

Astrocytes as Neuroimmunocytes in Alzheimer’s Disease: A Biochemical Tool in the Neuron–Glia Crosstalk along the Pathogenetic Pathways



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Background
2. Methods
3. Results and Discussion
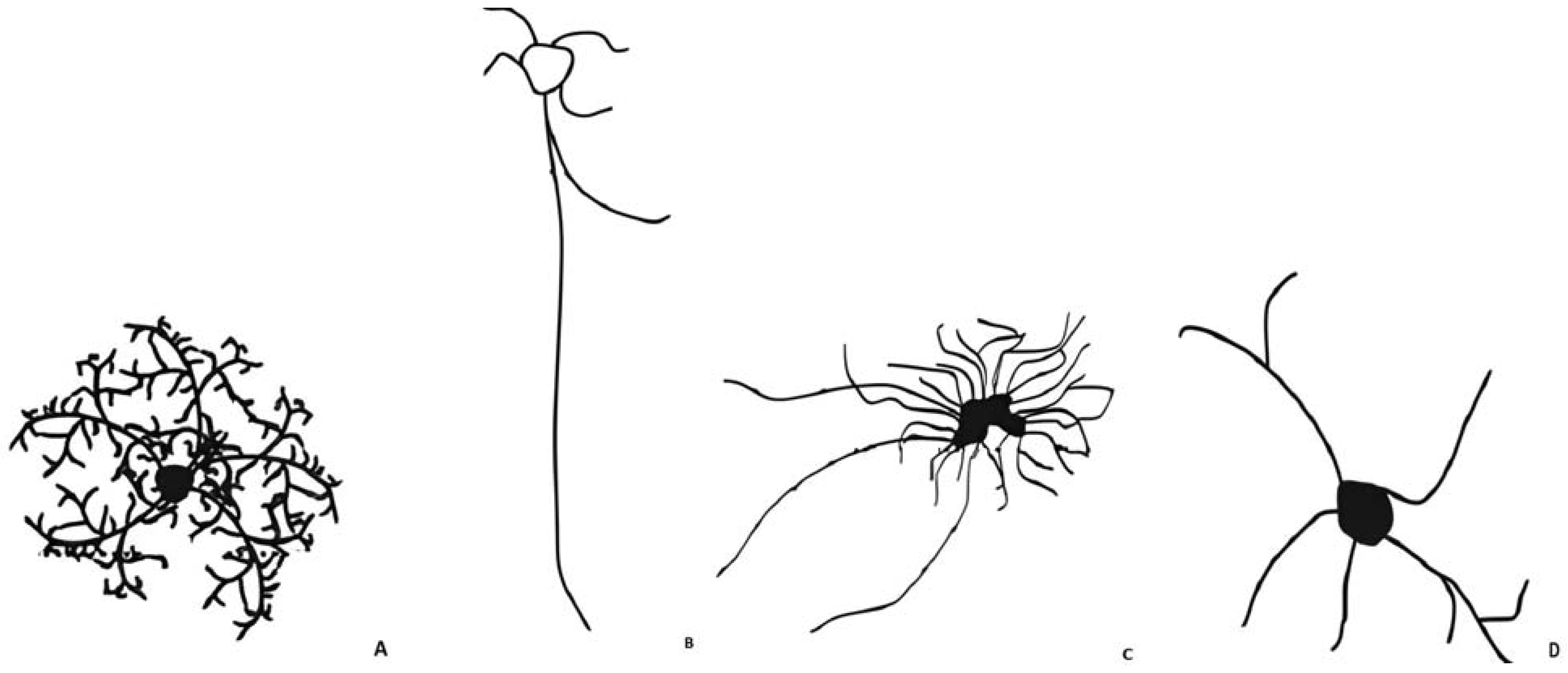
3.1. Astrocytes
- Protoplasmic astrocytes
- Interlaminar astrocytes
- Varicose-projection astrocytes
- Fibrous astrocytes
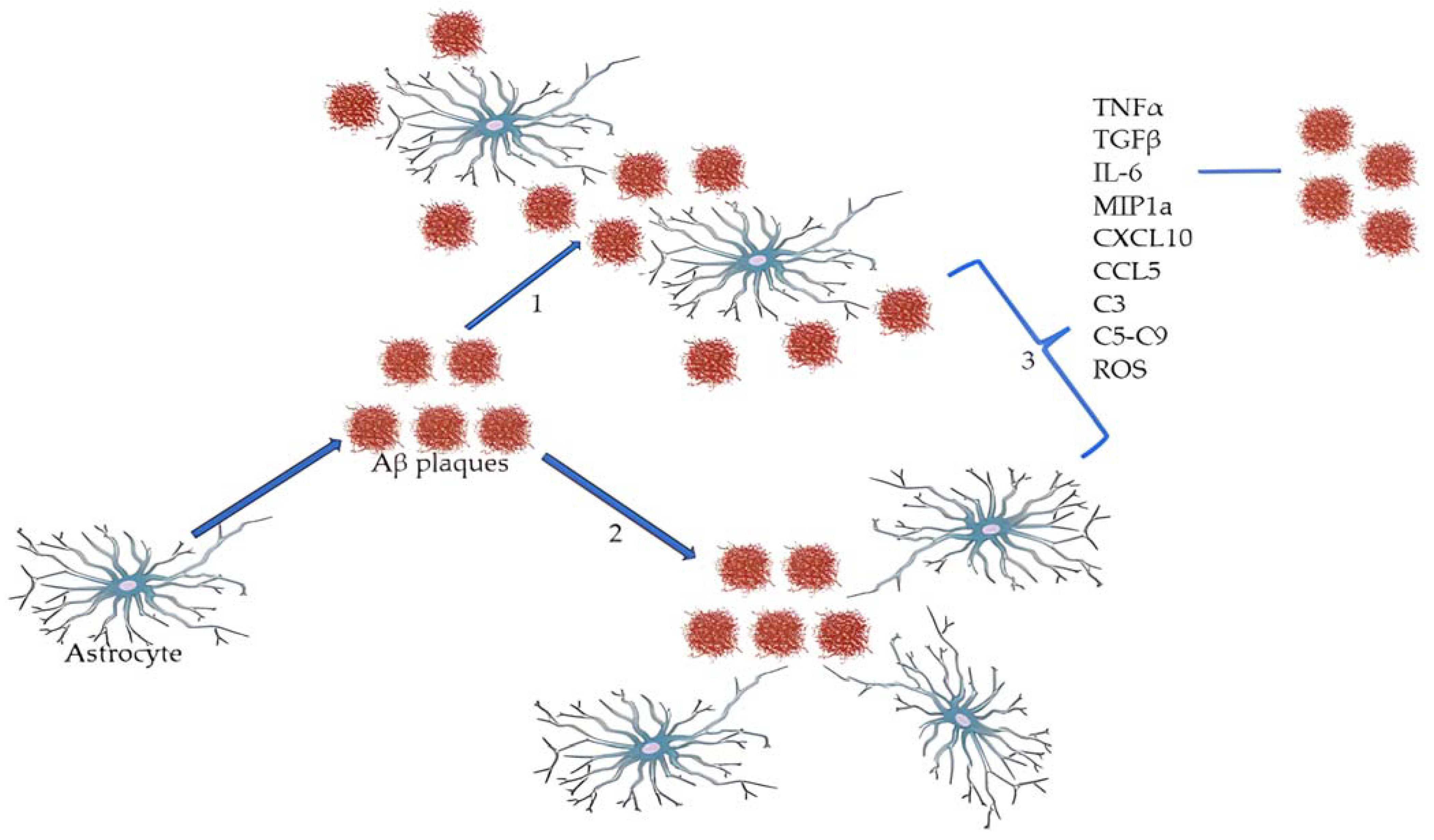
3.2. Astrocytes, Beta-Amyloid and Tau: The Story of Alzheimer’s Disease Begins with Aggregopathy Going on to Immunopathy
3.3. The Astrocyte Pathway toward Neurons in Alzheimer’s Disease
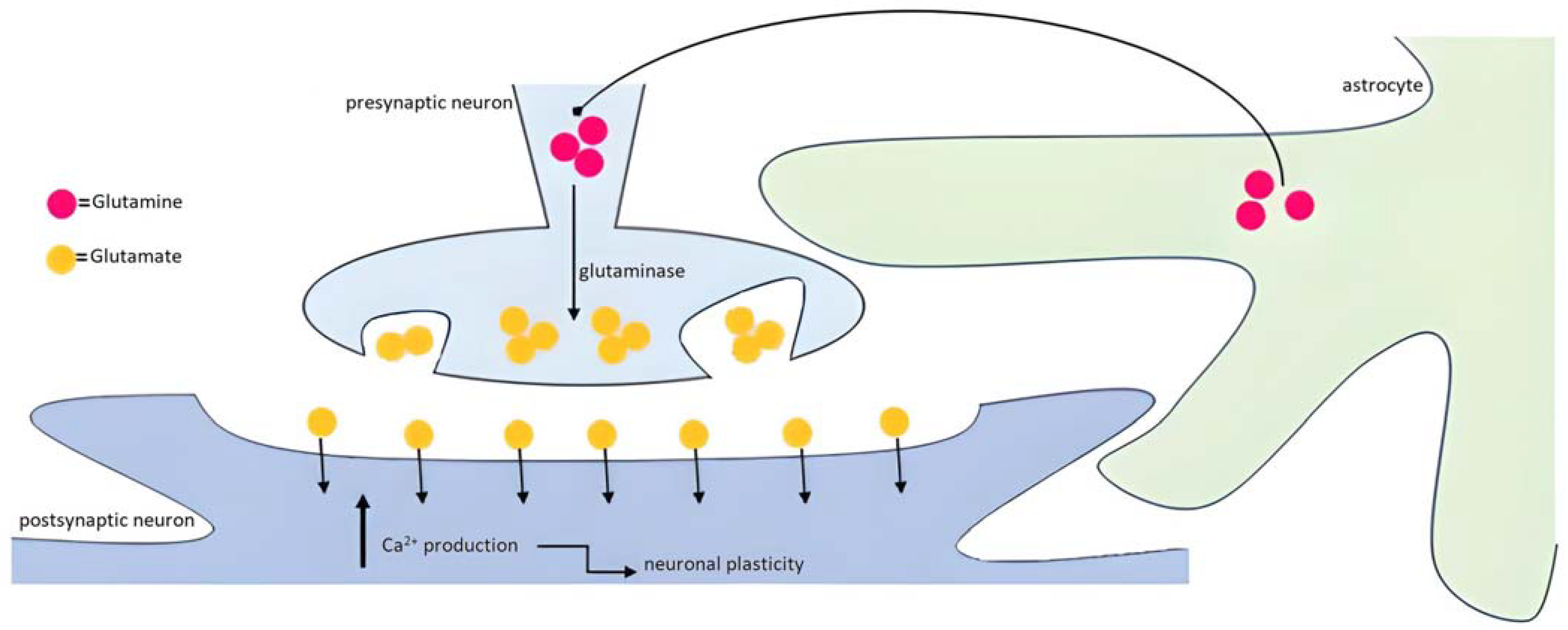
3.3.1. The Tripartite Synapse: The Neuron–Glia Close Contact
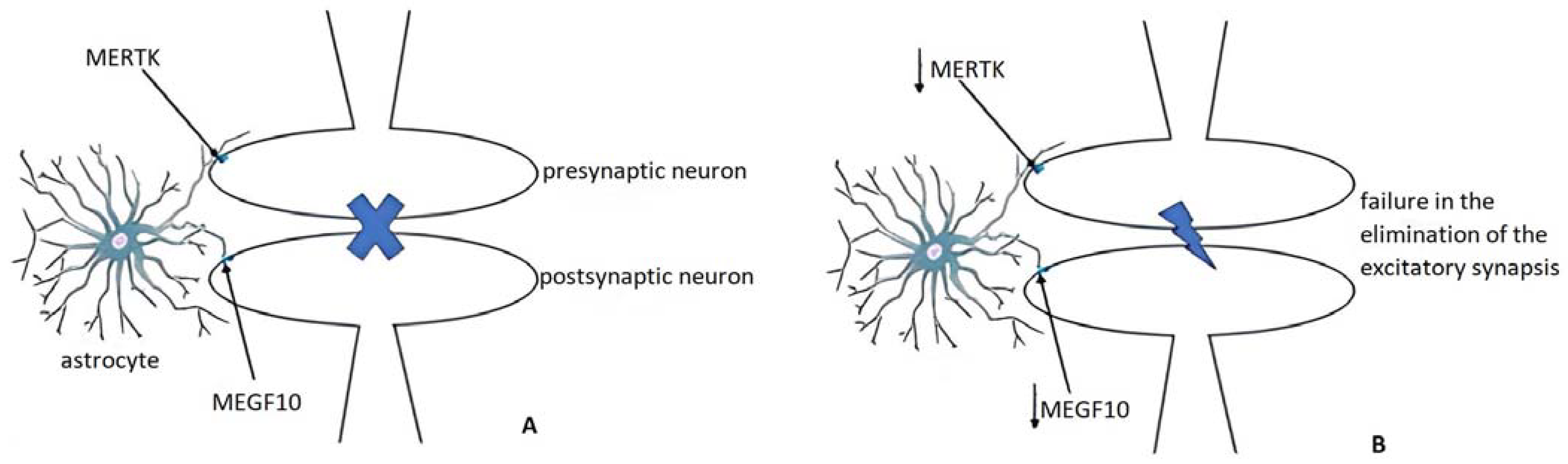
3.3.2. Pruning and Adult Remodeling: Astrocytes from Normality to Alzheimer’s Disease
3.3.3. Sprouting and the Building of New Synapses: Astrocytes from Normality to Alzheimer’s Disease
3.3.4. Astrocytes too, Not Only Neurons: What about the Clearance of Neurotransmitters in the Synaptic Cleft in Alzheimer’s Disease
3.3.5. Metabolism Impairment in the AD Astrocytes
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviation | Definition |
| CNS | central nervous system |
| AD | Alzheimer’s Disease |
| Aβ | amyloid-beta |
| APP | amyloid precursor protein |
| GFAP | glial fibrillary acidic protein |
| NFTs | neurofibrillary tangles |
| ApoE | apolipoprotein E |
| TNF | tumor necrosis factor |
| IL | interleukin |
| STAT3 | signal transducer and activator of transcription 3 |
| Glu | glutamate |
| TSP | thrombospondin |
References
- Sun, D.; Jakobs, T.C. Structural remodeling of astrocytes in the injured CNS. Neuroscientist 2012, 18, 567–588. [Google Scholar] [CrossRef] [PubMed]
- Parpura, V.; Verkhratsky, A. The astrocyte excitability brief: From receptors to gliotransmission. Neurochem. Int. 2012, 61, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Nakamura, Y.; Lo, E.H.; Hayakawa, K. Astrocyte signaling in the neurovascular unit after central nervous system injury. Int. J. Mol. Sci. 2019, 20, 282. [Google Scholar] [CrossRef]
- Lyon, K.A.; Allen, N.J. From Synapses to Circuits, Astrocytes Regulate Behavior. Front. Neural Circuits 2022, 15, 786293. [Google Scholar] [CrossRef] [PubMed]
- Hulshof, L.A.; van Nuijs, D.; Hol, E.M.; Middeldorp, J. The Role of Astrocytes in Synapse Loss in Alzheimer’s Disease: A Systematic Review. Front. Cell. Neurosci. 2022, 16, 899251. [Google Scholar] [CrossRef]
- Frost, G.R.; Li, Y.-M. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017, 7, 170228. [Google Scholar] [CrossRef]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef]
- Gehlot, P.; Kumar, S.; Vyas, V.K.; Choudhary, B.S.; Sharma, M.; Malik, R. Guanidine-based β amyloid precursor protein cleavage enzyme 1 (BACE-1) inhibitors for the Alzheimer’s disease (AD): A review. Bioorganic Med. Chem. 2022, 74, 117047. [Google Scholar] [CrossRef]
- Villegas, S.; Roda, A.; Serra-Mir, G.; Montoliu-Gaya, L.; Tiessler, L. Amyloid-beta peptide and tau protein crosstalk in Alzheimer’s disease. Neural Regen. Res. 2022, 17, 1666–1674. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D. If amyloid drives Alzheimer disease, why have anti-amyloid therapies not yet slowed cognitive decline? PLOS Biol. 2022, 20, e3001694. [Google Scholar] [CrossRef]
- Tolar, M.; Hey, J.; Power, A.; Abushakra, S. Neurotoxic Soluble Amyloid Oligomers Drive Alzheimer’s Pathogenesis and Represent a Clinically Validated Target for Slowing Disease Progression. Int. J. Mol. Sci. 2021, 22, 6355. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Bongioanni, P.; Del Carratore, R.; Corbianco, S.; Diana, A.; Cavallini, G.; Masciandaro, S.M.; Dini, M.; Buizza, R. Climate change and neurodegenerative diseases. Environ. Res. 2021, 201, 111511. [Google Scholar] [CrossRef]
- Guan, Y.; Zhang, L.; Wang, S.; Deng, Y.; Zhou, H.; Chen, D.; Zhang, L. The role of microglia in Alzheimer’s disease and progress of treatment. Ibrain 2022, 8, 37–47. [Google Scholar] [CrossRef]
- Preman, P.; Alfonso-Triguero, M.; Alberdi, E.; Verkhratsky, A.; Arranz, A.M. Astrocytes in Alzheimer’s disease: Pathological significance and molecular pathways. Cells 2021, 10, 540. [Google Scholar] [CrossRef]
- Monterey, M.D.; Wei, H.; Wu, X.; Wu, J.Q. The Many Faces of Astrocytes in Alzheimer’s Disease. Front. Neurol. 2021, 12, 619626. [Google Scholar] [CrossRef] [PubMed]
- Perez-Nievas, B.G.; Serrano-Pozo, A. Deciphering the astrocyte reaction in Alzheimer’s disease. Front. Aging Neurosci. 2018, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Beltran-Lobo, P.; Reid, M.J.; Jimenez-Sanchez, M.; Verkhratsky, A.; Perez-Nievas, B.G.; Noble, W. Astrocyte adaptation in Alzheimer’s disease: A focus on astrocytic P2X7R. Essays Biochem. 2023, 67, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.K.; Sato, K.; Sekino, Y. Neurons Induce Tiled Astrocytes with Branches That Avoid Each Other. Int. J. Mol. Sci. 2022, 23, 4161. [Google Scholar] [CrossRef]
- Price, B.R.; Johnson, L.A.; Norris, C.M. Reactive astrocytes: The nexus of pathological and clinical hallmarks of Alzheimer’s disease. Ageing Res. Rev. 2021, 68, 101335. [Google Scholar] [CrossRef]
- Brockett, A.T.; Kane, G.A.; Monari, P.K.; Briones, B.A.; Vigneron, P.-A.; Barber, G.A.; Bermudez, A.; Dieffenbach, U.; Kloth, A.D.; Buschman, T.J.; et al. Evidence supporting a role for astrocytes in the regulation of cognitive flexibility and neuronal oscillations through the Ca2+ binding protein S100β. PLoS ONE 2018, 13, e0195726. [Google Scholar] [CrossRef]
- Moloney, C.M.; Lowe, V.J.; Murray, M.E. Visualization of neurofibrillary tangle maturity in Alzheimer’s disease: A clinicopathologic perspective for biomarker research. Alzheimer’s Dement. 2021, 17, 1554–1574. [Google Scholar] [CrossRef] [PubMed]
- Balaban, D.; Miyawaki, E.K.; Bhattacharyya, S.; Torre, M. The phenomenon of clasmatodendrosis. Heliyon 2021, 7, e07605. [Google Scholar] [CrossRef] [PubMed]
- Koistinaho, M.; Lin, S.; Wu, X.; Esterman, M.; Koger, D.; Hanson, J.; Higgs, R.; Liu, F.; Malkani, S.; Bales, K.R.; et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat. Med. 2004, 10, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive astrocytes in neurodegenerative diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Jiwaji, Z.; Tiwari, S.S.; Avilés-Reyes, R.X.; Hooley, M.; Hampton, D.; Torvell, M.; Johnson, D.A.; McQueen, J.; Baxter, P.; Sabari-Sankar, K.; et al. Reactive astrocytes acquire neuroprotective as well as deleterious signatures in response to Tau and Aß pathology. Nat. Commun. 2022, 13, 135. [Google Scholar] [CrossRef] [PubMed]
- Dejanovic, B.; Wu, T.; Tsai, M.-C.; Graykowski, D.; Gandham, V.D.; Rose, C.M.; Bakalarski, C.E.; Ngu, H.; Wang, Y.; Pandey, S.; et al. Complement C1q-dependent excitatory and inhibitory synapse elimination by astrocytes and microglia in Alzheimer’s disease mouse models. Nat. Aging 2022, 2, 837–850. [Google Scholar] [CrossRef]
- Wu, T.; Dejanovic, B.; Gandham, V.D.; Gogineni, A.; Edmonds, R.; Schauer, S.; Srinivasan, K.; Huntley, M.A.; Wang, Y.; Wang, T.-M.; et al. Complement C3 Is Activated in Human AD Brain and Is Required for Neurodegeneration in Mouse Models of Amyloidosis and Tauopathy. Cell Rep. 2019, 28, 2111–2123.e6. [Google Scholar] [CrossRef]
- Crehan, H.; Hardy, J.; Pocock, J. Microglia, Alzheimer’s disease, and complement. Int. J. Alzheimer’s Dis. 2012, 2012, 983640. [Google Scholar] [CrossRef]
- Pekna, M.; Pekny, M. The Complement System: A Powerful modulator and effector of astrocyte function in the healthy and diseased central nervous system. Cells 2021, 10, 1812. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Schwartz, J.B.; Abner, E.L.; Jicha, G.A.; Kapogiannis, D. High complement levels in astrocyte-derived exosomes of Alzheimer disease. Ann. Neurol. 2018, 83, 544–552. [Google Scholar] [CrossRef]
- Lian, H.; Litvinchuk, A.; Chiang, A.C.-A.; Aithmitti, N.; Jankowsky, J.L.; Zheng, H. Astrocyte-Microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s disease. J. Neurosci. 2016, 36, 577–589. [Google Scholar] [CrossRef]
- Fleeman, R.M.; Proctor, E.A. Astrocytic Propagation of Tau in the Context of Alzheimer’s Disease. Front. Cell. Neurosci. 2021, 15, 645233. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.J.; Beltran-Lobo, P.; Johnson, L.; Perez-Nievas, B.G.; Noble, W. Astrocytes in Tauopathies. Front. Neurol. 2020, 11, 572850. [Google Scholar] [CrossRef] [PubMed]
- Reichenbach, N.; Delekate, A.; Plescher, M.; Schmitt, F.; Krauss, S.; Blank, N.; Halle, A.; Petzold, G.C. Inhibition of Stat3-mediated astrogliosis ameliorates pathology in an Alzheimer’s disease model. EMBO Mol. Med. 2019, 11, 201809665. [Google Scholar] [CrossRef] [PubMed]
- Millot, P.; San, C.; Bennana, E.; Hugon, J.; Paquet, C.; Hosten, B.; Mouton-Liger, F. STAT3 inhibition reverses neuroinflammation and Aβ metabolism induced by systemic inflammation. Alzheimer’s Dement. 2020, 16, e041019. [Google Scholar] [CrossRef]
- Kang, J.; Jiang, L.; Goldman, S.A.; Nedergaard, M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat. Neurosci. 1998, 1, 683–692. [Google Scholar] [CrossRef]
- Pasti, L.; Volterra, A.; Pozzan, T.; Carmignoto, G. Intracellular Calcium Oscillations in Astrocytes: A Highly Plastic, Bidirectional Form of Communication between Neurons and Astrocytes In Situ. J. Neurosci. 1997, 17, 7817–7830. [Google Scholar] [CrossRef]
- Guthrie, P.B.; Knappenberger, J.; Segal, M.; Bennett, M.V.L.; Charles, A.C.; Kater, S.B. ATP Released from Astrocytes Mediates Glial Calcium Waves. J. Neurosci. 1999, 19, 520–528. [Google Scholar] [CrossRef]
- Durkee, C.A.; Araque, A. Diversity and Specificity of Astrocyte–neuron Communication. Neuroscience 2019, 396, 73–78. [Google Scholar] [CrossRef]
- Harada, R.; Okamura, N.; Furumoto, S.; Furukawa, K.; Ishiki, A.; Tomita, N.; Tago, T.; Hiraoka, K.; Watanuki, S.; Shidahara, M.; et al. 18F-THK5351: A Novel PET Radiotracer for Imaging Neurofibrillary Pathology in Alzheimer Disease. J. Nucl. Med. 2015, 57, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Alvarez, A.; Araque, A. Astrocyte-Neuron Interaction at Tripartite Synapses. Curr. Drug Targets 2013, 14, 1220–1224. [Google Scholar] [CrossRef]
- Farhy-Tselnicker, I.; Allen, N.J. Astrocytes, neurons, synapses: A tripartite view on cortical circuit development. Neural Dev. 2018, 13, 7. [Google Scholar] [CrossRef]
- Lia, A.; Henriques, V.J.; Zonta, M.; Chiavegato, A.; Carmignoto, G.; Gómez-Gonzalo, M.; Losi, G. Calcium Signals in Astrocyte Microdomains, a Decade of Great Advances. Front. Cell. Neurosci. 2021, 15, 673433. [Google Scholar] [CrossRef] [PubMed]
- Bernardinelli, Y.; Randall, J.; Janett, E.; Nikonenko, I.; König, S.; Jones, E.V.; Flores, C.E.; Murai, K.K.; Bochet, C.G.; Holtmaat, A.; et al. Activity-Dependent Structural Plasticity of Perisynaptic Astrocytic Domains Promotes Excitatory Synapse Stability. Curr. Biol. 2014, 24, 1679–1688. [Google Scholar] [CrossRef]
- Santello, M.; Toni, N.; Volterra, A. Astrocyte function from information processing to cognition and cognitive impairment. Nat. Neurosci. 2019, 22, 154–166. [Google Scholar] [CrossRef]
- Ding, H.; Roncari, L.; Shannon, P.; Wu, X.; Lau, N.; Karaskova, J.; Gutmann, D.; Squire, J.; Nagy, A.; Guha, A. Astrocyte-specific expression of activated p21-ras results in malignant astrocytoma formation in a transgenic mouse model of human gliomas. Cancer Res. 2001, 61, 3826–3836. [Google Scholar]
- Dohm, C.; Kermer, P.; Bähr, M. Aggregopathy in Neurodegenerative Diseases: Mechanisms and Therapeutic Implication. Neurodegener. Dis. 2008, 5, 321–338. [Google Scholar] [CrossRef]
- Meyer, P.-F.; Savard, M.; Poirier, J.; Labonté, A.; Rosa-Neto, P.; Weitz, T.M.; Town, T.; Breitner, J.; Initiative, F.T.A.D.N.; the PREVENT-AD Research Group. Bi-directional Association of Cerebrospinal Fluid Immune Markers with Stage of Alzheimer’s Disease Pathogenesis. J. Alzheimer’s Dis. 2018, 63, 577–590. [Google Scholar] [CrossRef]
- Onyango, I.G.; Jauregui, G.V.; Čarná, M.; Bennett, J.P.; Stokin, G.B. Neuroinflammation in Alzheimer’s Disease. Biomedicines 2021, 9, 524. [Google Scholar] [CrossRef] [PubMed]
- Garland, E.F.; Hartnell, I.J.; Boche, D. Microglia and Astrocyte Function and Communication: What Do We Know in Humans? Front. Neurosci. 2022, 16, 824888. [Google Scholar] [CrossRef] [PubMed]
- Boche, D.; Perry, V.H.; Nicoll, J.A.R. Review: Activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 2013, 39, 3–18. [Google Scholar] [CrossRef]
- Li, J.; Shui, X.; Sun, R.; Wan, L.; Zhang, B.; Xiao, B.; Luo, Z. Microglial Phenotypic Transition: Signaling Pathways and Influencing Modulators Involved in Regulation in Central Nervous System Diseases. Front. Cell. Neurosci. 2021, 15, 736310. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.-Y.; Lang, G.-P.; Li, C. Rutin pretreatment promotes microglial M1 to M2 phenotype polarization. Neural Regen. Res. 2021, 16, 2499–2504. [Google Scholar] [CrossRef]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Sagara, Y.; Mallory, M.; Hashimoto, M.; Mucke, L. β-Amyloid peptides enhance α-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 12245–12250. [Google Scholar] [CrossRef]
- Yu, X. Less is more: A critical role of synapse pruning in neural circuit wiring. Nat. Rev. Neurosci. 2023, 24, 61. [Google Scholar] [CrossRef]
- Faust, T.E.; Gunner, G.; Schafer, D.P. Mechanisms governing activity-dependent synaptic pruning in the developing mammalian CNS. Nat. Rev. Neurosci. 2021, 22, 657–673. [Google Scholar] [CrossRef]
- Morizawa, Y.M.; Matsumoto, M.; Nakashima, Y.; Endo, N.; Aida, T.; Ishikane, H.; Beppu, K.; Moritoh, S.; Inada, H.; Osumi, N.; et al. Synaptic pruning through glial synapse engulfment upon motor learning. Nat. Neurosci. 2022, 25, 1458–1469. [Google Scholar] [CrossRef]
- Terni, B.; López-Murcia, F.J.; Llobet, A. Role of neuron-glia interactions in developmental synapse elimination. Brain Res. Bull. 2017, 129, 74–81. [Google Scholar] [CrossRef]
- DeWitt, D.A.; Perry, G.; Cohen, M.; Doller, C.; Silver, J. Astrocytes Regulate Microglial Phagocytosis of Senile Plaque Cores of Alzheimer’s Disease. Exp. Neurol. 1998, 149, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Geloso, M.C.; D’ambrosi, N. Microglial Pruning: Relevance for Synaptic Dysfunction in Multiple Sclerosis and Related Experimental Models. Cells 2021, 10, 686. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.-S.; Clarke, L.E.; Wang, G.X.; Stafford, B.K.; Sher, A.; Chakraborty, C.; Joung, J.; Foo, L.C.; Thompson, A.; Chen, C.; et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 2013, 504, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Soteros, B.M.; Sia, G.M. Complement and microglia dependent synapse elimination in brain development. WIREs Mech. Dis. 2022, 14, e1545. [Google Scholar] [CrossRef]
- Lee, J.-H.; Kim, J.-Y.; Noh, S.; Lee, H.; Lee, S.Y.; Mun, J.Y.; Park, H.; Chung, W.-S. Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis. Nature 2021, 590, 612–617. [Google Scholar] [CrossRef]
- Gomez-Arboledas, A.; Davila, J.C.; Sanchez-Mejias, E.; Navarro, V.; Nuñez-Diaz, C.; Sanchez-Varo, R.; Micó, M.V.S.; Trujillo-Estrada, L.; Fernandez-Valenzuela, J.J.; Vizuete, M.; et al. Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer’s disease. Glia 2018, 66, 637–653. [Google Scholar] [CrossRef]
- Sanchez-Mico, M.V.; Jimenez, S.; Gomez-Arboledas, A.; Muñoz-Castro, C.; Romero-Molina, C.; Navarro, V.; Sanchez-Mejias, E.; Nuñez-Diaz, C.; Sanchez-Varo, R.; Galea, E.; et al. Amyloid-β impairs the phagocytosis of dystrophic synapses by astrocytes in Alzheimer’s disease. Glia 2021, 69, 997–1011. [Google Scholar] [CrossRef]
- Masliah, E.; Mallory, M.; Hansen, L.; Alford, M.; Albright, T.; DeTeresa, R.; Terry, R.; Baudier, J.; Saitoh, T. Patterns of aberrant sprouting in alzheimer’s disease. Neuron 1991, 6, 729–739. [Google Scholar] [CrossRef]
- Masliah, E.; Mallory, M.; DeTeresa, R.; Alford, M.; Hansen, L. Differing patterns of aberrant neuronal sprouting in Alzheimer’s disease with and without Lewy bodies. Brain Res. 1993, 617, 258–266. [Google Scholar] [CrossRef]
- Allen, N.J.; Eroglu, C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017, 96, 697–708. [Google Scholar] [CrossRef]
- Baldwin, K.T.; Eroglu, C. Molecular mechanisms of astrocyte-induced synaptogenesis. Curr. Opin. Neurobiol. 2017, 45, 113–120. [Google Scholar] [CrossRef]
- Sadick, J.S.; Liddelow, S.A. Don’t forget astrocytes when targeting Alzheimer’s disease. Br. J. Pharmacol. 2019, 176, 3585–3598. [Google Scholar] [CrossRef]
- Christopherson, K.S.; Ullian, E.M.; Stokes, C.C.A.; Mullowney, C.E.; Hell, J.W.; Agah, A.; Lawler, J.; Mosher, D.F.; Bornstein, P.; Barres, B.A. Thrombospondins Are Astrocyte-Secreted Proteins that Promote CNS Synaptogenesis. Cell 2005, 120, 421–433. [Google Scholar] [CrossRef]
- Kang, S.; Byun, J.; Son, S.M.; Mook-Jung, I. Thrombospondin-1 protects against Aβ-induced mitochondrial fragmentation and dysfunction in hippocampal cells. Cell Death Discov. 2018, 4, 31. [Google Scholar] [CrossRef]
- Son, S.M.; Nam, D.W.; Cha, M.-Y.; Kim, K.H.; Byun, J.; Ryu, H.; Mook-Jung, I. Thrombospondin-1 prevents amyloid beta–mediated synaptic pathology in Alzheimer’s disease. Neurobiol. Aging 2015, 36, 3214–3227. [Google Scholar] [CrossRef]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. [Google Scholar] [CrossRef]
- Norenberg, M.D.; Martinez-Hernandez, A. Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res. 1979, 161, 303–310. [Google Scholar] [CrossRef]
- McKenna, M.C. Glutamate Pays Its Own Way in Astrocytes. Front. Endocrinol. 2013, 4, 191. [Google Scholar] [CrossRef]
- Schousboe, A.; Scafidi, S.; Bak, L.K.; Waagepetersen, H.S.; McKenna, M.C. Glutamate Metabolism in the Brain Focusing on Astrocytes. Adv. Neurobiol. 2014, 11, 13–30. [Google Scholar] [CrossRef]
- Kazama, M.; Kato, Y.; Kakita, A.; Noguchi, N.; Urano, Y.; Masui, K.; Niida-Kawaguchi, M.; Yamamoto, T.; Watabe, K.; Kitagawa, K.; et al. Astrocytes release glutamate via cystine/glutamate antiporter upregulated in response to increased oxidative stress related to sporadic amyotrophic lateral sclerosis. Neuropathology 2020, 40, 587–598. [Google Scholar] [CrossRef]
- Andersen, J.V.; Markussen, K.H.; Jakobsen, E.; Schousboe, A.; Waagepetersen, H.S.; Rosenberg, P.A.; Aldana, B.I. Glutamate metabolism and recycling at the excitatory synapse in health and neurodegeneration. Neuropharmacology 2021, 196, 108719. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.; Nakano, M.; Kubota, K.; Himuro, N.; Mizoguchi, S.; Chikenji, T.; Otani, M.; Mizue, Y.; Nagaishi, K.; Fujimiya, M. Activated forms of astrocytes with higher GLT-1 expression are associated with cognitive normal subjects with Alzheimer pathology in human brain. Sci. Rep. 2018, 8, 1712. [Google Scholar] [CrossRef]
- Matos, M.; Augusto, E.; Oliveira, C.; Agostinho, P. Amyloid-beta peptide decreases glutamate uptake in cultured astrocytes: Involvement of oxidative stress and mitogen-activated protein kinase cascades. Neuroscience 2008, 156, 898–910. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Rivas-Santisteban, R.; Casanovas, M.; Lillo, A.; Saura, C.A.; Navarro, G. Adenosine A2A Receptor Antagonists Affects NMDA Glutamate Receptor Function. Potential to Address Neurodegeneration in Alzheimer’s Disease. Cells 2020, 9, 1075. [Google Scholar] [CrossRef]
- Matos, M.; Augusto, E.; Machado, N.J.; dos Santos-Rodrigues, A.; Cunha, R.A.; Agostinho, P. Astrocytic Adenosine A2A Receptors Control the Amyloid-β Peptide-Induced Decrease of Glutamate Uptake. J. Alzheimer’s Dis. 2012, 31, 555–567. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef]
- Lee, M.-C.; Ting, K.K.; Adams, S.; Brew, B.J.; Chung, R.; Guillemin, G.J. Characterisation of the Expression of NMDA Receptors in Human Astrocytes. PLoS ONE 2010, 5, e14123. [Google Scholar] [CrossRef]
- Liu, P.-P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef]
- Li, Y.; Chang, L.; Song, Y.; Gao, X.; Roselli, F.; Liu, J.; Zhou, W.; Fang, Y.; Ling, W.; Li, H.; et al. Astrocytic GluN2A and GluN2B Oppose the Synaptotoxic Effects of Amyloid-β1-40 in Hippocampal Cells. J. Alzheimer’s Dis. 2016, 54, 135–148. [Google Scholar] [CrossRef]
- Charles, A.C.; Merrill, J.E.; Dirksen, E.R.; Sandersont, M.J. Intercellular Signaling in Glial Cells: Calcium Waves and Oscillations in Response to Mechanical Stimulation and Glutamate. Neuron 1991, 6, 983–992. [Google Scholar] [CrossRef]
- Cornell-Bell, A.H.; Finkbeiner, S.M.; Cooper, M.S.; Smith, S.J. Glutamate Induces Calcium Waves in Cultured Astrocytes: Long-Range Glial Signaling. Science (1979) 1990, 247, 470–473. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Kettenmann, H. Calcium signalling in glial cells. Trends Neurosci. 1996, 19, 346–352. [Google Scholar] [CrossRef]
- Aguado, F.; Espinosa-Parrilla, J.F.; Carmona, M.A.; Soriano, E. Neuronal Activity Regulates Correlated Network Properties of Spontaneous Calcium Transients in Astrocytes In Situ. J. Neurosci. 2002, 22, 9430–9444. [Google Scholar] [CrossRef]
- Perea, G.; Araque, A. Properties of Synaptically Evoked Astrocyte calcium signal reveal synaptic information processing by astrocytes. J. Neurosci. 2005, 25, 2192–2203. [Google Scholar] [CrossRef] [PubMed]
- Bushong, E.A.; Martone, M.E.; Jones, Y.Z.; Ellisman, M.H. Protoplasmic Astrocytes in CA1 Stratum Radiatum Occupy Separate Anatomical Domains. J. Neurosci. 2002, 22, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A. Astroglial Calcium signaling in aging and Alzheimer’s disease. Cold Spring Harb. Perspect. Biol. 2019, 11, a035188. [Google Scholar] [CrossRef]
- Oberheim, N.A.; Wang, X.; Goldman, S.; Nedergaard, M. Astrocytic complexity distinguishes the human brain. Trends Neurosci. 2006, 29, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Oberheim, N.A.; Takano, T.; Han, X.; He, W.; Lin, J.H.C.; Wang, F.; Xu, Q.; Wyatt, J.D.; Pilcher, W.; Ojemann, J.; et al. Uniquely Hominid Features of Adult Human Astrocytes. J. Neurosci. 2009, 29, 3276–3287. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.Y.-S.; Woo, J.; Sardar, D.; Lozzi, B.; Huerta, N.A.B.; Lin, C.-C.J.; Felice, D.; Jain, A.; Paulucci-Holthauzen, A.; Deneen, B. Region-Specific Transcriptional Control of Astrocyte Function Oversees Local Circuit Activities. Neuron 2020, 106, 992–1008.e9. [Google Scholar] [CrossRef]
- González, A.; Calfío, C.; Churruca, M.; Maccioni, R.B. Glucose metabolism and AD: Evidence for a potential diabetes type 3. Alzheimer’s Res. Ther. 2022, 14, 56. [Google Scholar] [CrossRef] [PubMed]
- Tomi, M.; Zhao, Y.; Thamotharan, S.; Shin, B.-C.; Devaskar, S.U. Early life nutrient restriction impairs blood–brain metabolic profile and neurobehavior predisposing to Alzheimer’s disease with aging. Brain Res. 2013, 1495, 61–75. [Google Scholar] [CrossRef]
- Winkler, E.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef]
- Bigl, M.; Arendt, T.; Bigl, V.; Eschrich, K.; Brückner, M.K. Activities of key glycolytic enzymes in the brains of patients with Alzheimer’s disease. J. Neural Transm. 1999, 106, 499–511. [Google Scholar] [CrossRef]
- Šalković-Petrišić, M.; Riederer, P. Brain glucose transporter protein 2 and sporadic Alzheimer’s disease. Transl. Neurosci. 2010, 1, 200–206. [Google Scholar] [CrossRef]
- Yu, L.; Jin, J.; Xu, Y.; Zhu, X. Aberrant energy metabolism in Alzheimer’s disease. J. Transl. Intern. Med. 2022, 10, 197–206. [Google Scholar] [CrossRef]
- Wang, X.F.; Cynader, M.S. Pyruvate Released by Astrocytes Protects Neurons from Copper-Catalyzed Cysteine Neurotoxicity. J. Neurosci. 2001, 21, 3322–3331. [Google Scholar] [CrossRef]
- Bonvento, G.; Bolaños, J.P. Astrocyte-neuron metabolic cooperation shapes brain activity. Cell Metab. 2021, 33, 1546–1564. [Google Scholar] [CrossRef]
- Bigl, M.; Apelt, J.; Eschrich, K.; Schliebs, R. Cortical glucose metabolism is altered in aged transgenic Tg2576 mice that demonstrate Alzheimer plaque pathology. J. Neural Transm. 2003, 110, 77–94. [Google Scholar] [CrossRef]
- Fu, W.; Shi, D.; Westaway, D.; Jhamandas, J.H. Bioenergetic mechanisms in astrocytes may contribute to amyloid plaque deposition and toxicity. J. Biol. Chem. 2015, 290, 12504–12513. [Google Scholar] [CrossRef]
- Staurenghi, E.; Cerrato, V.; Gamba, P.; Testa, G.; Giannelli, S.; Leoni, V.; Caccia, C.; Buffo, A.; Noble, W.; Perez-Nievas, B.G.; et al. Oxysterols present in Alzheimer’s disease brain induce synaptotoxicity by activating astrocytes: A major role for lipocalin-2. Redox Biol. 2020, 39, 101837. [Google Scholar] [CrossRef]
- Liu, C.-C.; Hu, J.; Zhao, N.; Wang, J.; Wang, N.; Cirrito, J.R.; Kanekiyo, T.; Holtzman, D.M.; Bu, G. Astrocytic LRP1 Mediates Brain Aβ Clearance and Impacts Amyloid Deposition. J. Neurosci. 2017, 37, 4023–4031. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. β-Amyloid Peptides Induce Mitochondrial Dysfunction and Oxidative Stress in Astrocytes and Death of Neurons through Activation of NADPH Oxidase. J. Neurosci. 2004, 24, 565–575. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- King, A.; Szekely, B.; Calapkulu, E.; Ali, H.; Rios, F.; Jones, S.; Troakes, C. The Increased Densities, But Different Distributions, of Both C3 and S100A10 Immunopositive Astrocyte-Like Cells in Alzheimer’s Disease Brains Suggest Possible Roles for Both A1 and A2 Astrocytes in the Disease Pathogenesis. Brain Sci. 2020, 10, 503. [Google Scholar] [CrossRef]
- Balu, D.T.; Pantazopoulos, H.; Huang, C.C.; Muszynski, K.; Harvey, T.L.; Uno, Y.; Rorabaugh, J.M.; Galloway, C.R.; Botz-Zapp, C.; Berretta, S.; et al. Neurotoxic astrocytes express the d-serine synthesizing enzyme, serine racemase, in Alzheimer’s disease. Neurobiol. Dis. 2019, 130, 104511. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stanca, S.; Rossetti, M.; Bongioanni, P. Astrocytes as Neuroimmunocytes in Alzheimer’s Disease: A Biochemical Tool in the Neuron–Glia Crosstalk along the Pathogenetic Pathways. Int. J. Mol. Sci. 2023, 24, 13880. https://doi.org/10.3390/ijms241813880
Stanca S, Rossetti M, Bongioanni P. Astrocytes as Neuroimmunocytes in Alzheimer’s Disease: A Biochemical Tool in the Neuron–Glia Crosstalk along the Pathogenetic Pathways. International Journal of Molecular Sciences. 2023; 24(18):13880. https://doi.org/10.3390/ijms241813880
Chicago/Turabian StyleStanca, Stefano, Martina Rossetti, and Paolo Bongioanni. 2023. "Astrocytes as Neuroimmunocytes in Alzheimer’s Disease: A Biochemical Tool in the Neuron–Glia Crosstalk along the Pathogenetic Pathways" International Journal of Molecular Sciences 24, no. 18: 13880. https://doi.org/10.3390/ijms241813880
APA StyleStanca, S., Rossetti, M., & Bongioanni, P. (2023). Astrocytes as Neuroimmunocytes in Alzheimer’s Disease: A Biochemical Tool in the Neuron–Glia Crosstalk along the Pathogenetic Pathways. International Journal of Molecular Sciences, 24(18), 13880. https://doi.org/10.3390/ijms241813880