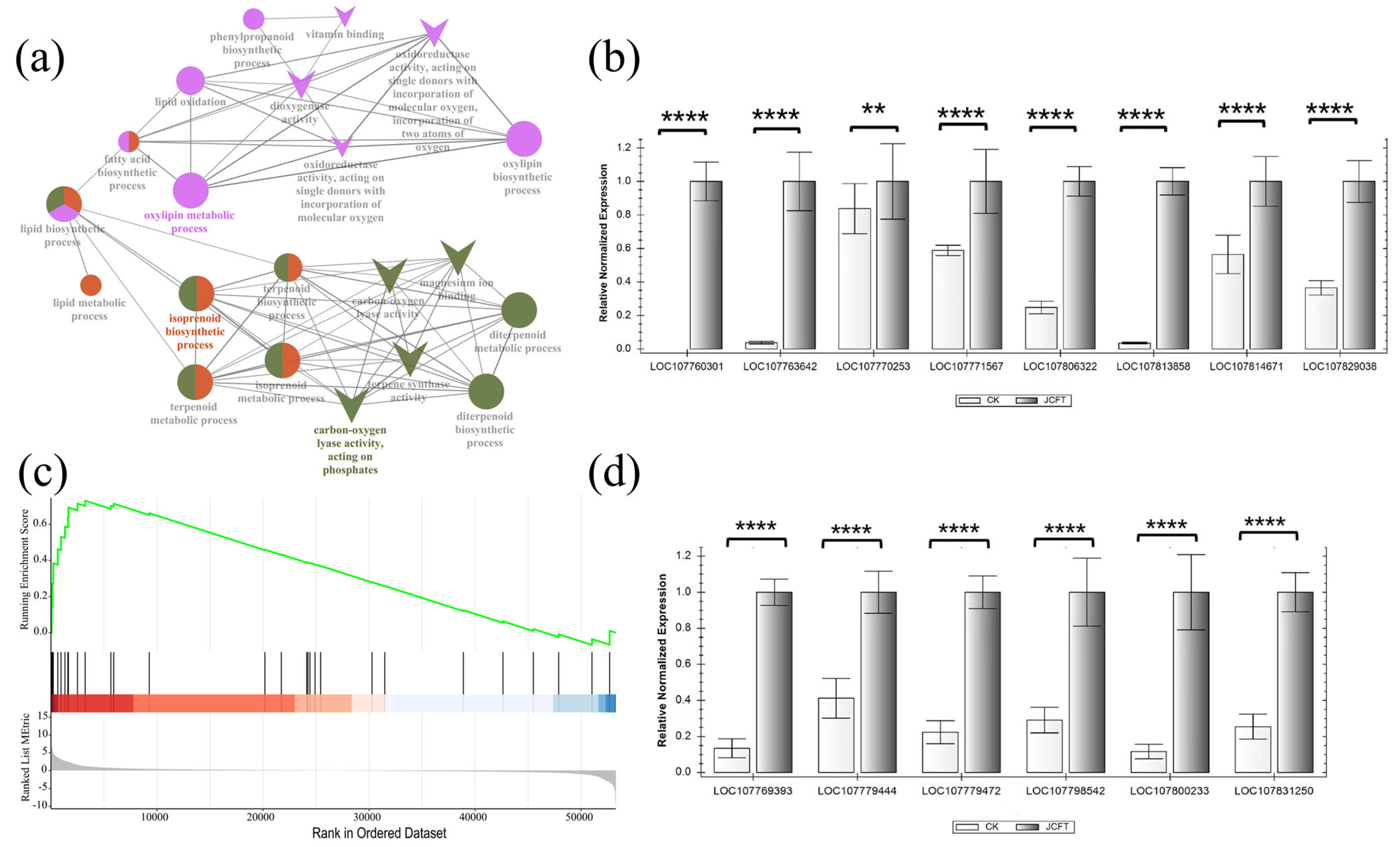
2.5. Effects of JcFTOE on Lipid Biosynthetic Process
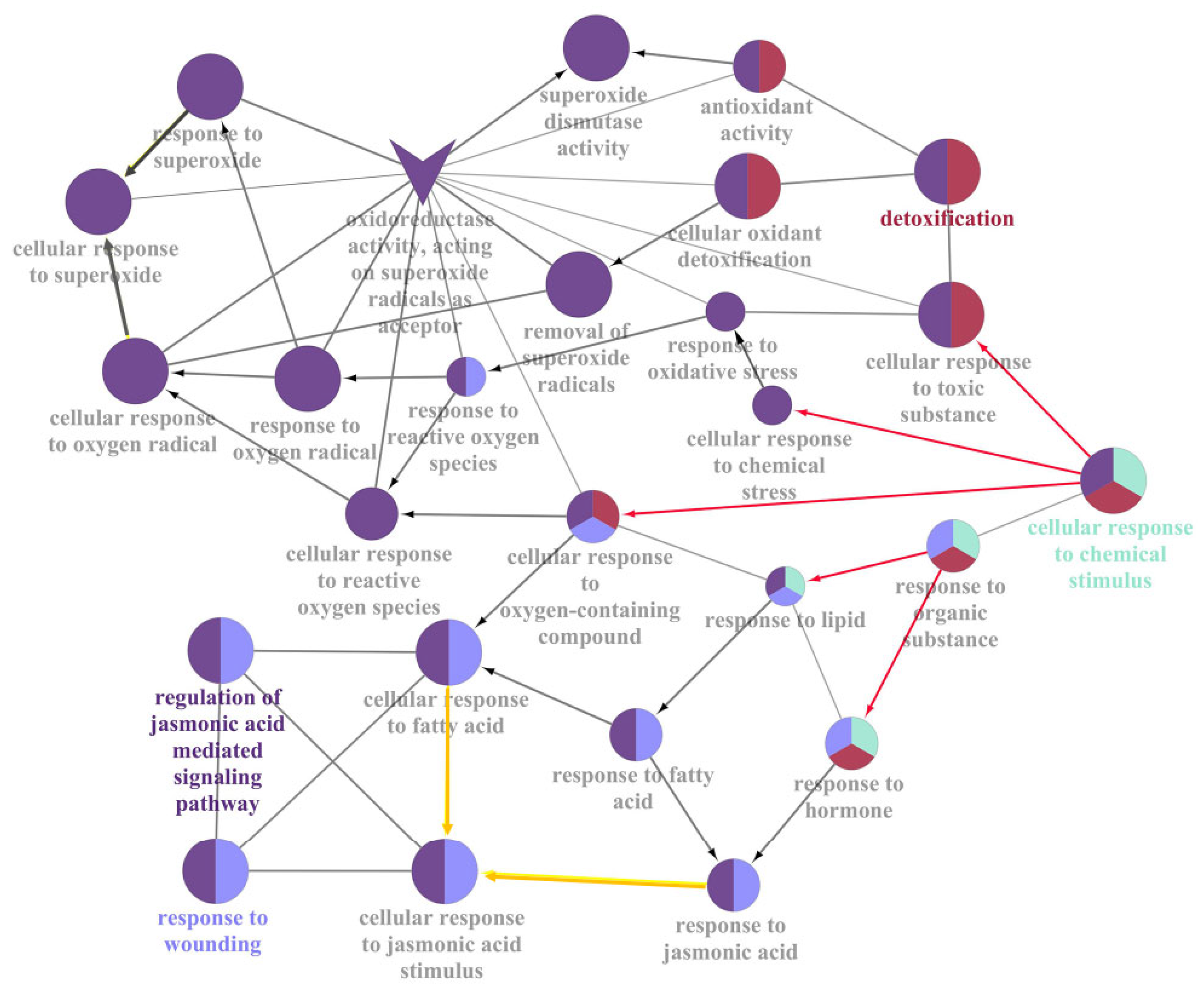
2.6. Stimulus Response Induced by JcFT Overexpression
3. Discussion
4. Materials and Methods
4.1. Vector Construction, Plant Transformation, and Cultivation
4.2. RNA Isolation, Library Construction, and Sequencing
4.3. Global and Differential Gene Expression Analysis of RNA-Seq Data
4.4. RT-qPCR Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shalit-Kaneh, A.; Eviatar-Ribak, T.; Horev, G.; Suss, N.; Aloni, R.; Eshed, Y.; Lifschitz, E. The flowering hormone florigen accelerates secondary cell wall biogenesis to harmonize vascular maturation with reproductive development. Proc. Natl. Acad. Sci. USA 2019, 116, 16127–16136. [Google Scholar] [CrossRef] [PubMed]
- Putterill, J.; Varkonyi-Gasic, E. FT and florigen long-distance flowering control in plants. Curr. Opin. Plant Biol. 2016, 33, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Wigge, P.A.; Kim, M.C.; Jaeger, K.E.; Busch, W.; Schmid, M.; Lohmann, J.U.; Weigel, D. Integration of spatial and temporal information during floral induction in Arabidopsis. Science 2005, 309, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Corbesier, L.; Vincent, C.; Jang, S.H.; Fornara, F.; Fan, Q.Z.; Slyle, I.; Giakountis, A.; Farrona, S.; Gissot, L.; Turnbull, C.; et al. FT protein movement contributes to long-distance signaling in floral induction of Arabidopsis. Science 2007, 316, 1030–1033. [Google Scholar] [CrossRef] [PubMed]
- Taoka, K.; Ohki, I.; Tsuji, H.; Furuita, K.; Hayashi, K.; Yanase, T.; Yamaguchi, M.; Nakashima, C.; Purwestri, Y.A.; Tamaki, S.; et al. 14-3-3 proteins act as intracellular receptors for rice Hd3a florigen. Nature 2011, 476, 332–335. [Google Scholar] [CrossRef] [PubMed]
- Turck, F.; Fornara, F.; Coupland, G. Regulation and identity of florigen: FLOWERING LOCUS T moves center stage. Annu. Rev. Plant Biol. 2008, 59, 573–594. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.W.H.; Weigel, D. Structural Features Determining Flower-Promoting Activity of Arabidopsis FLOWERING LOCUS T. Plant Cell 2014, 26, 552–564. [Google Scholar] [CrossRef]
- Zhu, Y.; Klasfeld, S.; Wagner, D. Molecular regulation of plant developmental transitions and plant architecture via PEPB family proteins: An update on mechanism of action. J. Exp. Bot. 2021, 72, 2301–2311. [Google Scholar] [CrossRef]
- Rinne, P.L.; Welling, A.; Vahala, J.; Ripel, L.; Ruonala, R.; Kangasjarvi, J.; van der Schoot, C. Chilling of dormant buds hyperinduces FLOWERING LOCUS T and recruits GA-inducible 1,3-beta-glucanases to reopen signal conduits and release dormancy in Populus. Plant Cell 2011, 23, 130–146. [Google Scholar] [CrossRef]
- Pin, P.A.; Nilsson, O. The multifaceted roles of FLOWERING LOCUS T in plant development. Plant Cell Environ. 2012, 35, 1742–1755. [Google Scholar] [CrossRef]
- Kinoshita, T.; Ono, N.; Hayashi, Y.; Morimoto, S.; Nakamura, S.; Soda, M.; Kato, Y.; Ohnishi, M.; Nakano, T.; Inoue, S.; et al. FLOWERING LOCUS T Regulates Stomatal Opening. Curr. Biol. 2011, 21, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- Shalit, A.; Rozman, A.; Goldshmidt, A.; Alvarez, J.P.; Bowman, J.L.; Eshed, Y.; Lifschitz, E. The flowering hormone florigen functions as a general systemic regulator of growth and termination. Proc. Natl. Acad. Sci. USA 2009, 106, 8392–8397. [Google Scholar] [CrossRef] [PubMed]
- Navarro, C.; Abelenda, J.A.; Cruz-Oro, E.; Cuellar, C.A.; Tamaki, S.; Silva, J.; Shimamoto, K.; Prat, S. Control of flowering and storage organ formation in potato by FLOWERING LOCUS T. Nature 2011, 478, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Izawa, T.; Oikawa, T.; Sugiyama, N.; Tanisaka, T.; Yano, M.; Shimamoto, K. Phytochrome mediates the external light signal to repress FT orthologs in photoperiodic flowering of rice. Gene Dev. 2002, 16, 2006–2020. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Muszynski, M.G.; Danilevskaya, O.N. The FT-Like ZCN8 Gene Functions as a Floral Activator and Is Involved in Photoperiod Sensitivity in Maize. Plant Cell 2011, 23, 942–960. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, Y.; Zhang, K.; Guo, D.; Cui, B.; Wang, X.; Huang, X. Promoting flowering, lateral shoot outgrowth, leaf development, and flower abscission in tobacco plants overexpressing cotton FLOWERING LOCUS T (FT)-like gene GhFT1. Front. Plant Sci. 2015, 6, 00454. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wu, Q.H.; Bo, Z.J.; Zhu, X.L.; Zhang, J.H.; Li, Q.Y.; Kong, W.Q. Comprehensive Effects of Flowering Locus T-Mediated Stem Growth in Tobacco. Front. Plant Sci. 2022, 13, 922919. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Yang, K.Z.; Wei, X.X.; Wang, X.Q. Revisiting the phosphatidylethanolamine-binding protein (PEBP) gene family reveals cryptic FLOWERING LOCUS T gene homologs in gymnosperms and sheds new light on functional evolution. New Phytol. 2016, 212, 730–744. [Google Scholar] [CrossRef] [PubMed]
- Blackman, B.K.; Strasburg, J.L.; Raduski, A.R.; Michaels, S.D.; Rieseberg, L.H. The role of recently derived FT paralogs in sunflower domestication. Curr. Biol. 2010, 20, 629–635. [Google Scholar] [CrossRef]
- Li, C.; Luo, L.; Fu, Q.; Niu, L.; Xu, Z.F. Isolation and functional characterization of JcFT, a FLOWERING LOCUS T (FT) homologous gene from the biofuel plant Jatropha curcas. BMC Plant Biol. 2014, 14, 1–12. [Google Scholar] [CrossRef]
- Nguyen, H.N.; Kambhampati, S.; Kisiala, A.; Seegobin, M.; Emery, R.J.N. The soybean (Glycine max L.) cytokinin oxidase/dehydrogenase multigene family; Identification of natural variations for altered cytokinin content and seed yield. Plant Direct 2021, 5, e00308. [Google Scholar] [CrossRef]
- Yamada, H.; Suzuki, T.; Terada, K.; Takei, K.; Ishikawa, K.; Miwa, K.; Yamashino, T.; Mizuno, T. The Arabidopsis AHK4 histidine kinase is a cytokinin-binding receptor that transduces cytokinin signals across the membrane. Plant Cell Physiol. 2001, 42, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, A.; Paul, L.K.; Sharma, E.; Jha, S.; Jain, M.; Khurana, J.P. OsRR6, a type-A response regulator in rice, mediates cytokinin, light and stress responses when over-expressed in Arabidopsis. Plant Physiol. Biochem. 2021, 161, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.F.; Li, H.; Hutchison, C.E.; Laskey, J.; Kieber, J.J. Biochemical and functional analysis of CTR1, a protein kinase that negatively regulates ethylene signaling in Arabidopsis. Plant J. 2003, 33, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wen, C.K. Arabidopsis ETR1 and ERS1 Differentially Repress the Ethylene Response in Combination with Other Ethylene Receptor Genes. Plant Physiol. 2012, 158, 1193–1207. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.N.; Sun, T.J.; Sun, Y.L.; Zhang, Y.J.; Radojicic, A.; Ding, Y.L.; Tian, H.N.; Huang, X.C.; Lan, J.M.; Chen, S.Y.; et al. Diverse Roles of the Salicylic Acid Receptors NPR1 and NPR3/NPR4 in Plant Immunity. Plant Cell 2020, 32, 4002–4016. [Google Scholar] [CrossRef] [PubMed]
- Avila, C.A.; Arevalo-Soliz, L.M.; Lorence, A.; Goggin, F.L. Expression of alpha-DIOXYGENASE 1 in tomato and Arabidopsis contributes to plant defenses against aphids. Mol. Plant Microbe Interact. 2013, 26, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Steppuhn, A.; Gaquerel, E.; Baldwin, I.T. The two alpha-dox genes of Nicotiana attenuata: Overlapping but distinct functions in development and stress responses. BMC Plant Biol. 2010, 10, 1–15. [Google Scholar] [CrossRef]
- Singh, P.; Arif, Y.; Miszczuk, E.; Bajguz, A.; Hayat, S. Specific Roles of Lipoxygenases in Development and Responses to Stress in Plants. Plants 2022, 11, 11070979. [Google Scholar] [CrossRef]
- Chini, A.; Fonseca, S.; Fernandez, G.; Adie, B.; Chico, J.M.; Lorenzo, O.; Garcia-Casado, G.; Lopez-Vidriero, I.; Lozano, F.M.; Ponce, M.R.; et al. The JAZ family of repressors is the missing link in jasmonate signalling. Nature 2007, 448, 666–671. [Google Scholar] [CrossRef]
- Saha, G.; Park, J.I.; Kayum, M.A.; Nou, I.S. A Genome-Wide Analysis Reveals Stress and Hormone Responsive Patterns of TIFY Family Genes in Brassica rapa. Front. Plant Sci. 2016, 7, 00936. [Google Scholar] [CrossRef] [PubMed]
- He, Y.H.; Chen, T.; Zeng, X.L. Genetic and Epigenetic Understanding of the Seasonal Timing of Flowering. Plant Commun. 2020, 1, 100008. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Harry, D.E.; Ma, C.; Yuceer, C.; Hsu, C.Y.; Vikram, V.; Shevchenko, O.; Etherington, E.; Strauss, S.H. Precocious flowering in trees: The FLOWERING LOCUS T gene as a research and breeding tool in Populus. J. Exp. Bot. 2010, 61, 2549–2560. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.J.; Zimmermann, M.M.; Wiedmann, D.R.; Lichtenauer, S.; Grundmann, L.; Muth, J.; Twyman, R.M.; Prüfer, D.; Noll, G.A. The major floral promoter NtFT5 in Tobacco (Nicotiana tabacum) is a promising target for crop improvement. Front. Plant Sci. 2020, 10, 01666. [Google Scholar] [CrossRef]
- Stortenbeker, N.; Bemer, M. The SAUR gene family: The plant’s toolbox for adaptation of growth and development. J. Exp. Bot. 2019, 70, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Wang, J.J.; Gao, Z.X.; Dong, J.; He, H.; Terzaghi, W.; Wei, N.; Deng, X.W.; Chen, H.D. Arabidopsis SAURs are critical for differential light regulation of the development of various organs. Proc. Natl. Acad. Sci. USA 2016, 113, 6071–6076. [Google Scholar] [CrossRef] [PubMed]
- Du, M.M.; Spalding, E.P.; Gray, W.M. Rapid Auxin-Mediated Cell Expansion. Annu. Rev. Plant Biol. 2020, 71, 379–402. [Google Scholar] [CrossRef] [PubMed]
- Dombrecht, B.; Xue, G.P.; Sprague, S.J.; Kirkegaard, J.A.; Ross, J.J.; Reid, J.B.; Fitt, G.P.; Sewelam, N.; Schenk, P.M.; Manners, J.M.; et al. MYC2 differentially modulates diverse jasmonate-dependent functions in Arabidopsis. Plant Cell 2007, 19, 2225–2245. [Google Scholar] [CrossRef]
- Breeze, E. Master MYCs: MYC2, the Jasmonate Signaling “Master Switch”. Plant Cell 2019, 31, 9–10. [Google Scholar] [CrossRef]
- Chung, H.S.; Howe, G.A. A Critical Role for the TIFY Motif in Repression of Jasmonate Signaling by a Stabilized Splice Variant of the JASMONATE ZIM-Domain Protein JAZ10 in Arabidopsis. Plant Cell 2009, 21, 131–145. [Google Scholar] [CrossRef]
- Saito, R.; Hayashi, K.; Nomoto, H.; Nakayama, M.; Takaoka, Y.; Saito, H.; Yamagami, S.; Muto, T.; Ueda, M. Extended JAZ degron sequence for plant hormone binding in jasmonate co-receptor of tomato SlCOI1-SlJAZ. Sci. Rep. 2021, 11, 13612. [Google Scholar] [CrossRef] [PubMed]
- Pasriga, R.; Yoon, J.; Cho, L.H.; An, G. Overexpression of RICE FLOWERING LOCUS T 1 (RFT1) Induces Extremely Early Flowering in Rice. Mol. Cells 2019, 42, 406–417. [Google Scholar] [PubMed]
- Lifschitz, E.; Eviatar, T.; Rozman, A.; Shalit, A.; Goldshmidt, A.; Amsellem, Z.; Alvarez, J.P.; Eshed, Y. The tomato FT ortholog triggers systemic signals that regulate growth and flowering and substitute for diverse environmental stimuli. Proc. Natl. Acad. Sci. USA 2006, 103, 6398–6403. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Walworth, A.E.; Mackie, C.; Song, G.Q. Overexpression of blueberry FLOWERING LOCUS T is associated with changes in the expression of phytohormone-related genes in blueberry plants. Hortic. Res. 2016, 3, 16053. [Google Scholar] [CrossRef] [PubMed]
- Adeyemo, O.S.; Chavarriaga, P.; Tohme, J.; Fregene, M.; Davis, S.J.; Setter, T.L. Overexpression of Arabidopsis FLOWERING LOCUS T (FT) gene improves floral development in cassava (Manihot esculenta, Crantz). PLoS ONE 2017, 12, e0181460. [Google Scholar] [CrossRef] [PubMed]
- Odipio, J.; Getu, B.; Chauhan, R.D.; Alicai, T.; Bart, R.; Nusinow, D.A.; Taylor, N.J. Transgenic overexpression of endogenous FLOWERING LOCUS T-like gene MeFT1 produces early flowering in cassava. PLoS ONE 2020, 15, e0227199. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, J.; Warthmann, N.; Kuttner, F.; Schmid, M. Export of FT protein from phloem companion cells is sufficient for floral induction in Arabidopsis. Curr. Biol. 2007, 17, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Lifschitz, E.; Ayre, B.G.; Eshed, Y. Florigen and anti-florigen-A systemic mechanism for coordinating growth and termination in flowering plants. Front. Plant Sci. 2014, 5, 00465. [Google Scholar] [CrossRef]
- Notaguchi, M.; Abe, M.; Kimura, T.; Daimon, Y.; Kobayashi, T.; Yamaguchi, A.; Tomita, Y.; Dohi, K.; Mori, M.; Araki, T. Long-Distance, Graft-Transmissible Action of Arabidopsis FLOWERING LOCUS T Protein to Promote Flowering. Plant Cell Physiol. 2008, 49, 1645–1658. [Google Scholar] [CrossRef]
- George, C.P.; Liradevito, L.M.; Wampler, S.L.; Kadonaga, J.T. A Spectrum of Mechanisms for the Assembly of the Rna-Polymerase-Ii Transcription Preinitiation Complex. Mol. Cell. Biol. 1995, 15, 1049–1059. [Google Scholar] [CrossRef]
- Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Landmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Schulze, S.K.; Kanwar, R.; Golzenleuchter, M.; Therneau, T.M.; Beutler, A.S. SERE: Single-parameter quality control and sample comparison for RNA-Seq. BMC Genom. 2012, 13, 524. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.H.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Wang, L.K.; Feng, Z.X.; Wang, X.; Wang, X.W.; Zhang, X.G. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.Y.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆CT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Schmidt, G.W.; Delaney, S.K. Stable internal reference genes for normalization of real-time RT-PCR in tobacco (Nicotiana tabacum) during development and abiotic stress. Mol. Genet. Genom. 2010, 283, 233–241. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Label of the Most Significant Term Per Group | Amount of DEGs | % Terms Per Group |
|---|---|---|
| Regulation of JA-mediated signaling pathway | 84 | 33.33 |
| Response to wounding | 78 | 15.69 |
| Carbon-oxygen lyase activity, acting on phosphates | 68 | 10.78 |
| Detoxification | 91 | 9.8 |
| Oxylipin metabolism | 75 | 9.8 |
| Isoprenoid biosynthetic process | 71 | 6.86 |
| Regulation of hormone levels | 21 | 3.92 |
| Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen | 55 | 3.92 |
| Cellular response to chemical stimulus | 86 | 3.92 |
| Carboxy-lyase activity | 11 | 0.98 |
| UDP-glycosyltransferase activity | 26 | 0.98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Q.; Zheng, D.; Lian, N.; Zhu, X.; Wu, J. Hormonal Regulation and Stimulation Response of Jatropha curcas L. Homolog Overexpression on Tobacco Leaf Growth by Transcriptome Analysis. Int. J. Mol. Sci. 2023, 24, 13183. https://doi.org/10.3390/ijms241713183
Wu Q, Zheng D, Lian N, Zhu X, Wu J. Hormonal Regulation and Stimulation Response of Jatropha curcas L. Homolog Overexpression on Tobacco Leaf Growth by Transcriptome Analysis. International Journal of Molecular Sciences. 2023; 24(17):13183. https://doi.org/10.3390/ijms241713183
Chicago/Turabian StyleWu, Qiuhong, Dongchao Zheng, Na Lian, Xuli Zhu, and Jun Wu. 2023. "Hormonal Regulation and Stimulation Response of Jatropha curcas L. Homolog Overexpression on Tobacco Leaf Growth by Transcriptome Analysis" International Journal of Molecular Sciences 24, no. 17: 13183. https://doi.org/10.3390/ijms241713183
APA StyleWu, Q., Zheng, D., Lian, N., Zhu, X., & Wu, J. (2023). Hormonal Regulation and Stimulation Response of Jatropha curcas L. Homolog Overexpression on Tobacco Leaf Growth by Transcriptome Analysis. International Journal of Molecular Sciences, 24(17), 13183. https://doi.org/10.3390/ijms241713183