
Lipopolysaccharide of Legionella pneumophila Serogroup 1 Facilitates Interaction with Host Cells

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
2.1. Phenotypical Screening
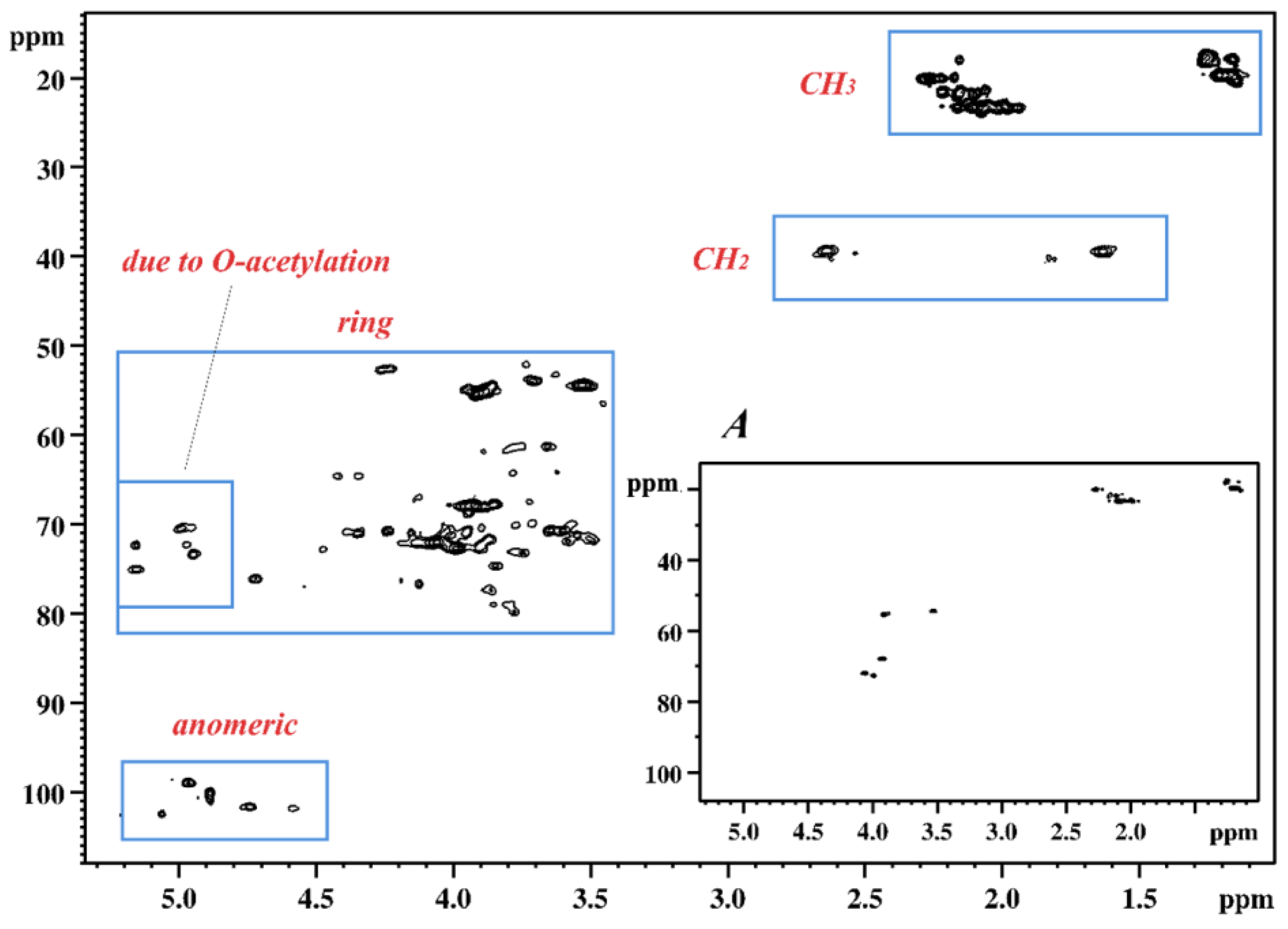
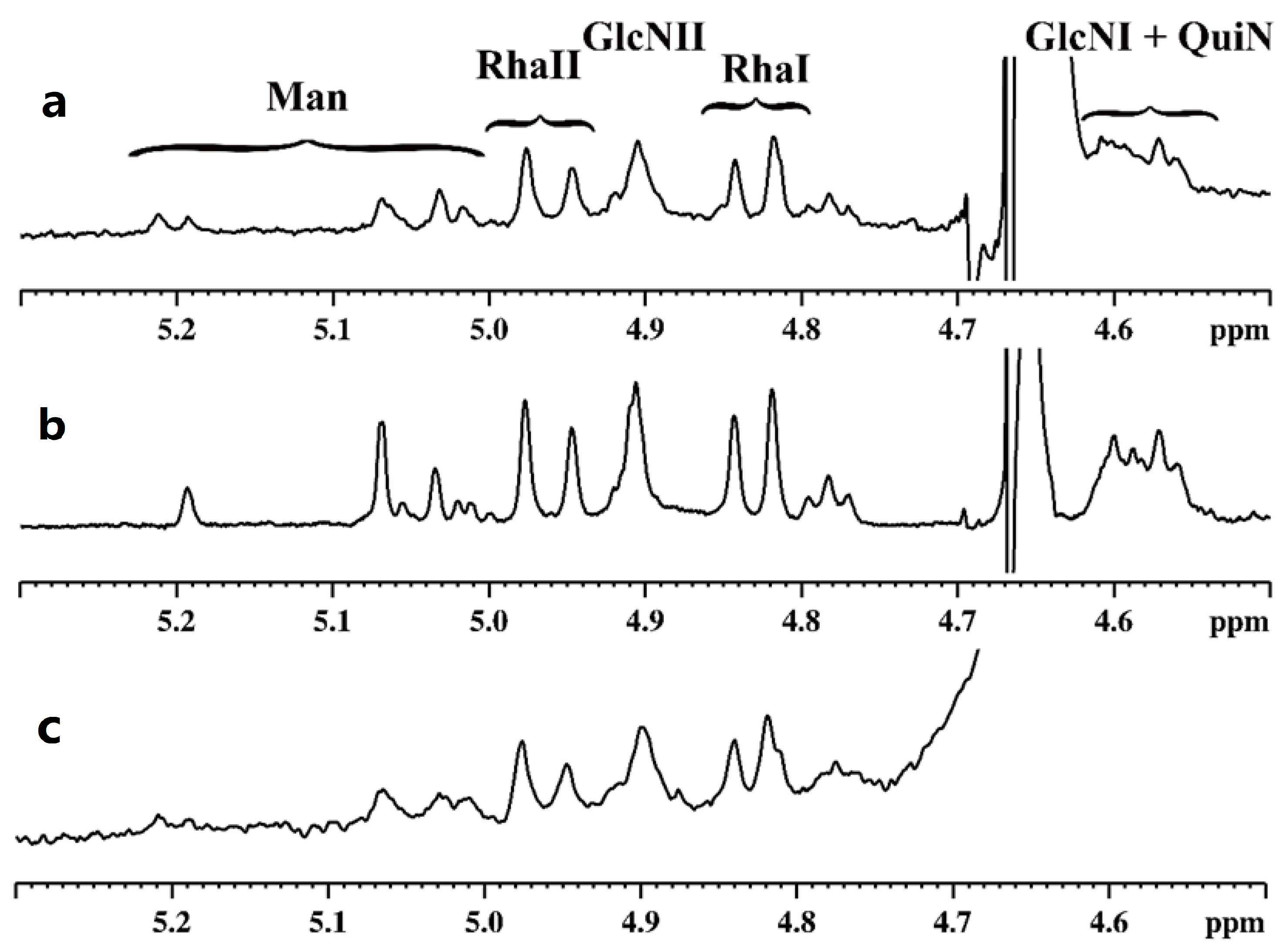
2.2. NMR Spectroscopy

2.3. A Mutation in the LPS Synthesis Gene Affects the Lipid Profile of L. pneumophila
2.4. FLIM-FRET Measurements
2.5. LPS Contributes to Host Cell Adherence and Invasion
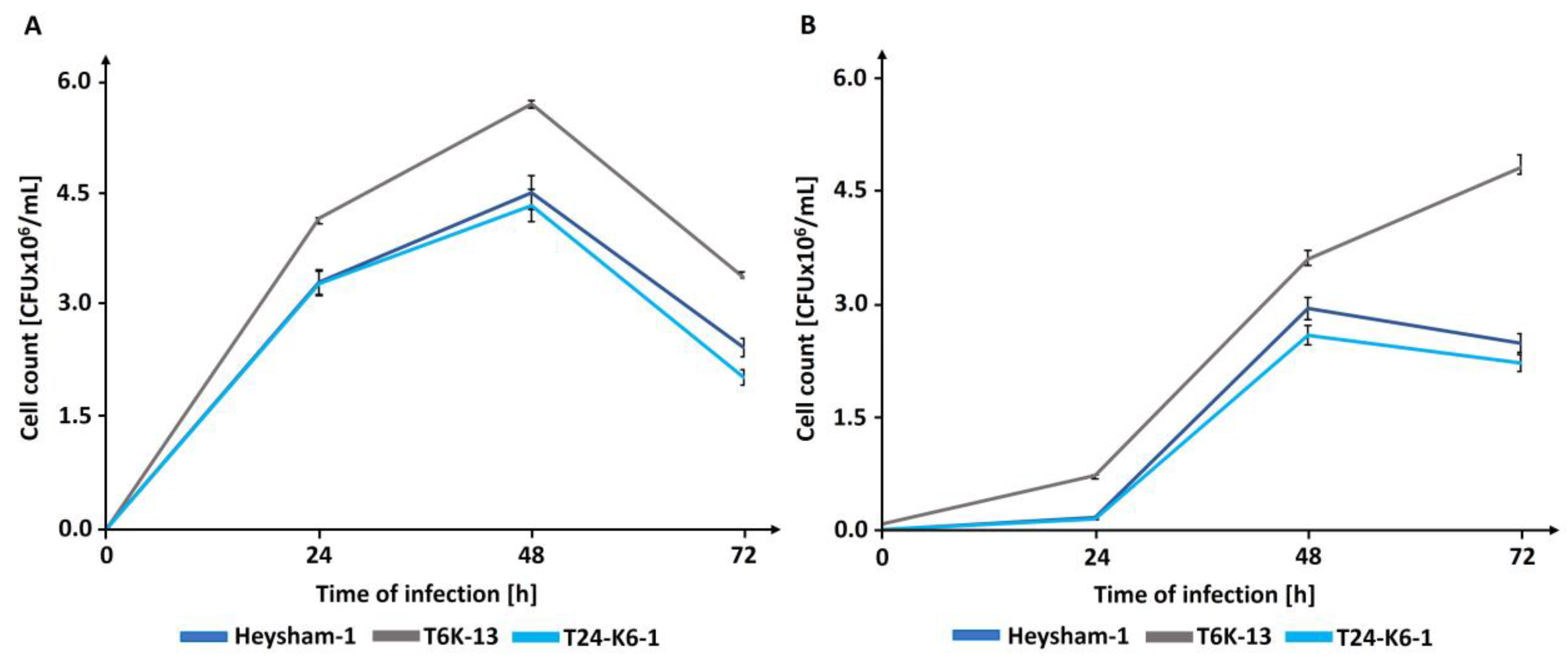
2.6. The T6-K13 Mutant-Type Exhibits a Higher Replication Efficiency within Host Cells than the Wild-Type Strain
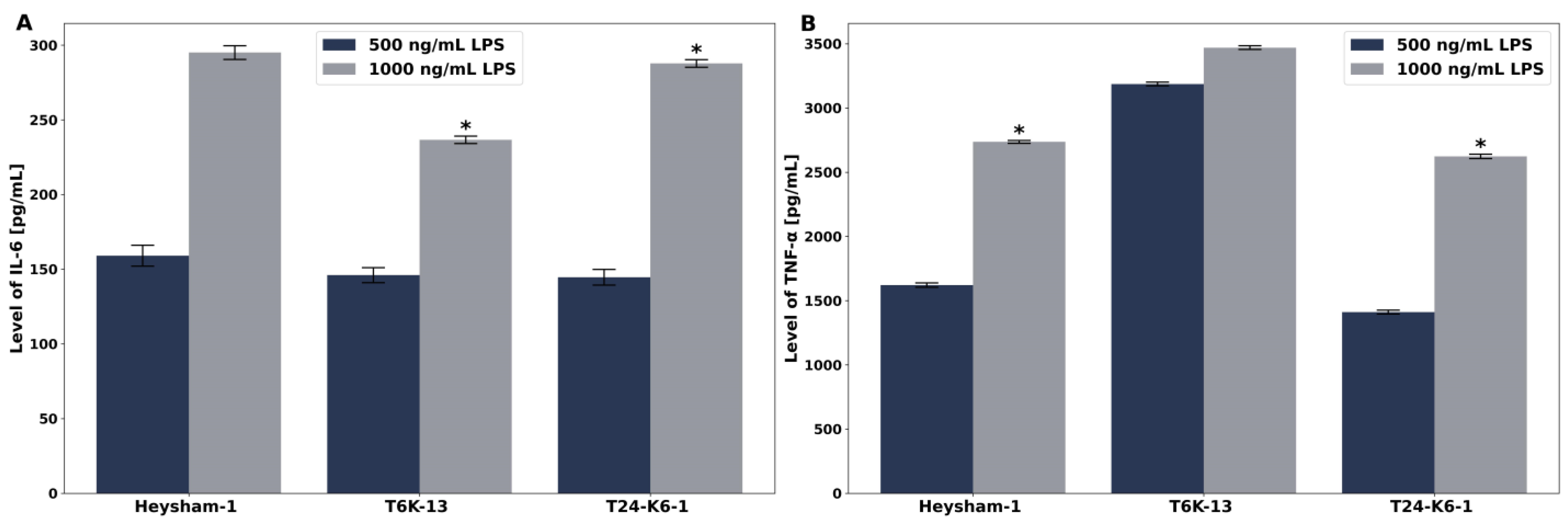
2.7. The Mutation in the Polysaccharide Region of L. pneumophila LPS Results in a More Potent Induction of Proinflammatory Cytokines
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Bacterial Strains and Growth Conditions
4.3. Construction of the L. pneumophila Mutant and Complementation
4.4. Cultivation of Eukaryotic Cells
4.4.1. Culture of Acanthamoeba castellanii
4.4.2. Growth and Differentiation of THP-1 Cells
4.5. In Vitro Cytokine Induction
4.6. Statistical Analysis
4.7. Lipid Isolation and Ultra-High Performance Liquid Chromatography/Mass Spectrometry (UHPLC-MS) Profiling of Lipids
4.8. Isolation, Degradation, and Electrodialysis of the Lipopolysaccharide
4.8.1. Extraction of LPS
4.8.2. Mild Acid Hydrolysis of LPS
4.8.3. Gel-Permeation Chromatography
4.8.4. Electrodialysis
4.9. NMR Spectroscopy
4.10. Infection Assays with THP-1 Macrophages and A. castellanii
4.11. Adhesin Test
4.12. Invasion Assay
4.13. Steady-State Spectroscopy
4.13.1. Microscopy Imaging
4.13.2. Microscopy Spectra
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nisar, M.A.; Ross, K.E.; Brown, M.H.; Bentham, R.; Whiley, H. Legionella pneumophila and protozoan hosts: Implications for the control of hospital and potable water systems. Pathogens 2020, 4, 286. [Google Scholar] [CrossRef]
- Mondino, S.; Schmidt, S.; Rolando, M.; Escoll, P.; Gomez-Valero, L.; Buchrieser, C. Legionnaires’ Disease: State of the Art Knowledge of Pathogenesis Mechanisms of Legionella. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 439–466. [Google Scholar] [CrossRef]
- Chauhan, D.; Shames, S.R. Pathogenicity and virulence of Legionella: Intracellular replication and host response. Virulence 2021, 1, 1122–1144. [Google Scholar] [CrossRef]
- Parte, A.C.; Sardà Carbasse, J.; Meier-Kolthoff, J.P.; Reimer, L.C.; Göker, M. List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 2020, 70, 5607–5612. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control. Legionnaires’ disease. In ECDC. Annual Epidemiological Report for 2019; ECDC: Stockholm, Sweden, 2021; pp. 1–10. [Google Scholar]
- Chahin, A.; Opal, S.M. Severe pneumonia caused by Legionella pneumophila: Differential diagnosis and therapeutic considerations. Infect. Dis. Clin. N. Am. 2017, 1, 111–121. [Google Scholar] [CrossRef]
- Arrigo, I.; Galia, E.; Fasciana, T.; Diquattro, O.; Tricoli, M.R.; Serra, N.; Palermo, M.; Giammanco, A. Four-Year Environmental Surveillance Program of Legionella spp. in One of Palermo’s Largest Hospitals. Microorganisms 2022, 10, 764. [Google Scholar] [CrossRef]
- Fasciana, T.; Mascarella, C.; Distefano, S.A.; Calà, C.; Capra, G.; Rampulla, A.; Di Carlo, P.; Palermo, M.; Giammanco, A. Cluster of Legionnaires’ Disease in an Italian Prison. Int. J. Environ. Res. Public Health 2019, 16, 2062. [Google Scholar] [CrossRef]
- Moll, H.; Sonesson, A.; Jantzen, E.; Marre, R.; Zähringer, U. Identification of 27-oxo-octacosanoic acid and heptacosane-1,27-dioic acid in Legionella pneumophila. FEMS Microbiol. Lett. 1992, 97, 1–6. [Google Scholar] [CrossRef]
- Zähringer, U.; Knirel, Y.A.; Lindner, B.; Helbig, J.H.; Sonesson, A.; Marre, R.; Rietschel, E.T. The lipopolysaccharide of Legionella pneumophila serogroup 1 (strain Philadelphia 1): Chemical structure and biological significance. Prog. Clin. Biol. Res. 1995, 392, 113–139. [Google Scholar]
- Knirel, Y.A.; Moll, H.; Zähringer, U. Structural study of a highly O-acetylated core of Legionella pneumophila serogroup 1 lipopolysaccharide. Carbohydr. Res. 1996, 293, 223–234. [Google Scholar] [CrossRef]
- Moll, H.; Knirel, Y.A.; Helbig, J.H.; Zähringer, U. Identification of an alpha-D-Manp-(1→8)-Kdo disaccharide in the inner core region and the structure of the complete core region of the Legionella pneumophila serogroup 1 lipopolysaccharide. Carbohydr. Res. 1997, 1, 91–95. [Google Scholar] [CrossRef]
- Knirel, Y.A.; Rietschel, E.T.; Marre, R.; Zähringer, U. The structure of the O-specific chain of Legionella pneumophila serogroup 1 lipopolysaccharide. Eur. J. Biochem. 1994, 221, 239–245. [Google Scholar] [CrossRef]
- Helbig, J.H.; Bernander, S.; Castellani Pastoris, M.; Etienne, J.; Gaia, V.; Lauwers, S.; Lindsay, D.; Lück, P.C.; Marques, T.; Mentula, S.; et al. Pan-European study on culture-proven Legionnaires’ disease: Distribution of Legionella pneumophila serogroups and monoclonal subgroups. Eur. J. Clin. Microbiol. Infect. Dis. 2002, 10, 710–716. [Google Scholar]
- Kooistra, O.; Lüneberg, E.; Lindner, B.; Knirel, Y.A.; Frosch, M.; Zähringer, U. Complex O-acetylation in Legionella pneumophila serogroup 1 lipopolysaccharide. Evidence for two genes involved in 8-O-acetylation of legionaminic acid. Biochemistry 2001, 40, 7630–7640. [Google Scholar] [CrossRef]
- Petzold, M.; Thürmer, A.; Menzel, S.; Mouton, J.W.; Heuner, K.; Lück, C. A structural comparison of lipopolysaccharide biosynthesis loci of Legionella pneumophila serogroup 1 strains. BMC Microbiol. 2013, 13, 198. [Google Scholar] [CrossRef]
- Wee, B.A.; Alves, J.; Lindsay, D.S.J.; Klatt, A.-B.; Sargison, F.A.; Cameron, R.L.; Pickering, A.; Gorzynski, J.; Corander, J.; Marttinen, P.; et al. Population analysis of Legionella pneumophila reveals a basis for resistance to complement-mediated killing. Nat. Commun. 2021, 1, 7165. [Google Scholar] [CrossRef]
- Jiang, L.; Amemura-Maekawa, J.; Ren, H.; Li, Y.; Sakata, M.; Zhou, H.; Murai, M.; Chang, B.; Ohnishi, M.; Qin, T. Distribution of lag-1 alleles, ORF7, and ORF8 genes of lipopolysaccharide and sequence-based types among Legionella pneumophila serogroup 1 isolates in Japan and China. Front. Cell. Infect. Microbiol. 2019, 9, 274. [Google Scholar] [CrossRef]
- Jepras, R.I.; Fitzgeorge, R.B.; Baskerville, A. A comparison of virulence of two strains of Legionella pneumophila based on experimental aerosol infection of guinea-pigs. J. Hyg. 1985, 95, 29–38. [Google Scholar] [CrossRef]
- Helbig, J.H.; Kurtz, J.B.; Pastoris, M.C.; Pelaz, C.; Lück, P.C. Antigenic lipopolysaccharide components of Legionella pneumophila recognized by monoclonal antibodies: Possibilities and limitations for division of the species into serogroups. J. Clin. Microbiol. 1997, 11, 2841–2845. [Google Scholar] [CrossRef]
- Knirel, Y.A.; Helbig, J.H.; Zähringer, U. Structure of a decasaccharide isolated by mild acid degradation and dephosphorylation of the lipopolysaccharide of Pseudomonas fluorescens strain ATCC 49271. Carbohydr. Res. 1996, 283, 129–139. [Google Scholar] [CrossRef]
- Knirel, Y.A.; Moll, H.; Helbig, J.H.; Zähringer, U. Chemical characterization of a new 5,7-diamino-3,5,7,9-tetradeoxynonulosonic acid released by mild acid hydrolysis of the Legionella pneumophila serogroup 1 lipopolysaccharide. Carbohydr. Res. 1997, 304, 77–79. [Google Scholar] [CrossRef]
- Edebrink, P.; Jansson, P.-E.; Bøgwald, J.; Hoffman, J. Structural studies of the Vibrio salmonicida lipopolysaccharide. Carbohydr. Res. 1996, 287, 225–245. [Google Scholar] [CrossRef]
- Stone, B.J.; Kwaik, Y.A. Expression of multiple pili by Legionella pneumophila: Identification and characterization of a type IV pilin gene and its role in adherence to mammalian and protozoan cells. Infect. Immun. 1998, 4, 1768–1775. [Google Scholar] [CrossRef]
- Aragon, V.; Kurtz, S.; Flieger, A.; Neumeister, B.; Cianciotto, N.P. Secreted enzymatic activities of wild-type and pilD-deficient Legionella pneumophila. Infect. Immun. 2000, 68, 1855–1863. [Google Scholar] [CrossRef]
- Aragon, V.; Rossier, O.; Cianciotto, N.P. Legionella pneumophila genes that encode lipase and phospolipase C activities. Microbiology 2002, 148, 2223–2231. [Google Scholar] [CrossRef]
- Palusinska-Szysz, M.; Luchowski, R.; Gruszecki, W.I.; Choma, A.; Szuster-Ciesielska, A.; Lück, C.; Petzold, M.; Sroka-Bartnicka, A.; Kowalczyk, B. The role of Legionella pneumophila serogroup 1 lipopolysaccharide in host-pathogen interaction. Front. Microbiol. 2019, 10, 2890. [Google Scholar] [CrossRef]
- Gosselin, F.; Duval, J.F.L.; Simonet, J.; Ginevra, C.; Gaboriaud, F.; Jarraud, S.; Mathieu, L. Impact of the virulence-associated MAb3/1 epitope on the physicochemical surface properties of Legionella pneumophila sg1: An issue to explain infection potential? Colloids Surf. B 2011, 82, 283–290. [Google Scholar] [CrossRef]
- Chmiel, E.; Galuska, C.E.; Koper, P.; Kowalczyk, B.; Urbanik-Sypniewska, T.; Palusińska-Szysz, M.; Fuchs, B. Unusual lipid components of Legionella gormanii membranes. Metabolites 2022, 12, 418. [Google Scholar] [CrossRef]
- Kawasaki, S.; Moriguchi, R.; Sekiya, K.; Nakai, T.; Ono, E.; Kume, K.; Kawahara, K. The cell envelope structure of the lipopolysaccharide-lacking gram-negative bacterium Sphingomonas paucimobilis. J. Bacteriol. 1994, 2, 284–290. [Google Scholar] [CrossRef]
- Keck, M.; Gisch, N.; Moll, H.; Vorhölter, F.J.; Gerth, K.; Kahmann, U.; Lissel, M.; Lindner, B.; Niehaus, K.; Holst, O. Unusual outer membrane lipid composition of the gram-negative, lipopolysaccharide-lacking myxobacterium Sorangium cellulosum So ce56. J. Biol. Chem. 2011, 15, 12850–12859. [Google Scholar] [CrossRef]
- Ogawa, S.; Tachimoto, H.; Kaga, T. Elevation of ceramide in Acetobacter malorum S24 by low pH stress and high temperature stress. J. Biosci. Bioeng. 2010, 109, 32–36. [Google Scholar] [CrossRef]
- Neumeister, B.; Faigle, M.; Sommer, M.; Zähringer, U.; Stelter, F.; Menzel, R.; Schütt, C.; Northoff, H. Low endotoxic potential of Legionella pneumophila lipopolysaccharide due to failure of interaction with the monocyte lipopolysaccharide receptor CD14. Infect. Immun. 1998, 9, 4151–4157. [Google Scholar] [CrossRef]
- Feeley, J.C.; Gibson, R.J.; Gorman, G.W.; Langford, N.C.; Rasheed, J.K.; Mackel, D.C.; Baineet, W.B. Charcoal-yeast extract agar: Primary isolation medium for Legionella pneumophila. J. Clin. Microbiol. 1979, 10, 437–441. [Google Scholar] [CrossRef]
- Unger, T.; Jacobovitch, Y.; Dantes, A.; Bernheim, R.; Peleg, Y. Applications of Restriction Free (RF) cloning procedure for molecular manipulations and protein expression. J. Struct. Biol. 2010, 172, 34–44. [Google Scholar] [CrossRef]
- Schunder, E.; Gillmaier, N.; Kutzner, E.; Eisenreich, W.; Herrmann, V.; Lautner, M.; Heuner, K. Amino acid uptake and metabolism of Legionella pneumophila hosted by Acanthamoeba castellanii. J. Biol. Chem. 2014, 30, 21040–21054. [Google Scholar] [CrossRef]
- Buchrieser, C.; Hilbi, H. Legionella: Methods and Protocols. Methods in Molecular Biology, 1st ed.; Humana Press: Clifton, NJ, USA, 2013; pp. 183–195. [Google Scholar]
- Pope, C.D.; Dhand, L.; Cianciotto, N.P. Random mutagenesis of Legionella pneumophila with mini-Tn10. FEMS Microbiol. Lett. 1994, 124, 107–111. [Google Scholar] [CrossRef]
- Shames, S.R. Screening targeted Legionella pneumophila mutant libraries in vivo using INSeq methods. Mol. Biol. 2019, 1921, 123–144. [Google Scholar]
- Band, R.N. Nutritional and related biological studies on the free-living soil amoeba, Hartmannella rhysodes. J. Gen. Microbiol. 1959, 21, 80–95. [Google Scholar] [CrossRef]
- Daigneault, M.; Preston, J.A.; Marriott, H.M.; Whyte, M.K.B.; Dockrell, D.H. The identification of markers of macrophage differentiation in PMA stimulated THP-1 cells and monocyte-derived macrophages. PLoS ONE 2010, 5, e8668. [Google Scholar] [CrossRef]
- Palusinska-Szysz, M.; Szuster-Ciesielska, A.; Janczarek, M.; Wdowiak-Wróbel, S.; Schiller, J.; Reszczyńska, E.; Gruszecki, W.I.; Fuchs, B. Genetic diversity of Legionella pcs and pmtA genes and the effect of utilization of choline by Legionella spp. on induction of proinflammatory cytokines. Pathog. Dis. 2019, 7, ftz065. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Palusińska-Szysz, M.; Zdybicka-Barabas, A.; Luchowski, R.; Reszczyńska, E.; Śmiałek, J.; Mak, P.; Gruszecki, W.I.; Cytryńska, M. Choline supplementation sensitizes Legionella dumoffii to Galleria mellonella apolipophorin III. Int. J. Mol. Sci. 2020, 21, 5818. [Google Scholar] [CrossRef]
- Westphal, O.; Jann, K. Bacterial lipopolysaccharide. Extraction with phenol-water and further applications of the procedure. Meth. Carbohydr. Chem. 1965, 5, 83–91. [Google Scholar]
- Galanos, C.; Lüderitz, O. Electrodialysis of lipopolysaccharides and their conversion to uniform salt forms. Eur. J. Biochem. 1975, 54, 60310. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue | Chemical Shifts (ppm) 1H and 13C | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| H1 C1 | H2 C2 | H3 C3 | H4 C4 | H5 C5 | H6 C6 | H7 C7 | H8 C8 | H9 C9 | |
| → 4)-α-Legp-(2 → | - 174.36 | - 101.87 | 2.635/1.630 39.51 | 4.057 72.06 | 3.529 54.46 | 3.994 72.72 | 3.913 55.43 | 3.920 67.91 | 1.202 19.72 |
| N-Acetyl | - 175.24 | 2.103 23.29 | |||||||
| N-Acetimidoyl | - 167.73 | 2.264 20.11 | |||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kowalczyk, B.; Petzold, M.; Kaczyński, Z.; Szuster-Ciesielska, A.; Luchowski, R.; Gruszecki, W.I.; Fuchs, B.; Galuska, C.E.; Choma, A.; Tarasiuk, J.; et al. Lipopolysaccharide of Legionella pneumophila Serogroup 1 Facilitates Interaction with Host Cells. Int. J. Mol. Sci. 2023, 24, 14602. https://doi.org/10.3390/ijms241914602
Kowalczyk B, Petzold M, Kaczyński Z, Szuster-Ciesielska A, Luchowski R, Gruszecki WI, Fuchs B, Galuska CE, Choma A, Tarasiuk J, et al. Lipopolysaccharide of Legionella pneumophila Serogroup 1 Facilitates Interaction with Host Cells. International Journal of Molecular Sciences. 2023; 24(19):14602. https://doi.org/10.3390/ijms241914602
Chicago/Turabian StyleKowalczyk, Bożena, Markus Petzold, Zbigniew Kaczyński, Agnieszka Szuster-Ciesielska, Rafał Luchowski, Wiesław I. Gruszecki, Beate Fuchs, Christina E. Galuska, Adam Choma, Jacek Tarasiuk, and et al. 2023. "Lipopolysaccharide of Legionella pneumophila Serogroup 1 Facilitates Interaction with Host Cells" International Journal of Molecular Sciences 24, no. 19: 14602. https://doi.org/10.3390/ijms241914602
APA StyleKowalczyk, B., Petzold, M., Kaczyński, Z., Szuster-Ciesielska, A., Luchowski, R., Gruszecki, W. I., Fuchs, B., Galuska, C. E., Choma, A., Tarasiuk, J., & Palusińska-Szysz, M. (2023). Lipopolysaccharide of Legionella pneumophila Serogroup 1 Facilitates Interaction with Host Cells. International Journal of Molecular Sciences, 24(19), 14602. https://doi.org/10.3390/ijms241914602