
FLII Modulates the Myogenic Differentiation of Progenitor Cells via Actin Remodeling-Mediated YAP1 Regulation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
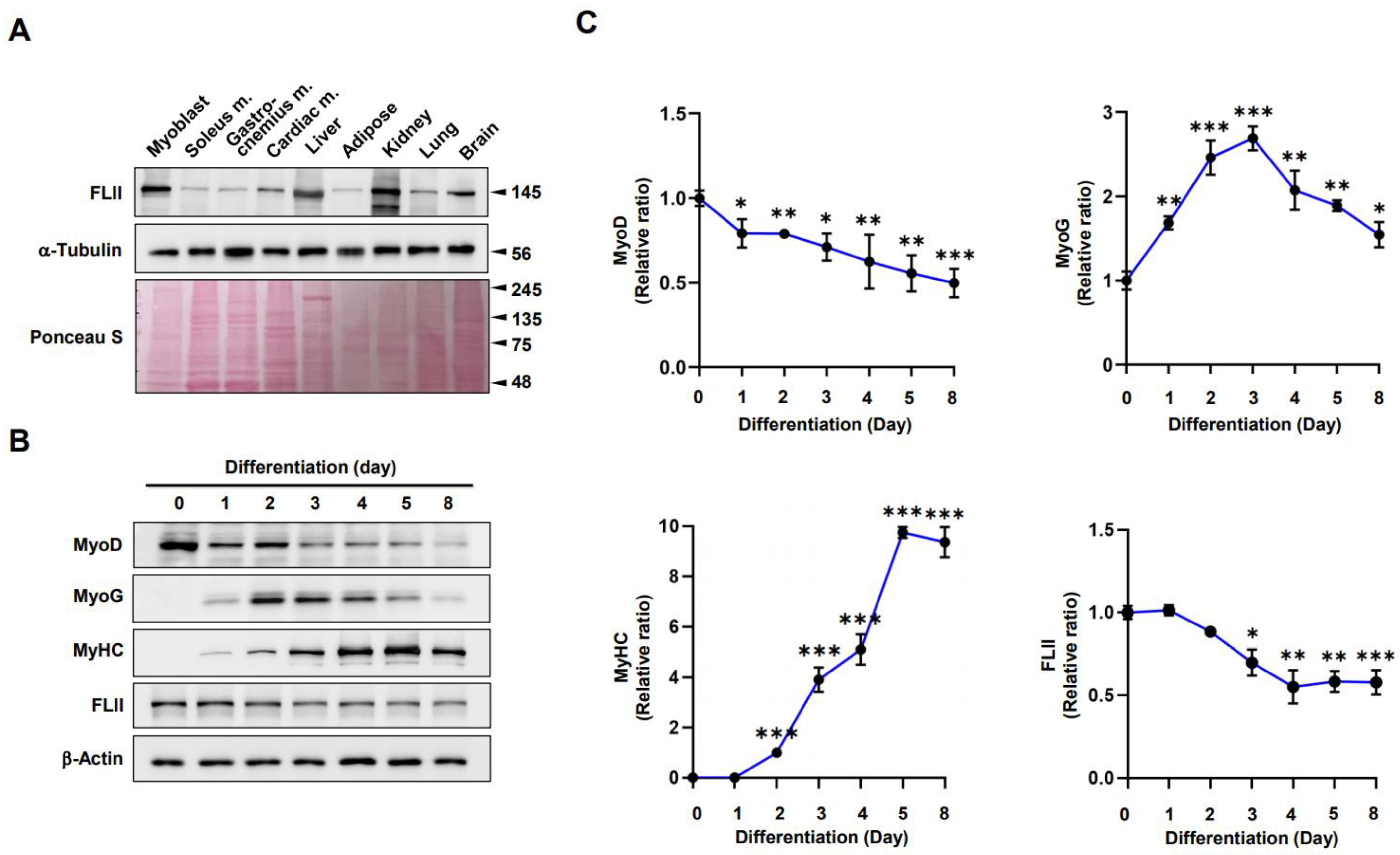
2.1. FLII Expression Is Modulated during Myoblast Differentiation
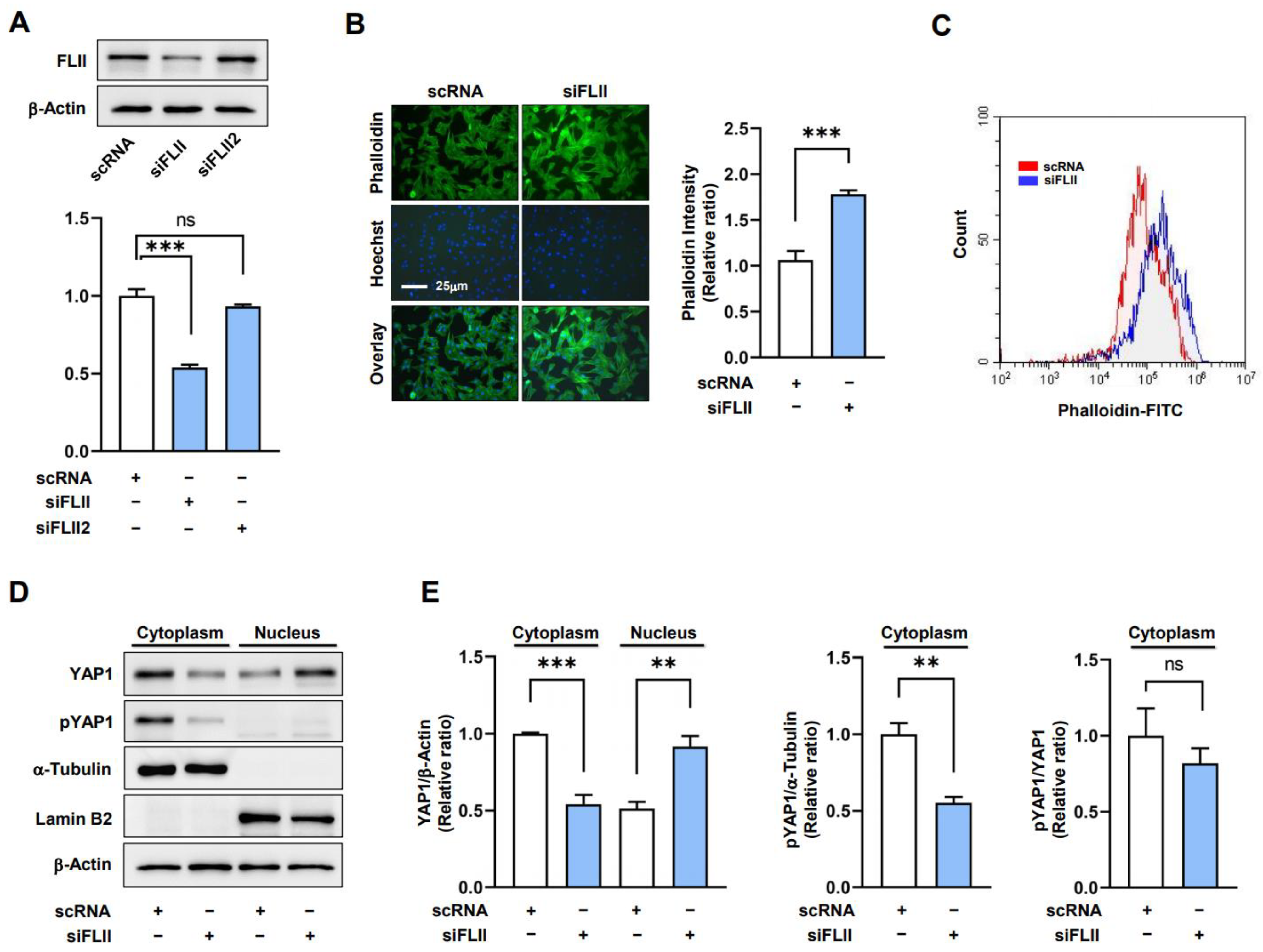
2.2. Knockdown of FLII Augments F-Actin and Nuclear YAP1 Levels in Myoblasts
2.3. FLII Knockdown Enhances Myoblast Proliferation
2.4. FLII Is Required for Myogenic Factors Expressions and Myoblast Differentiation
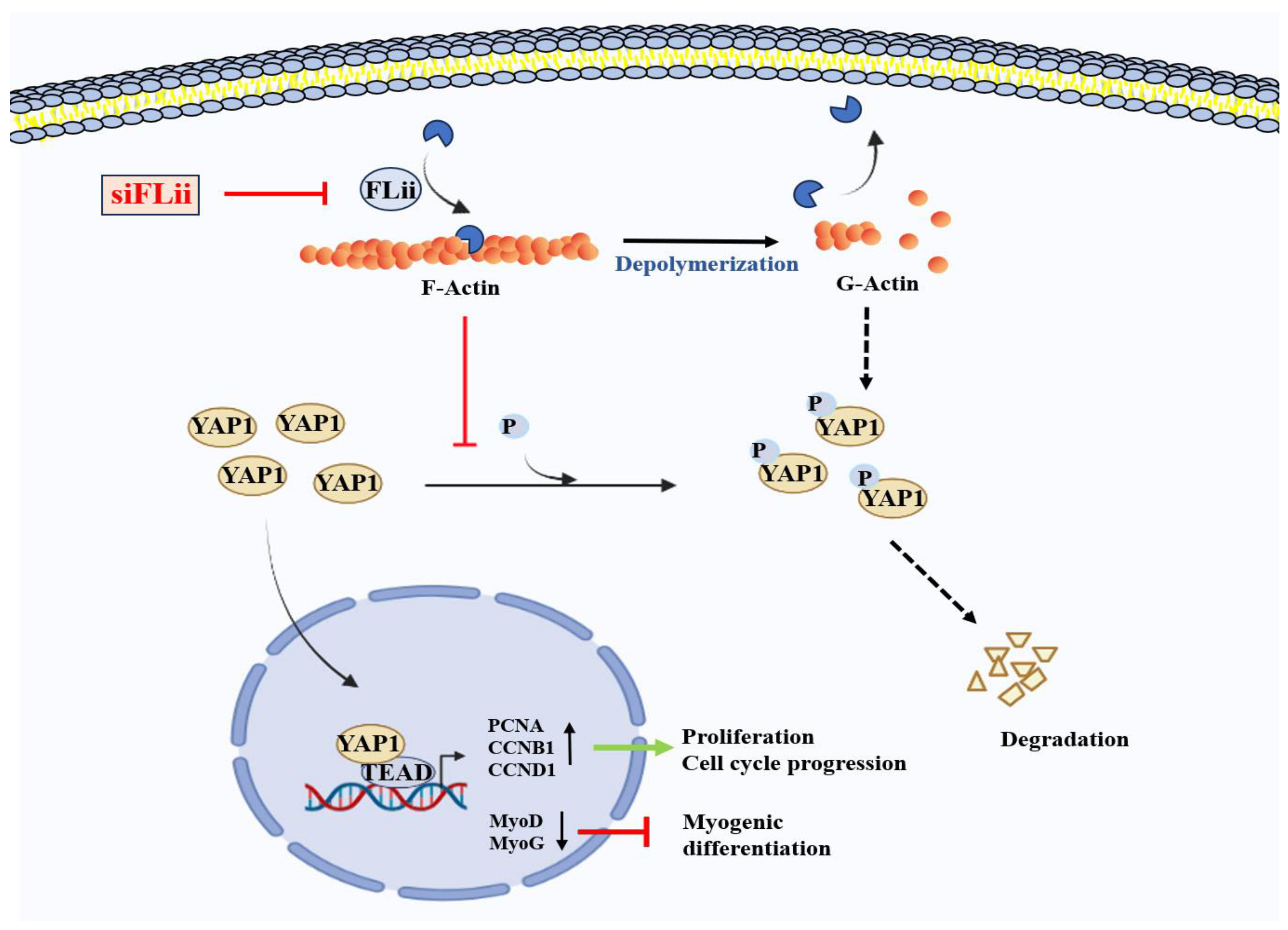
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Differentiation
4.2. Oligonucleotide Transfection
4.3. Real-Time Quantitative Reverse Transcription PCR (qRT-PCR)
4.4. Cytoplasmic and Nuclear Extraction
4.5. Immunoblot Analysis
4.6. Immunofluorescence Analysis
4.7. Cell Proliferation Assay
4.8. Cell Viability
4.9. Flow Cytometry
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mukund, K.; Subramaniam, S. Skeletal muscle: A review of molecular structure and function, in health and disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1462. [Google Scholar] [CrossRef] [PubMed]
- Chal, J.; Pourquie, O. Making muscle: Skeletal myogenesis in vivo and in vitro. Development 2017, 144, 2104–2122. [Google Scholar] [CrossRef] [PubMed]
- Sartori, R.; Romanello, V.; Sandri, M. Mechanisms of muscle atrophy and hypertrophy: Implications in health and disease. Nat. Commun. 2021, 12, 330. [Google Scholar] [CrossRef] [PubMed]
- Watt, K.I.; Goodman, C.A.; Hornberger, T.A.; Gregorevic, P. The Hippo Signaling Pathway in the Regulation of Skeletal Muscle Mass and Function. Exerc. Sport. Sci. Rev. 2018, 46, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Rikeit, P.; Knaus, P.; Coirault, C. YAP-Mediated Mechanotransduction in Skeletal Muscle. Front. Physiol. 2016, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Guerin, C.M.; Kramer, S.G. Cytoskeletal remodeling during myotube assembly and guidance: Coordinating the actin and microtubule networks. Commun. Integr. Biol. 2009, 2, 452–457. [Google Scholar] [CrossRef]
- Heng, Y.W.; Koh, C.G. Actin cytoskeleton dynamics and the cell division cycle. Int. J. Biochem. Cell Biol. 2010, 42, 1622–1633. [Google Scholar] [CrossRef]
- Pollard, T.D.; Borisy, G.G. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef]
- Nowak, S.J.; Nahirney, P.C.; Hadjantonakis, A.K.; Baylies, M.K. Nap1-mediated actin remodeling is essential for mammalian myoblast fusion. J. Cell Sci. 2009, 122, 3282–3293. [Google Scholar] [CrossRef]
- O’Connor, R.S.; Steeds, C.M.; Wiseman, R.W.; Pavlath, G.K. Phosphocreatine as an energy source for actin cytoskeletal rearrangements during myoblast fusion. J. Physiol. 2008, 586, 2841–2853. [Google Scholar] [CrossRef]
- Zhang, T.; Zaal, K.J.; Sheridan, J.; Mehta, A.; Gundersen, G.G.; Ralston, E. Microtubule plus-end binding protein EB1 is necessary for muscle cell differentiation, elongation and fusion. J. Cell Sci. 2009, 122, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.U.; Liang, V.R.; Wang, H.V. Actin-associated protein palladin is required for migration behavior and differentiation potential of C2C12 myoblast cells. Biochem. Biophys. Res. Commun. 2014, 452, 728–733. [Google Scholar] [CrossRef]
- Li, H.; Hou, L.; Zhang, Y.; Jiang, F.; Zhu, Y.; Li, Q.X.; Hu, C.Y.; Wang, C. PFN2a Suppresses C2C12 Myogenic Development by Inhibiting Proliferation and Promoting Apoptosis via the p53 Pathway. Cells 2019, 8, 959. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Min, K.H.; Kim, D.; Park, S.Y.; Lee, W. CFL2 is an essential mediator for myogenic differentiation in C2C12 myoblasts. Biochem. Biophys. Res. Commun. 2020, 533, 710–716. [Google Scholar] [CrossRef]
- Kopecki, Z.; Cowin, A. Flightless I: An actin-remodelling protein and an important negative regulator of wound repair. Int. J. Biochem. Cell Biol. 2008, 40, 1415–1419. [Google Scholar] [CrossRef]
- Liu, Y.-T.; Yin, H.L. Identification of the binding partners for flightless I, A novel protein bridging the leucine-rich repeat and the gelsolin superfamilies. J. Biol. Chem. 1998, 273, 7920–7927. [Google Scholar] [CrossRef]
- Davy, D.A.; Ball, E.E.; Matthaei, K.I.; Campbell, H.D.; Crouch, M.F. The flightless I protein localizes to actin-based structures during embryonic development. Immunol. Cell Biol. 2000, 78, 423–429. [Google Scholar] [CrossRef]
- Mohammad, I.; Arora, P.D.; Naghibzadeh, Y.; Wang, Y.; Li, J.; Mascarenhas, W.; Janmey, P.A.; Dawson, J.F.; McCulloch, C.A. Flightless I is a focal adhesion-associated actin-capping protein that regulates cell migration. FASEB J. 2012, 26, 3260–3272. [Google Scholar] [CrossRef]
- Deng, S.; Silimon, R.L.; Balakrishnan, M.; Bothe, I.; Juros, D.; Soffar, D.B.; Baylies, M.K. The actin polymerization factor Diaphanous and the actin severing protein Flightless I collaborate to regulate sarcomere size. Dev. Biol. 2021, 469, 12–25. [Google Scholar] [CrossRef]
- Schnorrer, F.; Schonbauer, C.; Langer, C.C.; Dietzl, G.; Novatchkova, M.; Schernhuber, K.; Fellner, M.; Azaryan, A.; Radolf, M.; Stark, A.; et al. Systematic genetic analysis of muscle morphogenesis and function in Drosophila. Nature 2010, 464, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S. Role of YAP/TAZ in cell-matrix adhesion-mediated signalling and mechanotransduction. Exp. Cell Res. 2016, 343, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Gu, W.; Zhang, Y.; Jiang, B.; Qiao, X.; Wen, Y. Activated Yes-Associated Protein Accelerates Cell Cycle, Inhibits Apoptosis, and Delays Senescence in Human Periodontal Ligament Stem Cells. Int. J. Med. Sci. 2018, 15, 1241–1250. [Google Scholar] [CrossRef]
- Kim, W.; Cho, Y.S.; Wang, X.; Park, O.; Ma, X.; Kim, H.; Gan, W.; Jho, E.H.; Cha, B.; Jeung, Y.J.; et al. Hippo signaling is intrinsically regulated during cell cycle progression by APC/C(Cdh1). Proc. Natl. Acad. Sci. USA 2019, 116, 9423–9432. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, M.L.; George, L.K.; Grant, G.D.; Perou, C.M. Common markers of proliferation. Nat. Rev. Cancer 2006, 6, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Campbell, H.D.; Schimansky, T.; Claudianos, C.; Ozsarac, N.; Kasprzak, A.B.; Cotsell, J.N.; Young, I.G.; de Couet, H.G.; Miklos, G.L. The Drosophila melanogaster flightless-I gene involved in gastrulation and muscle degeneration encodes gelsolin-like and leucine-rich repeat domains and is conserved in Caenorhabditis elegans and humans. Proc. Natl. Acad. Sci. USA 1993, 90, 11386–11390. [Google Scholar] [CrossRef]
- Bruyere, C.; Versaevel, M.; Mohammed, D.; Alaimo, L.; Luciano, M.; Vercruysse, E.; Gabriele, S. Actomyosin contractility scales with myoblast elongation and enhances differentiation through YAP nuclear export. Sci. Rep. 2019, 9, 15565. [Google Scholar] [CrossRef]
- Cowin, A.J.; Adams, D.H.; Strudwick, X.L.; Chan, H.; Hooper, J.A.; Sander, G.R.; Rayner, T.E.; Matthaei, K.I.; Powell, B.C.; Campbell, H.D. Flightless I deficiency enhances wound repair by increasing cell migration and proliferation. J. Pathol. 2007, 211, 572–581. [Google Scholar] [CrossRef]
- Kopecki, Z.; Arkell, R.; Powell, B.C.; Cowin, A.J. Flightless I regulates hemidesmosome formation and integrin-mediated cellular adhesion and migration during wound repair. J. Investig. Dermatol. 2009, 129, 2031–2045. [Google Scholar] [CrossRef]
- Wang, T.; Song, W.; Chen, Y.; Chen, R.; Liu, Z.; Wu, L.; Li, M.; Yang, J.; Wang, L.; Liu, J.; et al. Correction: Flightless I Homolog Represses Prostate Cancer Progression through Targeting Androgen Receptor Signaling. Clin. Cancer Res. 2022, 28, 2970. [Google Scholar] [CrossRef]
- Abramovici, H.; Gee, S.H. Morphological changes and spatial regulation of diacylglycerol kinase-zeta, syntrophins, and Rac1 during myoblast fusion. Cell Motil. Cytoskelet. 2007, 64, 549–567. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.G.; Janmey, P.A. Transcription factor regulation by mechanical stress. Int. J. Biochem. Cell Biol. 2012, 44, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Densham, R.M.; O’Neill, E.; Munro, J.; König, I.; Anderson, K.; Kolch, W.; Olson, M.F. MST Kinases Monitor Actin Cytoskeletal Integrity and Signal via c-Jun N-Terminal Kinase Stress-Activated Kinase to Regulate p21Waf1/Cip1 Stability. Mol. Cell. Biol. 2009, 29, 6380–6390. [Google Scholar] [CrossRef] [PubMed]
- Wada, K.-I.; Itoga, K.; Okano, T.; Yonemura, S.; Sasaki, H. Hippo pathway regulation by cell morphology and stress fibers. Development 2011, 138, 3907–3914. [Google Scholar] [CrossRef] [PubMed]
- Mana-Capelli, S.; Paramasivam, M.; Dutta, S.; McCollum, D. Angiomotins link F-actin architecture to Hippo pathway signaling. Mol. Biol. Cell 2014, 25, 1676–1685. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.W.; Bamburg, J.R. ADF/cofilin: A functional node in cell biology. Trends Cell Biol. 2010, 20, 187–195. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, M.T.; Ly, Q.K.; Kim, H.-J.; Lee, W. FLII Modulates the Myogenic Differentiation of Progenitor Cells via Actin Remodeling-Mediated YAP1 Regulation. Int. J. Mol. Sci. 2023, 24, 14335. https://doi.org/10.3390/ijms241814335
Nguyen MT, Ly QK, Kim H-J, Lee W. FLII Modulates the Myogenic Differentiation of Progenitor Cells via Actin Remodeling-Mediated YAP1 Regulation. International Journal of Molecular Sciences. 2023; 24(18):14335. https://doi.org/10.3390/ijms241814335
Chicago/Turabian StyleNguyen, Mai Thi, Quoc Kiet Ly, Hyun-Jung Kim, and Wan Lee. 2023. "FLII Modulates the Myogenic Differentiation of Progenitor Cells via Actin Remodeling-Mediated YAP1 Regulation" International Journal of Molecular Sciences 24, no. 18: 14335. https://doi.org/10.3390/ijms241814335
APA StyleNguyen, M. T., Ly, Q. K., Kim, H. -J., & Lee, W. (2023). FLII Modulates the Myogenic Differentiation of Progenitor Cells via Actin Remodeling-Mediated YAP1 Regulation. International Journal of Molecular Sciences, 24(18), 14335. https://doi.org/10.3390/ijms241814335