
The Bidirectional Interplay of α-Synuclein with Lipids in the Central Nervous System and Its Implications for the Pathogenesis of Parkinson’s Disease


Abstract
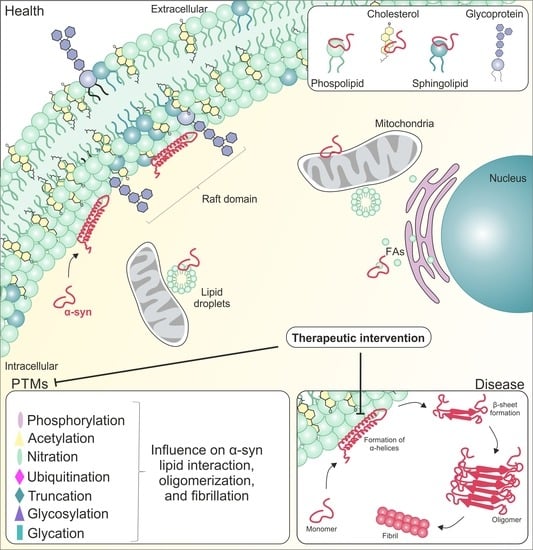
:
1. Introduction
2. Lipids and Lipid Metabolism
2.1. Lipid Metabolism in the Brain
2.1.1. Cholesterol
2.1.2. Fatty Acids
2.1.3. Sphingolipids
2.1.4. Phospholipids
3. α-syn and Lipids
3.1. Alterations of Lipids and Their Metabolism in PD
3.1.1. FA Metabolism
3.1.2. Cholesterol Metabolism
3.1.3. Sphingolipid Metabolism
3.1.4. Glycerophospholipid Metabolism
3.2. Effects of Missense Mutations on the Binding Capacity of α-syn to Lipids
3.3. Binding Capacity of Posttranslational Modified α-syn to Lipids
3.3.1. Phosphorylation
3.3.2. Acetylation
3.3.3. Nitration
3.3.4. Ubiquitination
3.3.5. Truncation
3.3.6. Glycosylation
3.3.7. Glycation
4. Therapeutic Potential
4.1. Enzymes Involved in Lipid Metabolism
4.2. Membrane Binding of α-syn
4.3. PTMs
4.4. Neuroprotective Lipids
4.5. Environmental Factors
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.; Flanagan, L.; De Silva, H.R.; Kittel, A.; Saitoh, T. The precursor protein of non-Aβ component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef]
- Gardai, S.J.; Mao, W.; Schüle, B.; Babcock, M.; Schoebel, S.; Lorenzana, C.; Alexander, J.; Kim, S.; Glick, H.; Hilton, K. Elevated alpha-synuclein impairs innate immune cell function and provides a potential peripheral biomarker for Parkinson’s disease. PLoS ONE 2013, 8, e71634. [Google Scholar] [CrossRef]
- Badawy, S.M.M.; Okada, T.; Kajimoto, T.; Hirase, M.; Matovelo, S.A.; Nakamura, S.; Yoshida, D.; Ijuin, T.; Nakamura, S.-I. Extracellular α-synuclein drives sphingosine 1-phosphate receptor subtype 1 out of lipid rafts, leading to impaired inhibitory G-protein signaling. J. Biol. Chem. 2018, 293, 8208–8216. [Google Scholar] [CrossRef]
- Bellani, S.; Sousa, V.L.; Ronzitti, G.; Valtorta, F.; Meldolesi, J.; Chieregatti, E. The regulation of synaptic function by α-synuclein. Commun. Integr. Biol. 2010, 3, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, G.; Puzzo, D.; Sant Angelo, A.; Trinchese, F.; Arancio, O. Alpha-synuclein: Between synaptic function and dysfunction. Histol. Histopathol. 2003, 18, 1257–1266. [Google Scholar]
- Cheng, F.; Vivacqua, G.; Yu, S. The role of alpha-synuclein in neurotransmission and synaptic plasticity. J. Chem. Neuroanat. 2011, 42, 242–248. [Google Scholar] [CrossRef]
- Baptista, M.J.; O’Farrell, C.; Daya, S.; Ahmad, R.; Miller, D.W.; Hardy, J.; Farrer, M.J.; Cookson, M.R. Co-ordinate transcriptional regulation of dopamine synthesis genes by α-synuclein in human neuroblastoma cell lines. J. Neurochem. 2003, 85, 957–968. [Google Scholar] [CrossRef]
- Fauvet, B.; Mbefo, M.K.; Fares, M.-B.; Desobry, C.; Michael, S.; Ardah, M.T.; Tsika, E.; Coune, P.; Prudent, M.; Lion, N. α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 2012, 287, 15345–15364. [Google Scholar] [CrossRef]
- Weinreb, P.H.; Zhen, W.; Poon, A.W.; Conway, K.A.; Lansbury, P.T. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef]
- Dettmer, U.; Newman, A.J.; Soldner, F.; Luth, E.S.; Kim, N.C.; von Saucken, V.E.; Sanderson, J.B.; Jaenisch, R.; Bartels, T.; Selkoe, D. Parkinson-causing alpha-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat. Commun. 2015, 6, 7314. [Google Scholar] [CrossRef]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef]
- Wang, W.; Perovic, I.; Chittuluru, J.; Kaganovich, A.; Nguyen, L.T.; Liao, J.; Auclair, J.R.; Johnson, D.; Landeru, A.; Simorellis, A.K. A soluble α-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. USA 2011, 108, 17797–17802. [Google Scholar] [CrossRef]
- Burre, J.; Sharma, M.; Sudhof, T.C. alpha-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. USA 2014, 111, E4274–E4283. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.W.; Drakulic, S.; Deas, E.; Ouberai, M.; Aprile, F.A.; Arranz, R.; Ness, S.; Roodveldt, C.; Guilliams, T.; De-Genst, E.J.; et al. Structural characterization of toxic oligomers that are kinetically trapped during alpha-synuclein fibril formation. Proc. Natl. Acad. Sci. USA 2015, 112, E1994–E2003. [Google Scholar] [CrossRef]
- Fusco, G.; Chen, S.W.; Williamson, P.T.; Cascella, R.; Perni, M.; Jarvis, J.A.; Cecchi, C.; Vendruscolo, M.; Chiti, F.; Cremades, N. Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science 2017, 358, 1440–1443. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef]
- Osterberg, V.R.; Spinelli, K.J.; Weston, L.J.; Luk, K.C.; Woltjer, R.L.; Unni, V.K. Progressive aggregation of alpha-synuclein and selective degeneration of lewy inclusion-bearing neurons in a mouse model of parkinsonism. Cell. Rep. 2015, 10, 1252–1260. [Google Scholar] [CrossRef]
- Coon, E.A.; Singer, W. Synucleinopathies. Continuum 2020, 26, 72–92. [Google Scholar] [CrossRef]
- Parkinson, J. An essay on the shaking palsy. Arch. Neurol. 1969, 20, 441–445. [Google Scholar] [CrossRef]
- National Collaborating Centre for Chronic Conditions. Parkinson’s Disease: National Clinical Guideline for Diagnosis and Management in Primary and Secondary Care; Royal College of Physicians of London: London, UK, 2006. [Google Scholar]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Damier, P.; Hirsch, E.; Agid, Y.; Graybiel, A. The substantia nigra of the human brain: II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 1999, 122, 1437–1448. [Google Scholar] [CrossRef]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. AlaSOPro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat. Genet 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B. The new mutation, E46K, of α-synuclein causes parkinson and Lewy body dementia. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- George, J.M.; Jin, H.; Woods, W.S.; Clayton, D.F. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 1995, 15, 361–372. [Google Scholar] [CrossRef]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of a-Synuclein Secondary Structure upon Binding to Synthetic Membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.A.; Ivanova, M.I.; Sawaya, M.R.; Cascio, D.; Reyes, F.E.; Shi, D.; Sangwan, S.; Guenther, E.L.; Johnson, L.M.; Zhang, M. Structure of the toxic core of α-synuclein from invisible crystals. Nature 2015, 525, 486–490. [Google Scholar] [CrossRef]
- Salveson, P.J.; Spencer, R.K.; Nowick, J.S. X-ray crystallographic structure of oligomers formed by a toxic β-hairpin derived from α-synuclein: Trimers and higher-order oligomers. J. Am. Chem. Soc. 2016, 138, 4458–4467. [Google Scholar] [CrossRef]
- Giasson, B.I.; Murray, I.V.; Trojanowski, J.Q.; Lee, V.M.-Y. A hydrophobic stretch of 12 amino acid residues in the middle of α-synuclein is essential for filament assembly. J. Biol. Chem. 2001, 276, 2380–2386. [Google Scholar] [CrossRef]
- Anderson, E.N.; Hirpa, D.; Zheng, K.H.; Banerjee, R.; Gunawardena, S. The non-amyloidal component region of α-synuclein is important for α-synuclein transport within axons. Front. Cell. Neurosci. 2020, 13, 540. [Google Scholar] [CrossRef]
- Park, S.M.; Jung, H.Y.; Chung, K.C.; Rhim, H.; Park, J.H.; Kim, J. Stress-Induced Aggregation Profiles of GST− α-Synuclein Fusion Proteins: Role of the C-Terminal Acidic Tail of α-Synuclein in Protein Thermosolubility and Stability. Biochemistry 2002, 41, 4137–4146. [Google Scholar] [CrossRef]
- Kim, T.D.; Paik, S.R.; Yang, C.-H. Structural and functional implications of C-terminal regions of α-synuclein. Biochemistry 2002, 41, 13782–13790. [Google Scholar] [CrossRef]
- Farzadfard, A.; Pedersen, J.N.; Meisl, G.; Somavarapu, A.K.; Alam, P.; Goksøyr, L.; Nielsen, M.A.; Sander, A.F.; Knowles, T.P.; Pedersen, J.S. The C-terminal tail of α-synuclein protects against aggregate replication but is critical for oligomerization. Commun. Biol. 2022, 5, 123. [Google Scholar] [CrossRef]
- Flagmeier, P.; Meisl, G.; Vendruscolo, M.; Knowles, T.P.; Dobson, C.M.; Buell, A.K.; Galvagnion, C. Mutations associated with familial Parkinson’s disease alter the initiation and amplification steps of α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2016, 113, 10328–10333. [Google Scholar] [CrossRef]
- Zunke, F.; Moise, A.C.; Belur, N.R.; Gelyana, E.; Stojkovska, I.; Dzaferbegovic, H.; Toker, N.J.; Jeon, S.; Fredriksen, K.; Mazzulli, J.R. Reversible Conformational Conversion of alpha-Synuclein into Toxic Assemblies by Glucosylceramide. Neuron 2018, 97, 92–107. [Google Scholar] [CrossRef]
- Tracey, T.; Kirk, S.; Steyn, F.; Ngo, S. The role of lipids in the central nervous system and their pathological implications in amyotrophic lateral sclerosis. Semin. Cell Dev. Biol. 2021, 112, 69–81. [Google Scholar] [CrossRef]
- Vandenheuvel, F.A. Study of biological structure at the molecular level with stereomodel projections I. The lipids in the myelin sheath of nerve. J. Am. Oil Chem. Soc. 1963, 40, 455–471. [Google Scholar] [CrossRef]
- Eliezer, D.; Kutluay, E.; Bussell, R., Jr.; Browne, G. Conformational properties of α-synuclein in its free and lipid-associated states. J. Mol. Biol. 2001, 307, 1061–1073. [Google Scholar]
- Bussell Jr, R.; Eliezer, D. A structural and functional role for 11-mer repeats in α-synuclein and other exchangeable lipid binding proteins. J. Mol. Biol. 2003, 329, 763–778. [Google Scholar] [CrossRef]
- Willingham, S.; Outeiro, T.F.; DeVit, M.J.; Lindquist, S.L.; Muchowski, P.J. Yeast genes that enhance the toxicity of a mutant huntingtin fragment or α-synuclein. Science 2003, 302, 1769–1772. [Google Scholar] [CrossRef]
- Nuber, S.; Nam, A.Y.; Rajsombath, M.M.; Cirka, H.; Hronowski, X.; Wang, J.; Hodgetts, K.; Kalinichenko, L.S.; Müller, C.P.; Lambrecht, V. A Stearoyl–Coenzyme A Desaturase Inhibitor Prevents Multiple Parkinson Disease Phenotypes in α-Synuclein Mice. Ann. Neurol. 2021, 89, 74–90. [Google Scholar] [CrossRef]
- Pranav, K.; Usha, M. Life Sciences Fundamental and Practice Part-1; Pathfinder Publishers: Sydney, Australia, 2014. [Google Scholar]
- Morell, P.; Toews, A.D. Biochemistry of lipids. Handb. Clin. Neurol. 1996, 22, 33–49. [Google Scholar]
- Williams, K.A.; Deber, C.M. The structure and function of central nervous system myelin. Crit. Rev. Clin. Lab. Sci. 1993, 30, 29–64. [Google Scholar] [CrossRef]
- Masoro, E.J. Lipids and Lipid Metabolism. Ann. Rev. Physioi. 1977, 39, 301–321. [Google Scholar] [CrossRef]
- Fahy, E.; Cotter, D.; Sud, M.; Subramaniam, S. Lipid classification, structures and tools. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2011, 1811, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Rustan, A.C.; Drevon, C.A. Fatty acids: Structures and properties. e LS 2001, 1–7. [Google Scholar] [CrossRef]
- Spector, R. Fatty acid transport through the blood-brain barrier. J. Neurochem. 1988, 50, 639–643. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Jurevics, H.; Morell, P. Cholesterol for synthesis of myelin is made locally, not imported into brain. J. Neurochem. 1995, 64, 895–901. [Google Scholar] [CrossRef]
- Saher, G.; Stumpf, S.K. Cholesterol in myelin biogenesis and hypomyelinating disorders. Biochim. Biophys. Acta 2015, 1851, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, K.; Rodriguez-Rodriguez, R.; Gaebler, A.; Casals, N.; Scheller, A.; Kuerschner, L. Astrocytes and oligodendrocytes in grey and white matter regions of the brain metabolize fatty acids. Sci. Rep. 2017, 7, 10779. [Google Scholar]
- van Deijk, A.L.F.; Camargo, N.; Timmerman, J.; Heistek, T.; Brouwers, J.F.; Mogavero, F.; Mansvelder, H.D.; Smit, A.B.; Verheijen, M.H. Astrocyte lipid metabolism is critical for synapse development and function in vivo. Glia 2017, 65, 670–682. [Google Scholar]
- Camargo, N.; Goudriaan, A.; van Deijk, A.-L.F.; Otte, W.M.; Brouwers, J.F.; Lodder, H.; Gutmann, D.H.; Nave, K.-A.; Dijkhuizen, R.M.; Mansvelder, H.D. Oligodendroglial myelination requires astrocyte-derived lipids. PLoS Biol. 2017, 15, e1002605. [Google Scholar]
- Chen, J.; Zhang, X.; Kusumo, H.; Costa, L.G.; Guizzetti, M. Cholesterol efflux is differentially regulated in neurons and astrocytes: Implications for brain cholesterol homeostasis. Biochim. Biophys. Acta 2013, 1831, 263–275. [Google Scholar] [CrossRef]
- Hirsch-Reinshagen, V.; Zhou, S.; Burgess, B.L.; Bernier, L.; McIsaac, S.A.; Chan, J.Y.; Tansley, G.H.; Cohn, J.S.; Hayden, M.R.; Wellington, C.L. Deficiency of ABCA1 impairs apolipoprotein E metabolism in brain. J. Biol. Chem. 2004, 279, 41197–41207. [Google Scholar]
- Swanson, L.W.; Simmons, D.M.; Hofmann, S.L.; Goldstein, J.L.; Brown, M.S. Localization of mRNA for low density lipoprotein receptor and a cholesterol synthetic enzyme in rabbit nervous system by in situ hybridization. Proc. Natl. Acad. Sci. USA 1988, 85, 9821–9825. [Google Scholar]
- Pitas, R.; Boyles, J.; Lee, S.; Hui, D.; Weisgraber, K. Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B, E (LDL) receptors in the brain. J. Biol. Chem. 1987, 262, 14352–14360. [Google Scholar]
- Zhao, S.; Hu, X.; Park, J.; Zhu, Y.; Zhu, Q.; Li, H.; Luo, C.; Han, R.; Cooper, N.; Qiu, M. Selective expression of LDLR and VLDLR in myelinating oligodendrocytes. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2007, 236, 2708–2712. [Google Scholar]
- Simons, M.; Krämer, E.-M.; Thiele, C.; Stoffel, W.; Trotter, J. Assembly of Myelin by Association of Proteolipid Protein with Cholesterol- and Galactosylceramid-rich Membrane Domains. J. Cell Biol. 2000, 151, 143–153. [Google Scholar]
- Werner, H.B.; Krämer-Albers, E.M.; Strenzke, N.; Saher, G.; Tenzer, S.; Ohno-Iwashita, Y.; De Monasterio-Schrader, P.; Möbius, W.; Moser, T.; Griffiths, I.R. A critical role for the cholesterol-associated proteolipids PLP and M6B in myelination of the central nervous system. Glia 2013, 61, 567–586. [Google Scholar]
- Garcia Corrales, A.V.; Haidar, M.; Bogie, J.F.J.; Hendriks, J.J.A. Fatty Acid Synthesis in Glial Cells of the CNS. Int. J. Mol. Sci. 2021, 22, 8159. [Google Scholar] [CrossRef]
- Moore, S.A. Polyunsaturated fatty acid synthesis and release by brain-derived cells in vitro. J. Mol. Neurosci. 2001, 16, 195–200. [Google Scholar]
- Bazan, N. Supply of n-3 polyunsaturated fatty acids and their significance in the central nervous system. Nutr. Brain 1990, 8, 1–24. [Google Scholar]
- Chandel, N.S. Lipid metabolism. Cold Spring Harb. Perspect. Biol. 2021, 13, a040576. [Google Scholar]
- Ralhan, I.; Chang, C.-L.; Lippincott-Schwartz, J.; Ioannou, M.S. Lipid droplets in the nervous system. J. Cell Biol. 2021, 220, e202102136. [Google Scholar]
- Unger, R.H.; Clark, G.O.; Scherer, P.E.; Orci, L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2010, 1801, 209–214. [Google Scholar]
- Rambold, A.S.; Cohen, S.; Lippincott-Schwartz, J. Fatty acid trafficking in starved cells: Regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 2015, 32, 678–692. [Google Scholar] [CrossRef]
- Cabodevilla, A.; Sánchez-Caballero, L.; Picatoste, F.; Gubern, A.; Claro, E. Cell survival during complete nutrient deprivation depends on lipid droplet-fueled β-oxidation of fatty acids (577.3). FASEB J. 2014, 28, 577-3. [Google Scholar]
- Edmond, J.; Robbins, R.; Bergstrom, J.; Cole, R.; De Vellis, J. Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J. Neurosci. Res. 1987, 18, 551–561. [Google Scholar]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Tracey, T.J.; Steyn, F.J.; Wolvetang, E.J.; Ngo, S.T. Neuronal Lipid Metabolism: Multiple Pathways Driving Functional Outcomes in Health and Disease. Front. Mol. Neurosci. 2018, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Burré, J. The synaptic function of α-synuclein. J. Park. Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.H.; Nielsen, M.S.; Jakes, R.; Dotti, C.G.; Goedert, M. Binding of α-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J. Biol. Chem. 1998, 273, 26292–26294. [Google Scholar] [CrossRef]
- Segrest, J.P.; Jackson, R.L.; Morrisett, J.D.; Gotto Jr, A.M. A molecular theory of lipid—Protein interactions in the plasma lipoproteins. FEBS Lett. 1974, 38, 247–253. [Google Scholar] [CrossRef]
- van Rooijen, B.D.; Claessens, M.M.; Subramaniam, V. Lipid bilayer disruption by oligomeric α-synuclein depends on bilayer charge and accessibility of the hydrophobic core. Biochim. Biophys. Acta (BBA)-Biomembr. 2009, 1788, 1271–1278. [Google Scholar] [CrossRef]
- Jo, E.; McLaurin, J.; Yip, C.M.; George-Hyslop, P.S.; Fraser, P.E. α-Synuclein membrane interactions and lipid specificity. J. Biol. Chem. 2000, 275, 34328–34334. [Google Scholar] [CrossRef]
- Rhoades, E.; Ramlall, T.F.; Webb, W.W.; Eliezer, D. Quantification of α-synuclein binding to lipid vesicles using fluorescence correlation spectroscopy. Biophys. J. 2006, 90, 4692–4700. [Google Scholar] [CrossRef]
- Narayanan, V.; Guo, Y.; Scarlata, S. Fluorescence studies suggest a role for α-synuclein in the phosphatidylinositol lipid signaling pathway. Biochemistry 2005, 44, 462–470. [Google Scholar] [CrossRef]
- Fantini, J.; Carlus, D.; Yahi, N. The fusogenic tilted peptide (67-78) of alpha-synuclein is a cholesterol binding domain. Biochim. Biophys. Acta 2011, 1808, 2343–2351. [Google Scholar] [CrossRef]
- Fantini, J.; Yahi, N. Molecular basis for the glycosphingolipid-binding specificity of alpha-synuclein: Key role of tyrosine 39 in membrane insertion. J. Mol. Biol. 2011, 408, 654–669. [Google Scholar] [CrossRef]
- Wang, G.F.; Li, C.; Pielak, G.J. 19F NMR studies of alpha-synuclein-membrane interactions. Protein Sci. 2010, 19, 1686–1691. [Google Scholar] [CrossRef] [PubMed]
- Fortin, D.L.; Troyer, M.D.; Nakamura, K.; Kubo, S.; Anthony, M.D.; Edwards, R.H. Lipid rafts mediate the synaptic localization of alpha-synuclein. J. Neurosci. 2004, 24, 6715–6723. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-W.; Yang, R.; Guo, J.-C.; Ren, H.-M.; Zha, X.-L.; Cheng, J.-S.; Cai, D.-F. Localization of α-synuclein to mitochondria within midbrain of mice. Neuroreport 2007, 18, 1543–1546. [Google Scholar] [CrossRef] [PubMed]
- Bodner, C.R.; Maltsev, A.S.; Dobson, C.M.; Bax, A. Differential phospholipid binding of α-synuclein variants implicated in Parkinson’s disease revealed by solution NMR spectroscopy. Biochemistry 2010, 49, 862–871. [Google Scholar] [CrossRef]
- Fusco, G.; De Simone, A.; Gopinath, T.; Vostrikov, V.; Vendruscolo, M.; Dobson, C.M.; Veglia, G. Direct observation of the three regions in α-synuclein that determine its membrane-bound behaviour. Nat. Commun. 2014, 5, 3827. [Google Scholar] [CrossRef]
- Bartels, T.; Ahlstrom, L.S.; Leftin, A.; Kamp, F.; Haass, C.; Brown, M.F.; Beyer, K. The N-terminus of the intrinsically disordered protein α-synuclein triggers membrane binding and helix folding. Biophys. J. 2010, 99, 2116–2124. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Fusco, G.; Pape, T.; Stephens, A.D.; Mahou, P.; Costa, A.R.; Kaminski, C.F.; Kaminski Schierle, G.S.; Vendruscolo, M.; Veglia, G.; Dobson, C.M. Structural basis of synaptic vesicle assembly promoted by α-synuclein. Nat. Commun. 2016, 7, 12563. [Google Scholar] [CrossRef] [PubMed]
- Cremades, N.; Cohen, S.I.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef]
- Grey, M.; Linse, S.; Nilsson, H.; Brundin, P.; Sparr, E. Membrane interaction of α-synuclein in different aggregation states. J. Park. Dis. 2011, 1, 359–371. [Google Scholar] [CrossRef]
- Fanning, S.; Selkoe, D.; Dettmer, U. Parkinson’s disease: Proteinopathy or lipidopathy? NPJ Park. Dis. 2020, 6, 3. [Google Scholar] [CrossRef]
- Klemann, C.; Martens, G.J.M.; Sharma, M.; Martens, M.B.; Isacson, O.; Gasser, T.; Visser, J.E.; Poelmans, G. Integrated molecular landscape of Parkinson’s disease. NPJ Park. Dis. 2017, 3, 14. [Google Scholar] [CrossRef]
- Golovko, M.Y.; Faergeman, N.J.; Cole, N.B.; Castagnet, P.I.; Nussbaum, R.L.; Murphy, E.J. α-synuclein gene deletion decreases brain palmitate uptake and alters the palmitate metabolism in the absence of α-synuclein palmitate binding. Biochemistry 2005, 44, 8251–8259. [Google Scholar] [CrossRef] [PubMed]
- Golovko, M.Y.; Rosenberger, T.A.; Faergeman, N.J.; Feddersen, S.; Cole, N.B.; Pribill, I.; Berger, J.; Nussbaum, R.L.; Murphy, E.J. Acyl-CoA synthetase activity links wild-type but not mutant α-synuclein to brain arachidonate metabolism. Biochemistry 2006, 45, 6956–6966. [Google Scholar] [CrossRef]
- Golovko, M.Y.; Rosenberger, T.A.; Feddersen, S.; Færgeman, N.J.; Murphy, E.J. α-Synuclein gene ablation increases docosahexaenoic acid incorporation and turnover in brain phospholipids. J. Neurochem. 2007, 101, 201–211. [Google Scholar] [CrossRef]
- Campos, S.S.; Alza, N.P.; Salvador, G.A. Lipid metabolism alterations in the neuronal response to A53T α-synuclein and Fe-induced injury. Arch. Biochem. Biophys. 2018, 655, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Barceló-Coblijn, G.; Golovko, M.Y.; Weinhofer, I.; Berger, J.; Murphy, E.J. Brain neutral lipids mass is increased in α-synuclein gene-ablated mice. J. Neurochem. 2007, 101, 132–141. [Google Scholar] [CrossRef]
- Ruipérez, V.; Darios, F.; Davletov, B. Alpha-synuclein, lipids and Parkinson’s disease. Prog. Lipid Res. 2010, 49, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Xicoy, H.; Wieringa, B.; Martens, G.J. The role of lipids in Parkinson’s disease. Cells 2019, 8, 27. [Google Scholar] [CrossRef]
- Jin, U.; Park, S.J.; Park, S.M. Cholesterol metabolism in the brain and its association with Parkinson’s disease. Exp. Neurobiol. 2019, 28, 554. [Google Scholar] [CrossRef]
- Huang, X.; Sterling, N.W.; Du, G.; Sun, D.; Stetter, C.; Kong, L.; Zhu, Y.; Neighbors, J.; Lewis, M.M.; Chen, H. Brain cholesterol metabolism and Parkinson’s disease. Mov. Disord. 2019, 34, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Indellicato, R.; Trinchera, M. The link between Gaucher disease and Parkinson’s disease sheds light on old and novel disorders of sphingolipid metabolism. Int. J. Mol. Sci. 2019, 20, 3304. [Google Scholar] [CrossRef] [PubMed]
- Quinville, B.M.; Deschenes, N.M.; Ryckman, A.E.; Walia, J.S. A comprehensive review: Sphingolipid metabolism and implications of disruption in sphingolipid homeostasis. Int. J. Mol. Sci. 2021, 22, 5793. [Google Scholar] [CrossRef] [PubMed]
- Fanning, S.; Haque, A.; Imberdis, T.; Baru, V.; Barrasa, M.I.; Nuber, S.; Termine, D.; Ramalingam, N.; Ho, G.P.H.; Noble, T.; et al. Lipidomic Analysis of alpha-Synuclein Neurotoxicity Identifies Stearoyl CoA Desaturase as a Target for Parkinson Treatment. Mol. Cell 2019, 73, 1001–1014. [Google Scholar] [CrossRef]
- Yoo, D.; Lim, Y.; Son, Y.; Rho, H.; Shin, C.; Ahn, T.-B. Dietary intake and plasma levels of polyunsaturated fatty acids in early-stage Parkinson’s disease. Sci. Rep. 2021, 11, 12489. [Google Scholar] [CrossRef]
- Assayag, K.; Yakunin, E.; Loeb, V.; Selkoe, D.J.; Sharon, R. Polyunsaturated fatty acids induce α-synuclein-related pathogenic changes in neuronal cells. Am. J. Pathol. 2007, 171, 2000–2011. [Google Scholar] [CrossRef]
- Sharon, R.; Bar-Joseph, I.; Mirick, G.E.; Serhan, C.N.; Selkoe, D.J. Altered fatty acid composition of dopaminergic neurons expressing α-synuclein and human brains with α-synucleinopathies. J. Biol. Chem. 2003, 278, 49874–49881. [Google Scholar] [CrossRef]
- Cheng, D.; Jenner, A.M.; Shui, G.; Cheong, W.F.; Mitchell, T.W.; Nealon, J.R.; Kim, W.S.; McCann, H.; Wenk, M.R.; Halliday, G.M. Lipid pathway alterations in Parkinson’s disease primary visual cortex. PLoS ONE 2011, 6, e17299. [Google Scholar] [CrossRef]
- García-Sanz, P.; Orgaz, L.; Fuentes, J.M.; Vicario, C.; Moratalla, R. Cholesterol and multilamellar bodies: Lysosomal dysfunction in GBA-Parkinson disease. Autophagy 2018, 14, 717–718. [Google Scholar] [CrossRef]
- Paul, R.; Choudhury, A.; Kumar, S.; Giri, A.; Sandhir, R.; Borah, A. Cholesterol contributes to dopamine-neuronal loss in MPTP mouse model of Parkinson’s disease: Involvement of mitochondrial dysfunctions and oxidative stress. PLoS ONE 2017, 12, e0171285. [Google Scholar] [CrossRef]
- Raju, A.; Jaisankar, P.; Borah, A.; Mohanakumar, K.P. 1-methyl-4-phenylpyridinium-induced death of differentiated SH-SY5Y neurons is potentiated by cholesterol. Ann. Neurosci. 2017, 24, 243–251. [Google Scholar] [CrossRef]
- Mullin, S.; Hughes, D.; Mehta, A.; Schapira, A. Neurological effects of glucocerebrosidase gene mutations. Eur. J. Neurol. 2019, 26, 388-e329. [Google Scholar] [CrossRef] [PubMed]
- McCampbell, A.; Truong, D.; Broom, D.C.; Allchorne, A.; Gable, K.; Cutler, R.G.; Mattson, M.P.; Woolf, C.J.; Frosch, M.P.; Harmon, J.M. Mutant SPTLC1 dominantly inhibits serine palmitoyltransferase activity in vivo and confers an age-dependent neuropathy. Hum. Mol. Genet 2005, 14, 3507–3521. [Google Scholar] [CrossRef]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Futerman, A.H.; Riezman, H. The ins and outs of sphingolipid synthesis. Trends Cell Biol. 2005, 15, 312–318. [Google Scholar] [CrossRef]
- Farmer, K.; Smith, C.A.; Hayley, S.; Smith, J. Major alterations of phosphatidylcholine and lysophosphotidylcholine lipids in the substantia nigra using an early stage model of Parkinson’s disease. Int. J. Mol. Sci. 2015, 16, 18865–18877. [Google Scholar] [CrossRef]
- Lobasso, S.; Tanzarella, P.; Vergara, D.; Maffia, M.; Cocco, T.; Corcelli, A. Lipid profiling of parkin-mutant human skin fibroblasts. J. Cell. Physiol. 2017, 232, 3540–3551. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, T.J.; Yao, L.; Lucena, A. Product inhibition of secreted phospholipase A2 may explain lysophosphatidylcholines’ unexpected therapeutic properties. J. Inflamm. 2008, 5, 17. [Google Scholar] [CrossRef]
- Riekkinen, P.; Rinne, U.K.; Pelliniemi, T.-T.; Sonninen, V. Interaction between dopamine and phospholipids: Studies of the substantia nigra in parkinson disease patients. Arch. Neurol. 1975, 32, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Hattingen, E.; Magerkurth, J.; Pilatus, U.; Mozer, A.; Seifried, C.; Steinmetz, H.; Zanella, F.; Hilker, R. Phosphorus and proton magnetic resonance spectroscopy demonstrates mitochondrial dysfunction in early and advanced Parkinson’s disease. Brain 2009, 132, 3285–3297. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, S.; Liou, L.-C.; Ren, Q.; Zhang, Z.; Caldwell, G.A.; Caldwell, K.A.; Witt, S.N. Phosphatidylethanolamine deficiency disrupts α-synuclein homeostasis in yeast and worm models of Parkinson disease. Proc. Natl. Acad. Sci. USA 2014, 111, E3976–E3985. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.L.; Tippireddy, S.; Feriante, J.; Woltjer, R.L. Augmented frontal cortex diacylglycerol levels in Parkinson’s disease and Lewy Body Disease. PLoS ONE 2018, 13, e0191815. [Google Scholar] [CrossRef]
- Nakamura, K.; Nemani, V.M.; Wallender, E.K.; Kaehlcke, K.; Ott, M.; Edwards, R.H. Optical reporters for the conformation of α-synuclein reveal a specific interaction with mitochondria. J. Neurosci. 2008, 28, 12305–12317. [Google Scholar] [CrossRef] [PubMed]
- Parihar, M.; Parihar, A.; Fujita, M.; Hashimoto, M.; Ghafourifar, P. Mitochondrial association of alpha-synuclein causes oxidative stress. Cell. Mol. Life Sci. 2008, 65, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, K.; Sinha, M.; Pham, C.L.L.; Jana, S.; Chanda, D.; Cappai, R.; Chakrabarti, S. α-Synuclein induced membrane depolarization and loss of phosphorylation capacity of isolated rat brain mitochondria: Implications in Parkinson’s disease. FEBS Lett. 2010, 584, 1571–1576. [Google Scholar] [CrossRef] [PubMed]
- Bodner, C.R.; Dobson, C.M.; Bax, A. Multiple tight phospholipid-binding modes of α-synuclein revealed by solution NMR spectroscopy. J. Mol. Biol. 2009, 390, 775–790. [Google Scholar] [CrossRef]
- Pancoe, S.X.; Wang, Y.J.; Shimogawa, M.; Perez, R.M.; Giannakoulias, S.; Petersson, E.J. Effects of Mutations and Post-Translational Modifications on α-Synuclein In Vitro Aggregation. J. Mol. Biol. 2022, 434, 167859. [Google Scholar] [CrossRef] [PubMed]
- Daida, K.; Shimonaka, S.; Shiba-Fukushima, K.; Ogata, J.; Yoshino, H.; Okuzumi, A.; Hatano, T.; Motoi, Y.; Hirunagi, T.; Katsuno, M. α-Synuclein V15A Variant in Familial Parkinson’s Disease Exhibits a Weaker Lipid-Binding Property. Mov. Disord. 2022, 37, 2075–2085. [Google Scholar] [CrossRef]
- Grassel, A.; Borland, C.; Bertolotti, F.; Osselborn, R.; Nassuna, T.; Zabat, B.; DebBurman, S. Insight into Parkinson’s Disease From a Yeast Model: How Three Alpha-Synuclein Mutants (A18T, A29S, & A53V) Generate Toxicity. FASEB J. 2022, 36. [Google Scholar] [CrossRef]
- Kim, Y.S.; Laurine, E.; Woods, W.; Lee, S.-J. A novel mechanism of interaction between α-synuclein and biological membranes. J. Mol. Biol. 2006, 360, 386–397. [Google Scholar] [CrossRef]
- Kuwahara, T.; Tonegawa, R.; Ito, G.; Mitani, S.; Iwatsubo, T. Phosphorylation of α-synuclein protein at Ser-129 reduces neuronal dysfunction by lowering its membrane binding property in Caenorhabditis elegans. J. Biol. Chem. 2012, 287, 7098–7109. [Google Scholar] [CrossRef]
- Ruf, V.C.; Nubling, G.S.; Willikens, S.; Shi, S.; Schmidt, F.; Levin, J.; Botzel, K.; Kamp, F.; Giese, A. Different Effects of alpha-Synuclein Mutants on Lipid Binding and Aggregation Detected by Single Molecule Fluorescence Spectroscopy and ThT Fluorescence-Based Measurements. ACS Chem. Neurosci. 2019, 10, 1649–1659. [Google Scholar] [CrossRef] [PubMed]
- Rovere, M.; Powers, A.E.; Jiang, H.; Pitino, J.C.; Fonseca-Ornelas, L.; Patel, D.S.; Achille, A.; Langen, R.; Varkey, J.; Bartels, T. E46K-like alpha-synuclein mutants increase lipid interactions and disrupt membrane selectivity. J. Biol. Chem. 2019, 294, 9799–9812. [Google Scholar] [CrossRef] [PubMed]
- Fredenburg, R.A.; Rospigliosi, C.; Meray, R.K.; Kessler, J.C.; Lashuel, H.A.; Eliezer, D.; Lansbury, P.T. The impact of the E46K mutation on the properties of α-synuclein in its monomeric and oligomeric states. Biochemistry 2007, 46, 7107–7118. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, O.; Fauvet, B.; Oueslati, A.; Dikiy, I.; Mahul-Mellier, A.L.; Ruggeri, F.S.; Mbefo, M.K.; Vercruysse, F.; Dietler, G.; Lee, S.J.; et al. The H50Q mutation enhances alpha-synuclein aggregation, secretion, and toxicity. J. Biol. Chem. 2014, 289, 21856–21876. [Google Scholar] [CrossRef]
- Mohite, G.M.; Kumar, R.; Panigrahi, R.; Navalkar, A.; Singh, N.; Datta, D.; Mehra, S.; Ray, S.; Gadhe, L.G.; Das, S. Comparison of kinetics, toxicity, oligomer formation, and membrane binding capacity of α-synuclein familial mutations at the A53 site, including the newly discovered A53V mutation. Biochemistry 2018, 57, 5183–5187. [Google Scholar] [CrossRef]
- Ghosh, D.; Sahay, S.; Ranjan, P.; Salot, S.; Mohite, G.M.; Singh, P.K.; Dwivedi, S.; Carvalho, E.; Banerjee, R.; Kumar, A. The newly discovered Parkinson’s disease associated Finnish mutation (A53E) attenuates α-synuclein aggregation and membrane binding. Biochemistry 2014, 53, 6419–6421. [Google Scholar] [CrossRef]
- Perissinotto, F.; Stani, C.; De Cecco, E.; Vaccari, L.; Rondelli, V.; Posocco, P.; Parisse, P.; Scaini, D.; Legname, G.; Casalis, L. Iron-mediated interaction of alpha synuclein with lipid raft model membranes. Nanoscale 2020, 12, 7631–7640. [Google Scholar] [CrossRef]
- Xiang, W.; Menges, S.; Schlachetzki, J.; Meixner, H.; Hoffmann, A.-C.; Schlötzer-Schrehardt, U.; Becker, C.-M.; Winkler, J.; Klucken, J. Posttranslational modification and mutation of histidine 50 trigger alpha synuclein aggregation and toxicity. Mol. Neurodegener. 2015, 10, 8. [Google Scholar] [CrossRef]
- Vilariño-Güell, C.; Wider, C.; Ross, O.A.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto-Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A. VPS35 mutations in Parkinson disease. Am. J. Hum. Genet 2011, 89, 162–167. [Google Scholar] [CrossRef]
- Patel, D.; Witt, S.N. Sorting Out the Role of α-Synuclein in Retromer-Mediated Endosomal Protein Sorting. J. Exp. Neurosci. 2018, 12, 1179069518796215. [Google Scholar] [CrossRef]
- Rajasekaran, S.; Peterson, P.P.; Liu, Z.; Robinson, L.C.; Witt, S.N. α-synuclein inhibits Snx3-retromer retrograde trafficking of the conserved membrane-bound proprotein convertase Kex2 in the secretory pathway of Saccharomyces cerevisiae. Hum. Mol. Genet 2022, 31, 705–717. [Google Scholar] [CrossRef]
- He, S.; Wang, F.; Yung, K.K.L.; Zhang, S.; Qu, S. Effects of α-Synuclein-associated post-translational modifications in Parkinson’s disease. ACS Chem. Neurosci. 2021, 12, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Schaffert, L.-N.; Carter, W.G. Do post-translational modifications influence protein aggregation in neurodegenerative diseases: A systematic review. Brain Sci. 2020, 10, 232. [Google Scholar] [CrossRef] [PubMed]
- Delenclos, M.; Burgess, J.D.; Lamprokostopoulou, A.; Outeiro, T.F.; Vekrellis, K.; McLean, P.J. Cellular models of alpha-synuclein toxicity and aggregation. J. Neurochem. 2019, 150, 566–576. [Google Scholar] [CrossRef]
- Gadhavi, J.; Patel, M.; Bhatia, D.; Gupta, S. Neurotoxic or neuroprotective: Post-translational modifications of α-synuclein at the cross-roads of functions. Biochimie 2022, 192, 38–50. [Google Scholar] [CrossRef]
- Oueslati, A.; Fournier, M.; Lashuel, H.A. Role of post-translational modifications in modulating the structure, function and toxicity of α-synuclein: Implications for Parkinson’s disease pathogenesis and therapies. Prog. Brain Res. 2010, 183, 115–145. [Google Scholar] [PubMed]
- Mahul-Mellier, A.-L.; Fauvet, B.; Gysbers, A.; Dikiy, I.; Oueslati, A.; Georgeon, S.; Lamontanara, A.J.; Bisquertt, A.; Eliezer, D.; Masliah, E. c-Abl phosphorylates α-synuclein and regulates its degradation: Implication for α-synuclein clearance and contribution to the pathogenesis of Parkinson’s disease. Hum. Mol. Genet 2014, 23, 2858–2879. [Google Scholar] [CrossRef]
- Paleologou, K.E.; Oueslati, A.; Shakked, G.; Rospigliosi, C.C.; Kim, H.-Y.; Lamberto, G.R.; Fernandez, C.O.; Schmid, A.; Chegini, F.; Gai, W.P. Phosphorylation at S87 is enhanced in synucleinopathies, inhibits α-synuclein oligomerization, and influences synuclein-membrane interactions. J. Neurosci. 2010, 30, 3184–3198. [Google Scholar] [CrossRef]
- Nubling, G.S.; Levin, J.; Bader, B.; Lorenzl, S.; Hillmer, A.; Hogen, T.; Kamp, F.; Giese, A. Modelling Ser129 phosphorylation inhibits membrane binding of pore-forming alpha-synuclein oligomers. PLoS ONE 2014, 9, e98906. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.-R.; Hu, Z.-W.; Zhao, Y.-F.; Chen, Y.-X.; Li, Y.-M. Phosphorylation induces distinct alpha-synuclein strain formation. Sci. Rep. 2016, 6, 37130. [Google Scholar] [CrossRef]
- Samuel, F.; Flavin, W.P.; Iqbal, S.; Pacelli, C.; Renganathan, S.D.S.; Trudeau, L.-E.; Campbell, E.M.; Fraser, P.E.; Tandon, A. Effects of serine 129 phosphorylation on α-synuclein aggregation, membrane association, and internalization. J. Biol. Chem. 2016, 291, 4374–4385. [Google Scholar] [CrossRef] [PubMed]
- Runfola, M.; De Simone, A.; Vendruscolo, M.; Dobson, C.M.; Fusco, G. The N-terminal acetylation of α-synuclein changes the affinity for lipid membranes but not the structural properties of the bound state. Sci. Rep. 2020, 10, 204. [Google Scholar] [CrossRef]
- Sevcsik, E.; Trexler, A.J.; Dunn, J.M.; Rhoades, E. Allostery in a disordered protein: Oxidative modifications to α-synuclein act distally to regulate membrane binding. J. Am. Chem. Soc. 2011, 133, 7152–7158. [Google Scholar] [CrossRef]
- Lewis, Y.E.; Abeywardana, T.; Lin, Y.H.; Galesic, A.; Pratt, M.R. Synthesis of a Bis-thio-acetone (BTA) Analogue of the Lysine Isopeptide Bond and its Application to Investigate the Effects of Ubiquitination and SUMOylation on α-Synuclein Aggregation and Toxicity. ACS Chem. Biol. 2016, 11, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Hejjaoui, M.; Haj-Yahya, M.; Kumar, K.A.; Brik, A.; Lashuel, H.A. Towards Elucidation of the Role of Ubiquitination in the Pathogenesis of Parkinson’s Disease with Semisynthetic Ubiquitinated α-Synuclein. Angew. Chem. Int. Ed. 2011, 50, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Caparotta, M.; Bustos, D.M.; Masone, D. Order–disorder skewness in alpha-synuclein: A key mechanism to recognize membrane curvature. Phys. Chem. Chem. Phys. 2020, 22, 5255–5263. [Google Scholar] [CrossRef]
- van der Wateren, I.M.; Knowles, T.P.; Buell, A.K.; Dobson, C.M.; Galvagnion, C. C-terminal truncation of α-synuclein promotes amyloid fibril amplification at physiological pH. Chem. Sci. 2018, 9, 5506–5516. [Google Scholar] [CrossRef]
- Flynn, J.D.; Gimmen, M.Y.; Dean, D.N.; Lacy, S.M.; Lee, J.C. Terminal Alkynes as Raman Probes of α-Synuclein in Solution and in Cells. ChemBioChem 2020, 21, 1582–1586. [Google Scholar] [CrossRef] [PubMed]
- Faustini, G.; Longhena, F.; Bruno, A.; Bono, F.; Grigoletto, J.; La Via, L.; Barbon, A.; Casiraghi, A.; Straniero, V.; Valoti, E. Alpha-synuclein/synapsin III pathological interplay boosts the motor response to methylphenidate. Neurobiol. Dis. 2020, 138, 104789. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Yang, C.; Zhang, X.; Li, Y.; Wang, S.; Zheng, L.; Huang, K. C-terminal truncation exacerbates the aggregation and cytotoxicity of α-Synuclein: A vicious cycle in Parkinson’s disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 3714–3725. [Google Scholar] [CrossRef]
- Levine, P.M.; Galesic, A.; Balana, A.T.; Mahul-Mellier, A.-L.; Navarro, M.X.; De Leon, C.A.; Lashuel, H.A.; Pratt, M.R. α-Synuclein O-GlcNAcylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2019, 116, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Uceda, A.B.; Frau, J.; Vilanova, B.; Adrover, M. Glycation of alpha-synuclein hampers its binding to synaptic-like vesicles and its driving effect on their fusion. Cell Mol. Life Sci. 2022, 79, 342. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Cohen, P. The structure and regulation of protein phosphatases. Annu. Rev. Biochem. 1989, 58, 453–508. [Google Scholar] [CrossRef]
- Cohen, P. The origins of protein phosphorylation. Nat. Cell Biol. 2002, 4, E127–E130. [Google Scholar] [CrossRef]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; De Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M. Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef]
- Kosten, J.; Binolfi, A.; Stuiver, M.; Verzini, S.; Theillet, F.-X.; Bekei, B.; van Rossum, M.; Selenko, P. Efficient modification of alpha-synuclein serine 129 by protein kinase CK1 requires phosphorylation of tyrosine 125 as a priming event. ACS Chem. Neurosci. 2014, 5, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhang, Z.; Ye, Y.; Wu, Q.; Liu, M.; Li, C. Phosphorylation dependent α-synuclein degradation monitored by in-cell NMR. Chem. Commun. 2019, 55, 11215–11218. [Google Scholar] [CrossRef] [PubMed]
- Schreurs, S.; Gerard, M.; Derua, R.; Waelkens, E.; Taymans, J.-M.; Baekelandt, V.; Engelborghs, Y. In vitro phosphorylation does not influence the aggregation kinetics of WT α-synuclein in contrast to its phosphorylation mutants. Int. J. Mol. Sci. 2014, 15, 1040–1067. [Google Scholar] [CrossRef] [PubMed]
- Brahmachari, S.; Ge, P.; Lee, S.H.; Kim, D.; Karuppagounder, S.S.; Kumar, M.; Mao, X.; Shin, J.H.; Lee, Y.; Pletnikova, O. Activation of tyrosine kinase c-Abl contributes to α-synuclein–induced neurodegeneration. J. Clin. Investig. 2016, 126, 2970–2988. [Google Scholar] [CrossRef]
- McFarland, N.R.; Fan, Z.; Xu, K.; Schwarzschild, M.A.; Feany, M.B.; Hyman, B.T.; McLean, P.J. α-Synuclein S129 phosphorylation mutants do not alter nigrostriatal toxicity in a rat model of parkinson disease. J. Neuropathol. Exp. Neurol. 2009, 68, 515–524. [Google Scholar] [CrossRef]
- Dikiy, I.; Eliezer, D. N-terminal acetylation stabilizes N-terminal helicity in lipid- and micelle-bound alpha-synuclein and increases its affinity for physiological membranes. J. Biol. Chem. 2014, 289, 3652–3665. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, P.; Öhrfelt, A.; Lashley, T.; Blennow, K.; Brinkmalm, A.; Zetterberg, H. Mass spectrometric analysis of Lewy body-enriched α-synuclein in Parkinson’s disease. J. Proteome Res. 2019, 18, 2109–2120. [Google Scholar] [CrossRef]
- Kang, L.; Moriarty, G.M.; Woods, L.A.; Ashcroft, A.E.; Radford, S.E.; Baum, J. N-Terminal acetylation of α-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Sci. 2012, 21, 911–917. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, E.I.; Jiang, Z.; Strub, M.-P.; Lee, J.C. Effects of phosphatidylcholine membrane fluidity on the conformation and aggregation of N-terminally acetylated α-synuclein. J. Biol. Chem. 2018, 293, 11195–11205. [Google Scholar] [CrossRef]
- Good, P.F.; Hsu, A.; Werner, P.; Perl, D.P.; Olanow, C.W. Protein nitration in Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1998, 57, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Paxinou, E.; Chen, Q.; Weisse, M.; Giasson, B.I.; Norris, E.H.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M.-Y.; Ischiropoulos, H. Induction of α-synuclein aggregation by intracellular nitrative insult. J. Neurosci. 2001, 21, 8053–8061. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef]
- Xiang, W.; Schlachetzki, J.C.; Helling, S.; Bussmann, J.C.; Berlinghof, M.; Schäffer, T.E.; Marcus, K.; Winkler, J.; Klucken, J.; Becker, C.-M. Oxidative stress-induced posttranslational modifications of alpha-synuclein: Specific modification of alpha-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol. Cell. Neurosci. 2013, 54, 71–83. [Google Scholar] [CrossRef]
- Liu, Y.; Qiang, M.; Wei, Y.; He, R. A novel molecular mechanism for nitrated α-synuclein-induced cell death. J. Mol. Cell. Biol. 2011, 3, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Xu, X.; Xiang, Z.; Zhou, J.; Zhang, Z.; Hu, C.; He, C. Nitrated α-synuclein induces the loss of dopaminergic neurons in the substantia nigra of rats. PLoS ONE 2010, 5, e9956. [Google Scholar] [CrossRef]
- Rape, M.; Komander, D. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar]
- Kuzuhara, S.; Mori, H.; Izumiyama, N.; Yoshimura, M.; Ihara, Y. Lewy bodies are ubiquitinated. Acta Neuropathol. 1988, 75, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Meier, F.; Abeywardana, T.; Dhall, A.; Marotta, N.P.; Varkey, J.; Langen, R.; Chatterjee, C.; Pratt, M.R. Semisynthetic, site-specific ubiquitin modification of α-synuclein reveals differential effects on aggregation. J. Am. Chem. Soc. 2012, 134, 5468–5471. [Google Scholar] [CrossRef]
- Iyer, A.; Claessens, M.M. Disruptive membrane interactions of alpha-synuclein aggregates. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2019, 1867, 468–482. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Choi, Y.R.; Park, J.-Y.; Lee, J.-H.; Kim, D.K.; Lee, S.-J.; Paik, S.R.; Jou, I.; Park, S.M. Proteolytic cleavage of extracellular α-synuclein by plasmin: Implications for Parkinson disease. J. Biol. Chem. 2012, 287, 24862–24872. [Google Scholar] [CrossRef] [PubMed]
- Iwata, A.; Maruyama, M.; Akagi, T.; Hashikawa, T.; Kanazawa, I.; Tsuji, S.; Nukina, N. Alpha-synuclein degradation by serine protease neurosin: Implication for pathogenesis of synucleinopathies. Hum. Mol. Genet 2003, 12, 2625–2635. [Google Scholar] [CrossRef]
- Sevlever, D.; Jiang, P.; Yen, S.-H.C. Cathepsin D is the main lysosomal enzyme involved in the degradation of α-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 2008, 47, 9678–9687. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Nguyen, L.T.; Burlak, C.; Chegini, F.; Guo, F.; Chataway, T.; Ju, S.; Fisher, O.S.; Miller, D.W.; Datta, D. Caspase-1 causes truncation and aggregation of the Parkinson’s disease-associated protein α-synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 9587–9592. [Google Scholar] [CrossRef]
- Diepenbroek, M.; Casadei, N.; Esmer, H.; Saido, T.C.; Takano, J.; Kahle, P.J.; Nixon, R.A.; Rao, M.V.; Melki, R.; Pieri, L. Overexpression of the calpain-specific inhibitor calpastatin reduces human alpha-Synuclein processing, aggregation and synaptic impairment in [A30P] αSyn transgenic mice. Hum. Mol. Genet 2014, 23, 3975–3989. [Google Scholar] [CrossRef] [PubMed]
- Terada, M.; Suzuki, G.; Nonaka, T.; Kametani, F.; Tamaoka, A.; Hasegawa, M. The effect of truncation on prion-like properties of α-synuclein. J. Biol. Chem. 2018, 293, 13910–13920. [Google Scholar] [CrossRef] [PubMed]
- Kellie, J.F.; Higgs, R.E.; Ryder, J.W.; Major, A.; Beach, T.G.; Adler, C.H.; Merchant, K.; Knierman, M.D. Quantitative measurement of intact alpha-synuclein proteoforms from post-mortem control and Parkinson’s disease brain tissue by intact protein mass spectrometry. Sci. Rep. 2014, 4, 5797. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, Z.A.; Giasson, B.I. The emerging role of α-synuclein truncation in aggregation and disease. J. Biol. Chem. 2020, 295, 10224–10244. [Google Scholar] [CrossRef] [PubMed]
- Öhrfelt, A.; Zetterberg, H.; Andersson, K.; Persson, R.; Secic, D.; Brinkmalm, G.; Wallin, A.; Mulugeta, E.; Francis, P.T.; Vanmechelen, E. Identification of novel α-synuclein isoforms in human brain tissue by using an online nanoLC-ESI-FTICR-MS method. Neurochem. Res. 2011, 36, 2029–2042. [Google Scholar] [CrossRef]
- Murray, I.V.; Giasson, B.I.; Quinn, S.M.; Koppaka, V.; Axelsen, P.H.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M.-Y. Role of α-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry 2003, 42, 8530–8540. [Google Scholar] [CrossRef] [PubMed]
- Michell, A.W.; Tofaris, G.; Gossage, H.; Tyers, P.; Spillantini, M.; Barker, R. The effect of truncated human α-synuclein (1–120) on dopaminergic cells in a transgenic mouse model of Parkinson’s disease. Cell Transplant. 2007, 16, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, Z.A.; Vijayaraghavan, N.; Gorion, K.-M.; Riffe, C.J.; Strang, K.H.; Caldwell, J.; Giasson, B.I. Physiological C-terminal truncation of α-synuclein potentiates the prion-like formation of pathological inclusions. J. Biol. Chem. 2018, 293, 18914–18932. [Google Scholar] [CrossRef]
- Torres, C.-R.; Hart, G.W. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J. Biol. Chem. 1984, 259, 3308–3317. [Google Scholar] [CrossRef]
- Wang, Z.; Park, K.; Comer, F.; Hsieh-Wilson, L.C.; Saudek, C.D.; Hart, G.W. Site-specific GlcNAcylation of human erythrocyte proteins: Potential biomarker (s) for diabetes. Diabetes 2009, 58, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Alfaro, J.F.; Gong, C.-X.; Monroe, M.E.; Aldrich, J.T.; Clauss, T.R.; Purvine, S.O.; Wang, Z.; Camp, D.G.; Shabanowitz, J.; Stanley, P. Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proc. Natl. Acad. Sci. USA 2012, 109, 7280–7285. [Google Scholar] [CrossRef]
- Wang, S.; Yang, F.; Petyuk, V.A.; Shukla, A.K.; Monroe, M.E.; Gritsenko, M.A.; Rodland, K.D.; Smith, R.D.; Qian, W.J.; Gong, C.X. Quantitative proteomics identifies altered O-GlcNAcylation of structural, synaptic and memory-associated proteins in Alzheimer’s disease. J. Pathol. 2017, 243, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Marotta, N.P.; Lin, Y.H.; Lewis, Y.E.; Ambroso, M.R.; Zaro, B.W.; Roth, M.T.; Arnold, D.B.; Langen, R.; Pratt, M.R. O-GlcNAc modification blocks the aggregation and toxicity of the protein α-synuclein associated with Parkinson’s disease. Nat. Chem. 2015, 7, 913–920. [Google Scholar] [CrossRef]
- Ryan, P.; Xu, M.; Davey, A.K.; Danon, J.J.; Mellick, G.D.; Kassiou, M.; Rudrawar, S. O-GlcNAc modification protects against protein misfolding and aggregation in neurodegenerative disease. ACS Chem. Neurosci. 2019, 10, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Salahuddin, P.; Rabbani, G.; Khan, R. The role of advanced glycation end products in various types of neurodegenerative disease: A therapeutic approach. Cell. Mol. Biol. Lett. 2014, 19, 407–437. [Google Scholar] [CrossRef]
- Choi, Y.-G.; Lim, S. Nε-(carboxymethyl) lysine linkage to α-synuclein and involvement of advanced glycation end products in α-synuclein deposits in an MPTP-intoxicated mouse model. Biochimie 2010, 92, 1379–1386. [Google Scholar] [CrossRef]
- Vicente Miranda, H.; Szegő, É.M.; Oliveira, L.M.; Breda, C.; Darendelioglu, E.; de Oliveira, R.M.; Ferreira, D.G.; Gomes, M.A.; Rott, R.; Oliveira, M. Glycation potentiates α-synuclein-associated neurodegeneration in synucleinopathies. Brain 2017, 140, 1399–1419. [Google Scholar] [CrossRef]
- Bar-On, P.; Crews, L.; Koob, A.O.; Mizuno, H.; Adame, A.; Spencer, B.; Masliah, E. Statins reduce neuronal α-synuclein aggregation in in vitro models of Parkinson’s disease. J. Neurochem. 2008, 105, 1656–1667. [Google Scholar] [CrossRef]
- Dai, L.; Wang, J.; He, M.; Xiong, M.; Tian, Y.; Liu, C.; Zhang, Z. Lovastatin Alleviates α-Synuclein Aggregation and Phosphorylation in Cellular Models of Synucleinopathy. Front. Mol. Neurosci. 2021, 14, 682320. [Google Scholar] [CrossRef] [PubMed]
- Koob, A.O.; Ubhi, K.; Paulsson, J.F.; Kelly, J.; Rockenstein, E.; Mante, M.; Adame, A.; Masliah, E. Lovastatin ameliorates α-synuclein accumulation and oxidation in transgenic mouse models of α-synucleinopathies. Exp. Neurol. 2010, 221, 267–274. [Google Scholar] [CrossRef]
- Selley, M.L. Simvastatin prevents 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine-induced striatal dopamine depletion and protein tyrosine nitration in mice. Brain Res. 2005, 1037, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Roy, A.; Matras, J.; Brahmachari, S.; Gendelman, H.E.; Pahan, K. Simvastatin inhibits the activation of p21ras and prevents the loss of dopaminergic neurons in a mouse model of Parkinson’s disease. J. Neurosci. 2009, 29, 13543–13556. [Google Scholar] [CrossRef] [PubMed]
- Mingione, A.; Pivari, F.; Plotegher, N.; Dei Cas, M.; Zulueta, A.; Bocci, T.; Trinchera, M.; Albi, E.; Maglione, V.; Caretti, A. Inhibition of Ceramide Synthesis Reduces α-Synuclein Proteinopathy in a Cellular Model of Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 6469. [Google Scholar] [CrossRef]
- Kumar, S.; Kumar, R.; Kumari, M.; Kumari, R.; Saha, S.; Bhavesh, N.S.; Maiti, T.K. Ellagic Acid Inhibits alpha-Synuclein Aggregation at Multiple Stages and Reduces Its Cytotoxicity. ACS Chem. Neurosci. 2021, 12, 1919–1930. [Google Scholar] [CrossRef]
- Perni, M.; Galvagnion, C.; Maltsev, A.; Meisl, G.; Müller, M.B.; Challa, P.K.; Kirkegaard, J.B.; Flagmeier, P.; Cohen, S.I.; Cascella, R. A natural product inhibits the initiation of α-synuclein aggregation and suppresses its toxicity. Proc. Natl. Acad. Sci. USA 2017, 114, E1009–E1017. [Google Scholar] [CrossRef] [PubMed]
- Hebron, M.L.; Lonskaya, I.; Moussa, C.E.-H. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of α-synuclein in Parkinson’s disease models. Hum. Mol. Genet 2013, 22, 3315–3328. [Google Scholar] [CrossRef]
- Collins, L.M.; Adriaanse, L.J.; Theratile, S.D.; Hegarty, S.V.; Sullivan, A.M.; O’Keeffe, G.W. Class-IIa histone deacetylase inhibition promotes the growth of neural processes and protects them against neurotoxic insult. Mol. Neurobiol. 2015, 51, 1432–1442. [Google Scholar] [CrossRef]
- Bassil, F.; Fernagut, P.-O.; Bezard, E.; Pruvost, A.; Leste-Lasserre, T.; Hoang, Q.Q.; Ringe, D.; Petsko, G.A.; Meissner, W.G. Reducing C-terminal truncation mitigates synucleinopathy and neurodegeneration in a transgenic model of multiple system atrophy. Proc. Natl. Acad. Sci. USA 2016, 113, 9593–9598. [Google Scholar] [CrossRef] [PubMed]
- Iljina, M.; Tosatto, L.; Choi, M.L.; Sang, J.C.; Ye, Y.; Hughes, C.D.; Bryant, C.E.; Gandhi, S.; Klenerman, D. Arachidonic acid mediates the formation of abundant alpha-helical multimers of alpha-synuclein. Sci. Rep. 2016, 6, 33928. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Bioenergetic approaches for neuroprotection in Parkinson’s disease. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child. Neurol. Soc. 2003, 53, S39–S48. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.W.; Bradbury, K.A.; Schneider, J.S. Neuroprotection in Parkinson models varies with toxin administration protocol. Eur. J. Neurosci. 2006, 24, 3174–3182. [Google Scholar] [CrossRef]
- Jia, H.; Li, X.; Gao, H.; Feng, Z.; Li, X.; Zhao, L.; Jia, X.; Zhang, H.; Liu, J. High doses of nicotinamide prevent oxidative mitochondrial dysfunction in a cellular model and improve motor deficit in a Drosophila model of Parkinson’s disease. J. Neurosci. Res. 2008, 86, 2083–2090. [Google Scholar] [CrossRef]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garçon, G.; Rouaix, N. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal. 2014, 21, 195–210. [Google Scholar] [CrossRef]
- Vincent, B.M.; Tardiff, D.F.; Piotrowski, J.S.; Aron, R.; Lucas, M.C.; Chung, C.Y.; Bacherman, H.; Chen, Y.; Pires, M.; Subramaniam, R. Inhibiting stearoyl-CoA desaturase ameliorates α-synuclein cytotoxicity. Cell Rep. 2018, 25, 2742–2754. E2731. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Strable, M.S.; Ntambi, J.M. Stearoyl CoA desaturase 1: Role in cellular inflammation and stress. Adv. Nutr. 2011, 2, 15–22. [Google Scholar] [CrossRef]
- Imberdis, T.; Negri, J.; Ramalingam, N.; Terry-Kantor, E.; Ho, G.P.; Fanning, S.; Stirtz, G.; Kim, T.-E.; Levy, O.A.; Young-Pearse, T.L. Cell models of lipid-rich α-synuclein aggregation validate known modifiers of α-synuclein biology and identify stearoyl-CoA desaturase. Proc. Natl. Acad. Sci. USA 2019, 116, 20760–20769. [Google Scholar] [CrossRef]
- Hubler, Z.; Allimuthu, D.; Bederman, I.; Elitt, M.S.; Madhavan, M.; Allan, K.C.; Shick, H.E.; Garrison, E.; Karl, M.T.; Factor, D.C. Accumulation of 8, 9-unsaturated sterols drives oligodendrocyte formation and remyelination. Nature 2018, 560, 372–376. [Google Scholar] [CrossRef]
- Alberts, A.; Chen, J.; Kuron, G.; Hunt, V.; Huff, J.; Hoffman, C.; Rothrock, J.; Lopez, M.; Joshua, H.; Harris, E. Mevinolin: A highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc. Natl. Acad. Sci. USA 1980, 77, 3957–3961. [Google Scholar] [CrossRef]
- Moore, K.S.; Wehrli, S.; Roder, H.; Rogers, M.; Forrest Jr, J.N.; McCrimmon, D.; Zasloff, M. Squalamine: An aminosterol antibiotic from the shark. Proc. Natl. Acad. Sci. USA 1993, 90, 1354–1358. [Google Scholar] [CrossRef]
- Selinsky, B.S.; Smith, R.; Frangiosi, A.; Vonbaur, B.; Pedersen, L. Squalamine is not a proton ionophore. Biochim. Biophys. Acta (BBA)-Biomembr. 2000, 1464, 135–141. [Google Scholar] [CrossRef]
- Guttuso Jr, T.; Andrzejewski, K.L.; Lichter, D.G.; Andersen, J.K. Targeting kinases in Parkinson’s disease: A mechanism shared by LRRK2, neurotrophins, exenatide, urate, nilotinib and lithium. J. Neurol. Sci. 2019, 402, 121–130. [Google Scholar] [CrossRef]
- Braithwaite, S.P.; Voronkov, M.; Stock, J.B.; Mouradian, M.M. Targeting phosphatases as the next generation of disease modifying therapeutics for Parkinson’s disease. Neurochem. Int. 2012, 61, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Braithwaite, S.P.; Stock, J.B.; Mouradian, M.M. α-Synuclein phosphorylation as a therapeutic target in Parkinson’s disease. Rev. Neurosci. 2012, 23, 191–198. [Google Scholar] [CrossRef]
- Bell, R.; Vendruscolo, M. Modulation of the Interactions Between alpha-Synuclein and Lipid Membranes by Post-translational Modifications. Front. Neurol. 2021, 12, 661117. [Google Scholar] [CrossRef]
- Lee, K.-W.; Chen, W.; Junn, E.; Im, J.-Y.; Grosso, H.; Sonsalla, P.K.; Feng, X.; Ray, N.; Fernandez, J.R.; Chao, Y. Enhanced phosphatase activity attenuates α-synucleinopathy in a mouse model. J. Neurosci. 2011, 31, 6963–6971. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Xu, J.; Xue, S.; Liu, Y.; Zhang, Y.; Zhang, X.; Wang, X.; Wu, F.; Li, X. The E3 ubiquitin ligase seven in absentia homolog 1 may be a potential new therapeutic target for Parkinson’s disease. Neural Regen. Res. 2015, 10, 1286. [Google Scholar] [CrossRef] [PubMed]
- Games, D.; Valera, E.; Spencer, B.; Rockenstein, E.; Mante, M.; Adame, A.; Patrick, C.; Ubhi, K.; Nuber, S.; Sacayon, P. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J. Neurosci. 2014, 34, 9441–9454. [Google Scholar] [CrossRef]
- Tavassoly, O.; Yue, J.; Vocadlo, D.J. Pharmacological inhibition and knockdown of O-GlcNAcase reduces cellular internalization of α-synuclein preformed fibrils. FEBS J. 2021, 288, 452–470. [Google Scholar] [CrossRef]
- Lee, B.E.; Kim, H.Y.; Kim, H.-J.; Jeong, H.; Kim, B.-G.; Lee, H.-E.; Lee, J.; Kim, H.B.; Lee, S.E.; Yang, Y.R. O-GlcNAcylation regulates dopamine neuron function, survival and degeneration in Parkinson disease. Brain 2020, 143, 3699–3716. [Google Scholar] [CrossRef]
- Li, P.; Song, C. Potential treatment of Parkinson’s disease with omega-3 polyunsaturated fatty acids. Nutr. Neurosci. 2022, 25, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, M.; De Groot, H. NAD (P) H, a directly operating antioxidant? FASEB J. 2001, 15, 1569–1574. [Google Scholar] [CrossRef] [PubMed]
- Higdon, J. An Evidence-Based Approach to Vitamins and Minerals Health Benefits and Intake Recommendations; Thieme Medical Publishers, Inc.: New York, NY, USA, 2003. [Google Scholar]
- Schoeler, M.; Caesar, R. Dietary lipids, gut microbiota and lipid metabolism. Rev. Endocr. Metab. Disord. 2019, 20, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.-Y.; Yin, X.-X.; Guan, Q.-W.; Xia, Q.-X.; Yang, N.; Zhou, H.-H.; Liu, Z.-Q.; Jin, W.-L. Dietary nutrition for neurological disease therapy: Current status and future directions. Pharmacol. Ther. 2021, 226, 107861. [Google Scholar] [CrossRef]
- Zhao, B. Natural antioxidants protect neurons in Alzheimer’s disease and Parkinson’s disease. Neurochem. Res. 2009, 34, 630–638. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Effects on Lipid Membranes | Ref. |
|---|---|---|
| V15A |
| [132] |
| A18T |
| [133] |
| A29S |
| [133] |
| A30P |
| [85,134,135,136] |
| E46K |
| [137,138] |
| H50Q |
| [139] |
| G51D |
| [136] |
| A53E |
| [140] |
| A53T |
| [85,136,141,142] |
| A53V |
| [133,140] |
| PTM | Position | Effects on Membranes | Ref. |
|---|---|---|---|
| Phosphorylation | Y39 |
| [152] |
| S87 |
| [153] | |
| S129 |
| [154,155,156] | |
| Acetylation | M1 |
| [157] |
| Nitration | Y39 |
| [158] |
| Y125 |
| ||
| Y133, Y136 |
| ||
| Ubiquitination | K6, K23, K43, K96 |
| [159,160] |
| Truncation | 1–100 |
| [161] |
| 1–103 |
| [162] | |
| 1–115 |
| [163] | |
| 1–119 |
| [162] | |
| 1–120 |
| [164] | |
| 1–121 |
| [165] | |
| Glycosylation | T72 |
| [166] |
| T75 |
| ||
| T81 |
| ||
| S87 |
| ||
| T72, T75, and T81 |
| ||
| Glycation | Lysine |
| [167] |
| Compound | Target | Effect | Clinical Trial | Clinical Trial PD | Ref. |
|---|---|---|---|---|---|
| Lovastatin | HMG-CoA reductase | reduces α-syn accumulation and its phosphorylation in vitro in HEK293 cells, SH-SY5Y cells, and in primary human neurons and in vivo in different transgenic mouse models that neuronally overexpress human α-syn | rheumatoid arthritis, cancer, etc. | Phase II | [212,213,214] |
| Simvastatin | HMG-CoA reductase | prevents MPTP-induced striatal dopamine depletion and protein tyrosine nitration in mice, and protects dopaminergic neurons in the substantia nigra, attenuates the expression of proinflammatory molecules, and improves motor deficits in the MPTP model of PD | hyper-lipidemia, diabetes, MS, etc. | Phase II | [212,215,216] |
| Myriocin | de novo ceramide synthesis | reduced oxidative stress and inflammation and increased vesicular trafficking in SH-SY5Y cells treated with α-syn fibrils | no | no | [217] |
| Ellagic acid | α-syn | polyphenolic compound that has an inhibitory effect toward oligomerization and fibrillation of α-syn in vitro, reduces α-syn aggregation, and increases cell survival | prostate cancer phase III | no | [218] |
| Squalamine | competitive of α-syn | specifically inhibits the initiation of aggregation of α-syn and alleviates its toxicity in neuronal cells and in a Caenorhabditis elegans model of PD | macular degeneration phase II and III | no | [219] |
| Nilotinib | α-syn kinase c-Abl | enhanced clearance of α-syn, reduced neurotoxicity, and improved motor behavior in a mouse model of PD | AD phase 3, leukemia, etc. | no | [220] |
| MC1568 | class IIa histone deacetylases | increased neurite density and cell survival and protected against the neurotoxin-treated SY5Y cells | cancer | no | [221] |
| VX-765 | caspase-1 | reduces neurodegeneration, motor symptoms, and neuroinflammation in a mouse model of MSA | no | no | [222] |
| Arachidonic acid | α-syn | essential FA that induces the formation of ordered, α-helical structured α-syn multimers being resistant to fibrillation | autism, fibrosis, diabetes, etc. | no | [223] |
| Niacin/Nicotin-amide | Poly (ADP-ribose) polymerase | precursor of NADH and cofactor of mitochondrial enzymes that protects from MPTP-induced neurotoxicity in mice and prevents mitochondrial dysfunction in a cellular model and improves motor behavior in a Drosophila model of PD | hyperlipidemia, myopathy, etc. | interventional study | [224,225,226] |
| Deferiprone | ferric ions | iron chelator that reduces iron depositions in the substantia nigra accompanied by alleviated motor deficits in a clinical trial in early PD | HIV, ALS, heart disease, etc. | failed | [227] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Battis, K.; Xiang, W.; Winkler, J. The Bidirectional Interplay of α-Synuclein with Lipids in the Central Nervous System and Its Implications for the Pathogenesis of Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 13270. https://doi.org/10.3390/ijms241713270
Battis K, Xiang W, Winkler J. The Bidirectional Interplay of α-Synuclein with Lipids in the Central Nervous System and Its Implications for the Pathogenesis of Parkinson’s Disease. International Journal of Molecular Sciences. 2023; 24(17):13270. https://doi.org/10.3390/ijms241713270
Chicago/Turabian StyleBattis, Kristina, Wei Xiang, and Jürgen Winkler. 2023. "The Bidirectional Interplay of α-Synuclein with Lipids in the Central Nervous System and Its Implications for the Pathogenesis of Parkinson’s Disease" International Journal of Molecular Sciences 24, no. 17: 13270. https://doi.org/10.3390/ijms241713270
APA StyleBattis, K., Xiang, W., & Winkler, J. (2023). The Bidirectional Interplay of α-Synuclein with Lipids in the Central Nervous System and Its Implications for the Pathogenesis of Parkinson’s Disease. International Journal of Molecular Sciences, 24(17), 13270. https://doi.org/10.3390/ijms241713270