

Using Mendelian Randomization to Study the Role of Iron in Health and Disease


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Iron Requirements
3. The Duality of Iron Levels
4. Iron Overload Genes
5. Low Iron Genes
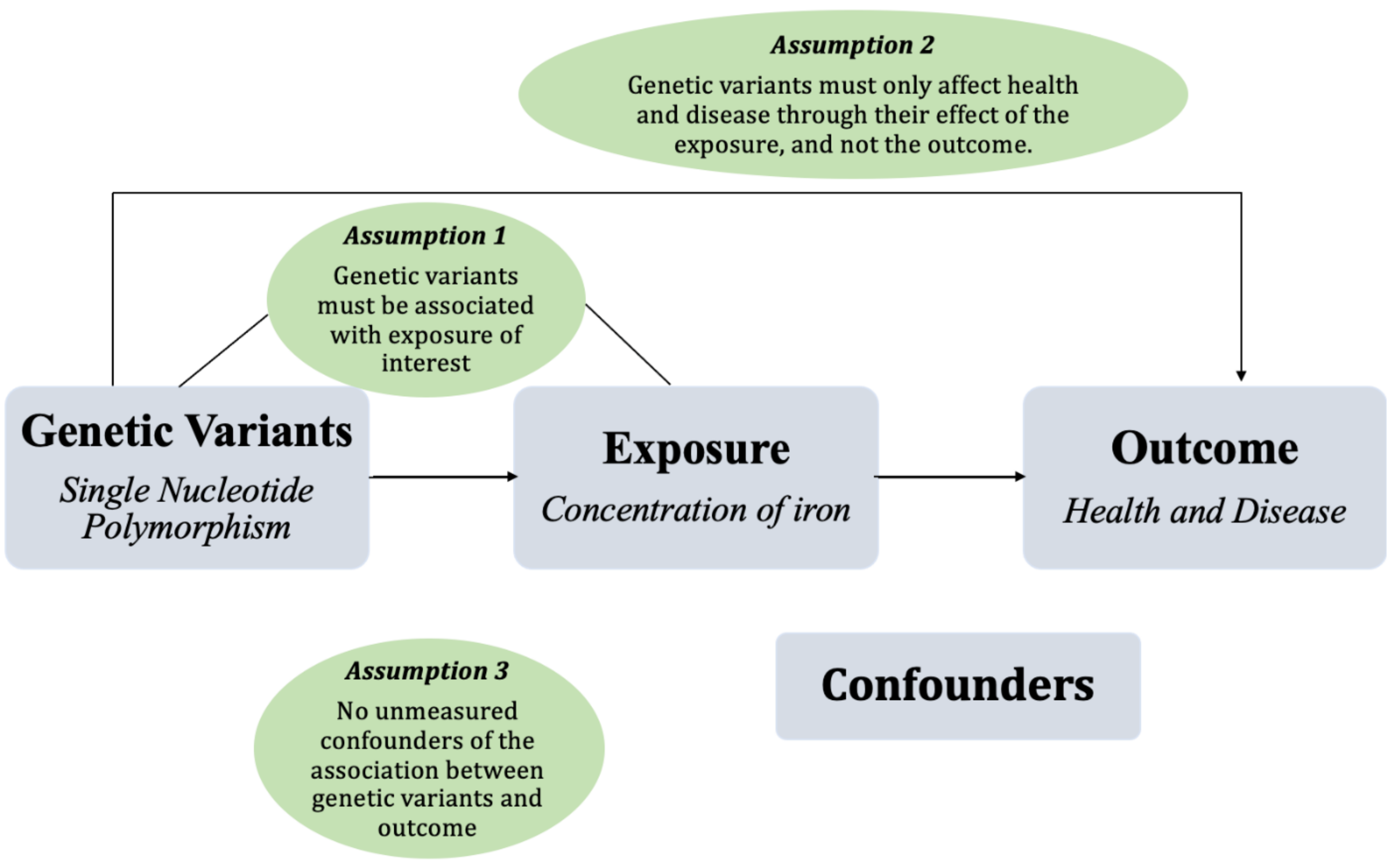
6. Mendelian Randomization
7. Mendelian Randomization, Iron, and Health
8. Cardiometabolic Disorders
8.1. Type 2 Diabetes, Obesity and Hypertension
8.2. Cholesterol
8.3. Heart Failure and Stroke
8.4. Vascular Disease
9. Neurological Disorders
10. Inflammation and Arthritis
11. Oncology
12. Other Conditions
13. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Hurrell, R.; Egli, I. Iron bioavailability and dietary reference values. Am. J. Clin. Nutr. 2010, 91, 1461s–1467s. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, S.; Mostofsky, D.I. Mostofsky, Iron Deficiency and Overload: From Basic Biology to Clinical Medicine; Humana: Totowa, NJ, USA, 2010. [Google Scholar]
- Milic, S.; Mikolasevic, I.; Orlic, L.; Devcic, E.; Starcevic-Cizmarevic, N.; Stimac, D.; Kapovic, M.; Ristic, S. The Role of Iron and Iron Overload in Chronic Liver Disease. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2016, 22, 2144–2151. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Haemoglobin Concentrations for the Diagnosis of Anaemia and Assessment of Severity; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Willett, W. Nutritional Epidemiology; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Smith, G.D.; Ebrahim, S. ‘Mendelian randomization’: Can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol. 2003, 32, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine Panel on Micronutrient. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc; National Academies Press: Washington, DC, USA, 2001. [Google Scholar]
- Jehn, M.L.; Guallar, E.; Clark, J.M.; Couper, D.; Duncan, B.B.; Ballantyne, C.M.; Hoogeveen, R.C.; Harris, Z.L.; Pankow, J.S. A prospective study of plasma ferritin level and incident diabetes: The Atherosclerosis Risk in Communities (ARIC) Study. Am. J. Epidemiol. 2007, 165, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Chavarro, J.E.; Rich-Edwards, J.W.; Rosner, B.A.; Willett, W.C. Iron intake and risk of ovulatory infertility. Obstet. Gynecol. 2006, 108, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Harrison-Findik, D.D. Gender-related variations in iron metabolism and liver diseases. World J. Hepatol. 2010, 2, 302–310. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Brown, K.E.; Ahn, J.; Sundaram, V. ACG Clinical Guideline: Hereditary Hemochromatosis. Off. J. Am. Coll. Gastroenterol. 2019, 114, 1202–1218. [Google Scholar] [CrossRef]
- Alexander, J.; Kowdley, K.V. HFE-associated hereditary hemochromatosis. Genet. Med. 2009, 11, 307–313. [Google Scholar] [CrossRef]
- Hsu, C.C.; Senussi, N.H.; Fertrin, K.Y.; Kowdley, K.V. Iron overload disorders. Hepatol. Commun. 2022, 6, 1842–1854. [Google Scholar] [CrossRef]
- Warne, C.D.; Zaloumis, S.G.; Bertalli, N.A.; Delatycki, M.B.; Nicoll, A.J.; McLaren, C.E.; Hopper, J.L.; Giles, G.G.; Anderson, G.J.; Olynyk, J.K.; et al. HFE p.C282Y homozygosity predisposes to rapid serum ferritin rise after menopause: A genotype-stratified cohort study of hemochromatosis in Australian women. J. Gastroenterol. Hepatol. 2017, 32, 797–802. [Google Scholar] [CrossRef]
- Powell, L.W.; Dixon, J.L.; Ramm, G.A.; Purdie, D.M.; Lincoln, D.J.; Anderson, G.J.; Subramaniam, V.N.; Hewett, D.G.; Searle, J.W.; Fletcher, L.M.; et al. Screening for hemochromatosis in asymptomatic subjects with or without a family history. Arch. Intern. Med. 2006, 166, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Patterson, A.J.; Brown, W.J.; Powers, J.R.; Roberts, D.C. Iron deficiency, general health and fatigue: Results from the Australian Longitudinal Study on Women’s Health. Qual. Life Res. 2000, 9, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Johnson-Wimbley, T.D.; Graham, D.Y. Diagnosis and management of iron deficiency anemia in the 21st century. Ther. Adv. Gastroenterol. 2011, 4, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C. Iron-deficiency anemia. N. Engl. J. Med. 2015, 372, 1832–1843. [Google Scholar] [CrossRef] [PubMed]
- Finberg, K.E.; Heeney, M.M.; Campagna, D.R.; Aydinok, Y.; Pearson, H.A.; Hartman, K.R.; Mayo, M.M.; Samuel, S.M.; Strouse, J.J.; Markianos, K.; et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat. Genet. 2008, 40, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.; Jeffrey, G.P. Clinical penetrance of C282Y homozygous HFE haemochromatosis. Clin. Biochem. Rev. 2004, 25, 183–190. [Google Scholar] [PubMed]
- Barton, J.C.; Edwards, C.Q.; Acton, R.T. HFE gene: Structure, function, mutations, and associated iron abnormalities. Gene 2015, 574, 179–192. [Google Scholar] [CrossRef]
- Powell, L.W.; Seckington, R.C.; Deugnier, Y. Haemochromatosis. Lancet 2016, 388, 706–716. [Google Scholar] [CrossRef]
- Allen, K.J.; Gurrin, L.C.; Constantine, C.C.; Osborne, N.J.; Delatycki, M.B.; Nicoll, A.J.; McLaren, C.E.; Bahlo, M.; Nisselle, A.E.; Vulpe, C.D.; et al. Iron-Overload–Related Disease in HFE Hereditary Hemochromatosis. N. Engl. J. Med. 2008, 358, 221–230. [Google Scholar] [CrossRef]
- Koller, D.L.; Imel, E.A.; Lai, D.; Padgett, L.R.; Acton, D.; Gray, A.; Peacock, M.; Econs, M.J.; Foroud, T. Genome-wide association study of serum iron phenotypes in premenopausal women of European descent. Blood Cells Mol. Dis. 2016, 57, 50–53. [Google Scholar] [CrossRef]
- Bacon, B.R.; Powell, L.W.; Adams, P.C.; Kresina, T.F.; Hoofnagle, J.H. Molecular medicine and hemochromatosis: At the crossroads. Gastroenterology 1999, 116, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, S.K.; Zakai, N.A.; van Rooij, F.J.; Soranzo, N.; Smith, A.V.; Nalls, M.A.; Chen, M.H.; Kottgen, A.; Glazer, N.L.; Dehghan, A.; et al. Multiple loci influence erythrocyte phenotypes in the CHARGE Consortium. Nat. Genet. 2009, 41, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Merryweather-Clarke, A.T.; Pointon, J.J.; Jouanolle, A.M.; Rochette, J.; Robson, K.J. Geography of HFE C282Y and H63D mutations. Genet. Test. 2000, 4, 183–198. [Google Scholar] [CrossRef]
- Hanson, E.H.; Imperatore, G.; Burke, W. HFE gene and hereditary hemochromatosis: A HuGE review. Human Genome Epidemiology. Am. J. Epidemiol. 2001, 154, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Bacon, B.R.; Adams, P.C.; Kowdley, K.V.; Powell, L.W.; Tavill, A.S. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011, 54, 328–343. [Google Scholar] [CrossRef] [PubMed]
- Mura, C.; Raguenes, O.; Férec, C. HFE mutations analysis in 711 hemochromatosis probands: Evidence for S65C implication in mild form of hemochromatosis. Blood 1999, 93, 2502–2505. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. Genetics, Genetic Testing, and Management of Hemochromatosis: 15 Years Since Hepcidin. Gastroenterology 2015, 149, 1240–1251.e4. [Google Scholar] [CrossRef]
- Whitlock, E.P.; Garlitz, B.A.; Harris, E.L.; Beil, T.L.; Smith, P.R. Screening for hereditary hemochromatosis: A systematic review for the U.S. Preventive Services Task Force. Ann. Intern. Med. 2006, 145, 209–223. [Google Scholar] [CrossRef]
- Benyamin, B.; Ferreira, M.A.; Willemsen, G.; Gordon, S.; Middelberg, R.P.; McEvoy, B.P.; Hottenga, J.J.; Henders, A.K.; Campbell, M.J.; Wallace, L.; et al. Common variants in TMPRSS6 are associated with iron status and erythrocyte volume. Nat. Genet. 2009, 41, 1173–1175. [Google Scholar] [CrossRef]
- Chambers, J.C.; Zhang, W.; Li, Y.; Sehmi, J.; Wass, M.N.; Zabaneh, D.; Hoggart, C.; Bayele, H.; McCarthy, M.I.; Peltonen, L. Genome-wide association study identifies variants in TMPRSS6 associated with hemoglobin levels. Nat. Genet. 2009, 41, 1170–1172. [Google Scholar] [CrossRef]
- Delbini, P.; Vaja, V.; Graziadei, G.; Duca, L.; Nava, I.; Refaldi, C.; Cappellini, M.D. Genetic variability of TMPRSS6 and its association with iron deficiency anaemia. Br. J. Haematol. 2010, 151, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Zhang, A.S.; Enns, C.A. Iron regulation by hepcidin. J. Clin. Investig. 2013, 123, 2337–2343. [Google Scholar] [CrossRef] [PubMed]
- Benyamin, B.; McRae, A.F.; Zhu, G.; Gordon, S.; Henders, A.K.; Palotie, A.; Peltonen, L.; Martin, N.G.; Montgomery, G.W.; Whitfield, J.B.; et al. Variants in TF and HFE Explain ∼40% of Genetic Variation in Serum-Transferrin Levels. Am. J. Hum. Genet. 2009, 84, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Dopsaj, V.; Topić, A.; Savković, M.; Milinković, N.; Novaković, I.; Ćujić, D.; Simić-Ogrizović, S. Associations of Common Variants in HFE and TMPRSS6 Genes with Hepcidin-25 and Iron Status Parameters in Patients with End-Stage Renal Disease. Dis. Markers 2019, 2019, 4864370. [Google Scholar] [CrossRef] [PubMed]
- Pei, S.N.; Ma, M.C.; You, H.L.; Fu, H.C.; Kuo, C.Y.; Rau, K.M.; Wang, M.C.; Lee, C.T. TMPRSS6 rs855791 polymorphism influences the susceptibility to iron deficiency anemia in women at reproductive age. Int. J. Med. Sci. 2014, 11, 614–619. [Google Scholar] [CrossRef] [PubMed]
- Gan, W.; Guan, Y.; Wu, Q.; An, P.; Zhu, J.; Lu, L.; Jing, L.; Yu, Y.; Ruan, S.; Xie, D.; et al. Association of TMPRSS6 polymorphisms with ferritin, hemoglobin, and type 2 diabetes risk in a Chinese Han population. Am. J. Clin. Nutr. 2011, 95, 626–632. [Google Scholar] [CrossRef] [PubMed]
- An, P.; Wu, Q.; Wang, H.; Guan, Y.; Mu, M.; Liao, Y.; Zhou, D.; Song, P.; Wang, C.; Meng, L.; et al. TMPRSS6, but not TF, TFR2 or BMP2 variants are associated with increased risk of iron-deficiency anemia. Hum. Mol. Genet. 2012, 21, 2124–2131. [Google Scholar] [CrossRef] [PubMed]
- Davey Smith, G.; Hemani, G. Mendelian randomization: Genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 2014, 23, R89–R98. [Google Scholar] [CrossRef]
- Evans, D.M.; Davey Smith, G. Mendelian Randomization: New Applications in the Coming Age of Hypothesis-Free Causality. Annu. Rev. Genom. Hum. Genet. 2015, 16, 327–350. [Google Scholar] [CrossRef]
- Didelez, V.; Sheehan, N. Mendelian randomization as an instrumental variable approach to causal inference. Stat. Methods Med. Res. 2007, 16, 309–330. [Google Scholar] [CrossRef]
- Smith, G.D.; Ebrahim, S. Mendelian randomization: Prospects, potentials, and limitations. Int. J. Epidemiol. 2004, 33, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.M.; Holmes, M.V.; Davey Smith, G. Reading Mendelian randomisation studies: A guide, glossary, and checklist for clinicians. BMJ 2018, 362, k601. [Google Scholar] [CrossRef]
- Hellwege, J.N.; Keaton, J.M.; Giri, A.; Gao, X.; Velez Edwards, D.R.; Edwards, T.L. Population Stratification in Genetic Association Studies. Curr. Protoc. Hum. Genet. 2017, 95, 1.22.1–1.22.23. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.D.; Ebrahim, S. Mendelian randomization: Genetic variants as instruments for strengthening causal inference in observational studies. In Biosocial Surveys; National Academies Press: Washington, DC, USA, 2008. [Google Scholar]
- Daghlas, I.; Gill, D. Genetically predicted iron status and life expectancy. Clin. Nutr. 2021, 40, 2456–2459. [Google Scholar] [CrossRef] [PubMed]
- Moksnes, M.R.; Graham, S.E.; Wu, K.H.; Hansen, A.F.; Gagliano Taliun, S.A.; Zhou, W.; Thorstensen, K.; Fritsche, L.G.; Gill, D.; Mason, A.; et al. Genome-wide meta-analysis of iron status biomarkers and the effect of iron on all-cause mortality in HUNT. Commun. Biol. 2022, 5, 591. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R. Heart disease and stroke statistics—2019 update: A report from the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Roa-Díaz, Z.M.; Raguindin, P.F.; Bano, A.; Laine, J.E.; Muka, T.; Glisic, M. Menopause and cardiometabolic diseases: What we (don’t) know and why it matters. Maturitas 2021, 152, 48–56. [Google Scholar] [CrossRef]
- Das De, S.; Krishna, S.; Jethwa, A. Iron status and its association with coronary heart disease: Systematic review and meta-analysis of prospective studies. Atherosclerosis 2015, 238, 296–303. [Google Scholar] [CrossRef]
- Wilman, H.R.; Parisinos, C.A.; Atabaki-Pasdar, N.; Kelly, M.; Thomas, E.L.; Neubauer, S.; Mahajan, A.; Hingorani, A.D.; Patel, R.S.; Hemingway, H.; et al. Genetic studies of abdominal MRI data identify genes regulating hepcidin as major determinants of liver iron concentration. J. Hepatol. 2019, 71, 594–602. [Google Scholar] [CrossRef]
- Wang, X.; Fang, X.; Zheng, W.; Zhou, J.; Song, Z.; Xu, M.; Min, J.; Wang, F. Genetic Support of A Causal Relationship Between Iron Status and Type 2 Diabetes: A Mendelian Randomization Study. J. Clin. Endocrinol. Metab. 2021, 106, e4641–e4651. [Google Scholar] [CrossRef]
- Wang, T.; Cheng, J.; Wang, Y. Genetic support of a causal relationship between iron status and atrial fibrillation: A Mendelian randomization study. Genes Nutr. 2022, 17, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liu, C.; Francis, M.; Sun, Y.; Ryu, M.S.; Grider, A.; Ye, K. The Causal Effects of Blood Iron and Copper on Lipid Metabolism Diseases: Evidence from Phenome-Wide Mendelian Randomization Study. Nutrients 2020, 12, 3174. [Google Scholar] [CrossRef]
- Wang, X.; Wang, X.; Gong, Y.; Chen, X.; Zhong, D.; Zhu, J.; Zhuang, L.; Gao, J.; Fu, G.; Lu, X.; et al. Appraising the Causal Association between Systemic Iron Status and Heart Failure Risk: A Mendelian Randomisation Study. Nutrients 2022, 14, 3258. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.; Del Greco, M.F.; Walker, A.P.; Srai, S.K.S.; Laffan, M.A.; Minelli, C. The Effect of Iron Status on Risk of Coronary Artery Disease: A Mendelian Randomization Study-Brief Report. Arter. Thromb. Vasc. Biol. 2017, 37, 1788–1792. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.; Monori, G.; Tzoulaki, I.; Dehghan, A. Iron Status and Risk of Stroke. Stroke 2018, 49, 2815–2821. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Bao, Q.; Wang, Z.; Ma, M.; Shen, J.; Ye, F.; Xie, X. Sex-Specific Genetically Predicted Iron Status in relation to 12 Vascular Diseases: A Mendelian Randomization Study in the UK Biobank. Biomed. Res. Int. 2020, 2020, 6246041. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Bruzelius, M.; Damrauer, S.M.; Larsson, S.C. Cardiometabolic, Lifestyle, and Nutritional Factors in Relation to Varicose Veins: A Mendelian Randomization Study. J. Am. Heart Assoc. 2021, 10, e022286. [Google Scholar] [CrossRef]
- Gill, D.; Brewer, C.F.; Monori, G.; Trégouët, D.A.; Franceschini, N.; Giambartolomei, C.; Tzoulaki, I.; Dehghan, A. Effects of Genetically Determined Iron Status on Risk of Venous Thromboembolism and Carotid Atherosclerotic Disease: A Mendelian Randomization Study. J. Am. Heart Assoc. 2019, 8, e012994. [Google Scholar] [CrossRef]
- Elliott, L.T.; Sharp, K.; Alfaro-Almagro, F.; Shi, S.; Miller, K.L.; Douaud, G.; Marchini, J.; Smith, S.M. Genome-wide association studies of brain imaging phenotypes in UK Biobank. Nature 2018, 562, 210–216. [Google Scholar] [CrossRef]
- Georgieff, M.K. Long-term brain and behavioral consequences of early iron deficiency. Nutr. Rev. 2011, 69 (Suppl. 1), S43–S48. [Google Scholar] [CrossRef]
- Yuan, S.; Tomson, T.; Larsson, S.C. Modifiable risk factors for epilepsy: A two-sample Mendelian randomization study. Brain Behav. 2021, 11, e02098. [Google Scholar] [CrossRef] [PubMed]
- Pichler, I.; Del Greco, M.F.; Gögele, M.; Lill, C.M.; Bertram, L.; Do, C.B.; Eriksson, N.; Foroud, T.; Myers, R.H.; Nalls, M.; et al. Serum iron levels and the risk of Parkinson disease: A Mendelian randomization study. PLoS Med. 2013, 10, e1001462. [Google Scholar] [CrossRef]
- Zeitoun, T.; Dehghan Noudeh, N.; Garcia-Bailo, B.; El-Sohemy, A. Genetics of Iron Metabolism and Premenstrual Symptoms: A Mendelian Randomization Study. J. Nutr. 2021, 151, 1747–1754. [Google Scholar] [CrossRef] [PubMed]
- Moen, I.W.; Bergholdt, H.K.M.; Mandrup-Poulsen, T.; Nordestgaard, B.G.; Ellervik, C. Increased Plasma Ferritin Concentration and Low-Grade Inflammation—A Mendelian Randomization Study. Clin. Chem. 2018, 64, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Larsson, S. Causal associations of iron status with gout and rheumatoid arthritis, but not with inflammatory bowel disease. Clin. Nutr. 2020, 39, 3119–3124. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, S.; Tian, Y.; Si, H.; Zeng, Y.; Wu, Y.; Liu, Y.; Li, M.; Sun, K.; Wu, L.; et al. Genetic Causal Association between Iron Status and Osteoarthritis: A Two-Sample Mendelian Randomization. Nutrients 2022, 14, 3683. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Yang, F.; Hong, J.; Wang, W.; Li, S.; Jiang, G.; Yan, S. Causal relationship of serum nutritional factors with osteoarthritis: A Mendelian randomization study. Rheumatology 2020, 60, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wu, J.; Xu, M.; Zhu, T.; Sun, Y.; Chen, H.; Wu, L.; Chen, C. Causal associations of iron status and back pain risk: A Mendelian randomization study. Front. Nutr. 2022, 9, 923590. [Google Scholar] [CrossRef]
- Fonseca-Nunes, A.; Jakszyn, P.; Agudo, A. Iron and cancer risk—A systematic review and meta-analysis of the epidemiological evidence. Cancer Epidemiol. Biomark. Prev. 2014, 23, 12–31. [Google Scholar] [CrossRef]
- Lv, Y.F.; Chang, X.; Hua, R.X.; Yan, G.N.; Meng, G.; Liao, X.Y.; Zhang, X.; Guo, Q.N. The risk of new-onset cancer associated with HFE C282Y and H63D mutations: Evidence from 87,028 participants. J. Cell. Mol. Med. 2016, 20, 1219–1233. [Google Scholar] [CrossRef]
- Wang, K.; Yang, F.; Zhang, P.; Yang, Y.; Jiang, L. Genetic effects of iron levels on liver injury and risk of liver diseases: A two-sample Mendelian randomization analysis. Front. Nutr. 2022, 9, 964163. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Xiao, F.; Li, H.; Ding, D.; Dong, W.; Hou, G.; Zhao, L.; Yang, Y.; Yang, Y.; Zhou, W. Association between serum iron status and primary liver cancer risk: A Mendelian randomization analysis. Ann. Transl. Med. 2021, 9, 1533. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Carter, P.; Vithayathil, M.; Kar, S.; Giovannucci, E.; Mason, A.M.; Burgess, S.; Larsson, S.C. Iron Status and Cancer Risk in UK Biobank: A Two-Sample Mendelian Randomization Study. Nutrients 2020, 12, 526. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Zeng, W.; Lou, Y. Mendelian randomization study indicates lack of causal associations between iron status and lung cancer. Medicine 2022, 101, e29879. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Hou, Q.; Xie, X.; Wang, H.; Chen, Y.; Lu, T.; Wu, Q.; Liang, Y.; Hu, Y.; Mao, Y. Serum iron status and the risk of breast cancer in the European population: A two-sample Mendelian randomisation study. Genes Nutr. 2021, 16, 9. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Lu, Y.; Jin, H. Appraising the role of circulating concentrations of micro-nutrients in epithelial ovarian cancer risk: A Mendelian randomization analysis. Sci. Rep. 2020, 10, 7356. [Google Scholar] [CrossRef] [PubMed]
- Kazmi, N.; Haycock, P.; Tsilidis, K.; Lynch, B.M.; Truong, T.; The PRACTICAL Consortium, CRUK, BPC3, CAPS, PEGASUS; Martin, R.M.; Lewis, S.J. Appraising causal relationships of dietary, nutritional and physical-activity exposures with overall and aggressive prostate cancer: Two-sample Mendelian-randomization study based on 79,148 prostate-cancer cases and 61,106 controls. Int. J. Epidemiol. 2019, 49, 587–596. [Google Scholar] [CrossRef]
- Del Greco, M.F.; Foco, L.; Pichler, I.; Eller, P.; Eller, K.; Benyamin, B.; Whitfield, J.B.; Pramstaller, P.P.; Thompson, J.R.; Pattaro, C.; et al. Serum iron level and kidney function: A Mendelian randomization study. Nephrol. Dial. Transpl. 2017, 32, 273–278. [Google Scholar] [CrossRef]
- Sha, T.; Li, W.; He, H.; Wu, J.; Wang, Y.; Li, H. Causal Relationship of Genetically Predicted Serum Micronutrients Levels With Sarcopenia: A Mendelian Randomization Study. Front. Nutr. 2022, 9, 913155. [Google Scholar] [CrossRef]
- Hermine, O.; Dine, G.; Genty, V.; Marquet, L.-A.; Fumagalli, G.; Tafflet, M.; Guillem, F.; Van Lierde, F.; Rousseaux-Blanchi, M.-P.; Palierne, C. Eighty percent of French sport winners in Olympic, World and Europeans competitions have mutations in the hemochromatosis HFE gene. Biochimie 2015, 119, 1–5. [Google Scholar] [CrossRef]
- Thakkar, D.; Sicova, M.; Guest, N.S.; Garcia-Bailo, B.; El-Sohemy, A. HFE Genotype and Endurance Performance in Competitive Male Athletes. Med. Sci. Sports Exerc. 2021, 53, 1385–1390. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeitoun, T.; El-Sohemy, A. Using Mendelian Randomization to Study the Role of Iron in Health and Disease. Int. J. Mol. Sci. 2023, 24, 13458. https://doi.org/10.3390/ijms241713458
Zeitoun T, El-Sohemy A. Using Mendelian Randomization to Study the Role of Iron in Health and Disease. International Journal of Molecular Sciences. 2023; 24(17):13458. https://doi.org/10.3390/ijms241713458
Chicago/Turabian StyleZeitoun, Tara, and Ahmed El-Sohemy. 2023. "Using Mendelian Randomization to Study the Role of Iron in Health and Disease" International Journal of Molecular Sciences 24, no. 17: 13458. https://doi.org/10.3390/ijms241713458
APA StyleZeitoun, T., & El-Sohemy, A. (2023). Using Mendelian Randomization to Study the Role of Iron in Health and Disease. International Journal of Molecular Sciences, 24(17), 13458. https://doi.org/10.3390/ijms241713458