
Genome-Wide DNA Methylation Profiles in Whole-Blood and Buccal Samples—Cross-Sectional, Longitudinal, and across Platforms
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
2.1. Longitudinal Comparison
2.2. Cross-Tissue Comparison
2.3. Cross-Platform Comparison
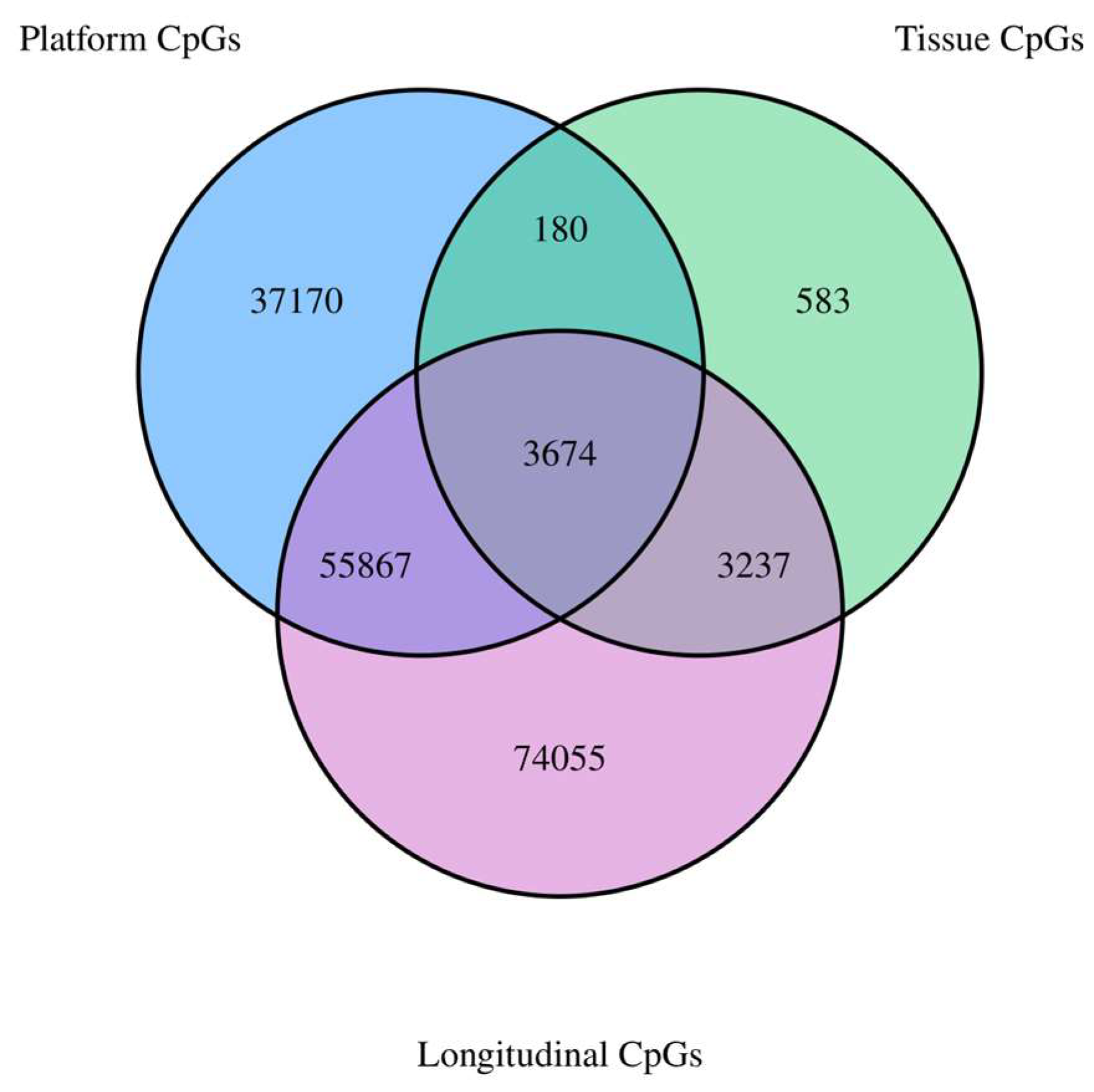
2.4. Overlap of Reliable CpGs Significantly and Strongly Correlated across Time and Tissue
2.5. EWAS Atlas and BBMRI mQTL Database Query
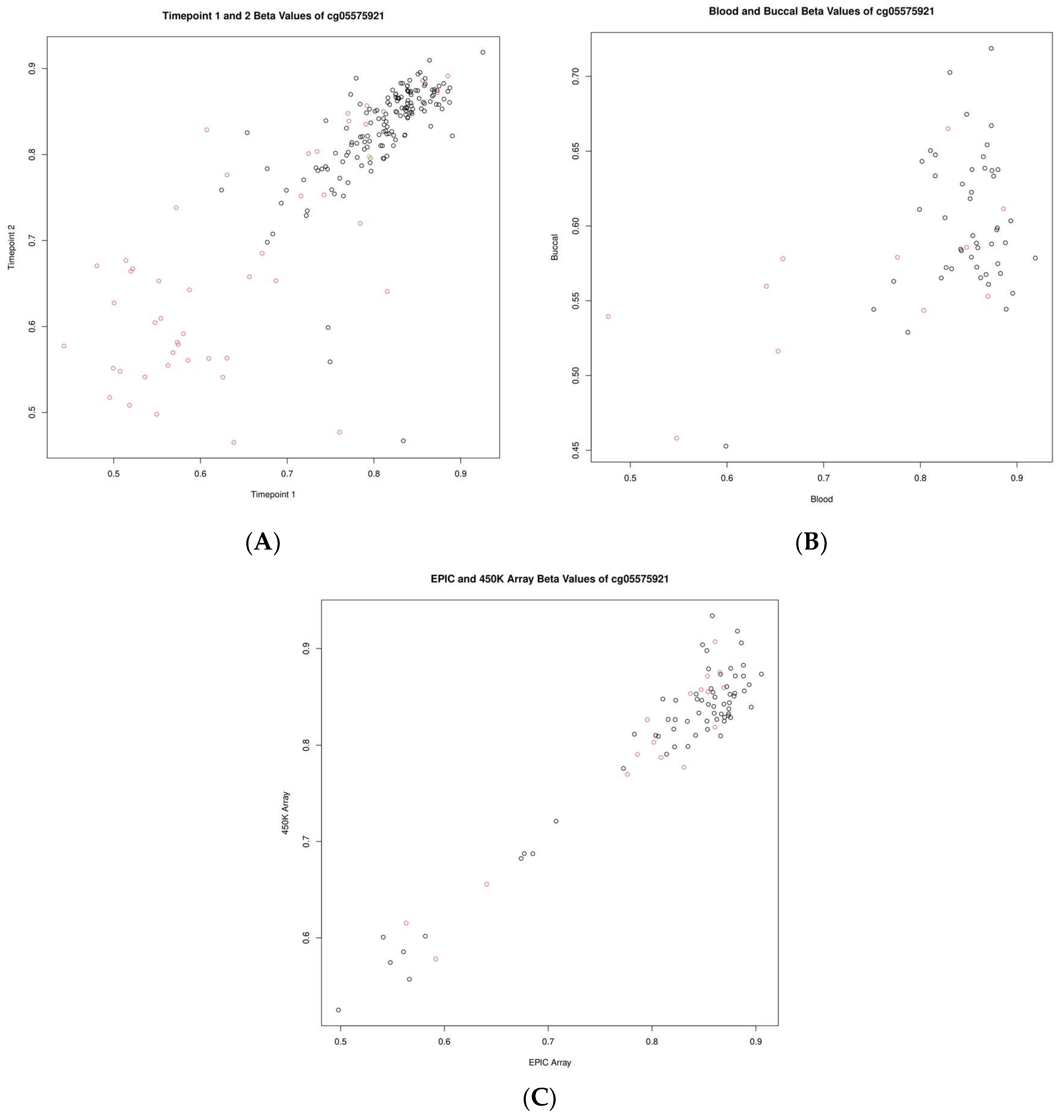
2.6. Investigation of Individual CpGs of Interest
3. Discussion
3.1. Longitudinal Comparison
3.2. Cross-Tissue Comparison
3.3. Cross-Platform Comparison
3.4. Interrogation of the Overlapping CpGs
3.5. EWAS Atlas and BBMRI
3.6. Individual CpG Assessment
3.7. Limitations
4. Materials and Methods
4.1. Overview
4.2. Sample Collection
4.3. DNA Extraction
4.4. Genotyping
4.5. DNA Methylation Assessment
4.6. DNA Methylation Data Quality Control
4.7. Statistical Analyses
4.8. Methylation Data Annotation
4.9. DNA Methylation Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, mechanisms and clinical perspective. Semin. Reprod. Med. 2009, 27, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.G.; Lowe, R.; Adams, P.D.; Baccarelli, A.A.; Beck, S.; Bell, J.T.; Christensen, B.C.; Gladyshev, V.N.; Heijmans, B.T.; Horvath, S.; et al. DNA methylation aging clocks: Challenges and recommendations. Genome Biol. 2019, 20, 249. [Google Scholar] [CrossRef] [PubMed]
- Unnikrishnan, A.; Freeman, W.M.; Jackson, J.; Wren, J.D.; Porter, H.; Richardson, A. The role of DNA methylation in epigenetics of aging. Pharmacol. Ther. 2019, 195, 172–185. [Google Scholar] [CrossRef]
- Belsky, D.W.; Moffitt, T.E.; Cohen, A.A.; Corcoran, D.L.; Levine, M.E.; Prinz, J.A.; Schaefer, J.; Sugden, K.; Williams, B.; Poulton, R.; et al. Eleven Telomere, Epigenetic Clock, and Biomarker-Composite Quantifications of Biological Aging: Do They Measure the Same Thing? Am. J. Epidemiol. 2018, 187, 1220–1230. [Google Scholar] [CrossRef]
- Marioni, R.E.; Suderman, M.; Chen, B.H.; Horvath, S.; Bandinelli, S.; Morris, T.; Beck, S.; Ferrucci, L.; Pedersen, N.L.; Relton, C.L.; et al. Tracking the Epigenetic Clock Across the Human Life Course: A Meta-analysis of Longitudinal Cohort Data. J. Gerontol. Ser. A 2019, 74, 57–61. [Google Scholar] [CrossRef]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Zheng, S.C. Cell-type deconvolution in epigenome-wide association studies: A review and recommendations. Epigenomics 2017, 9, 757–768. [Google Scholar] [CrossRef]
- Slieker, R.C.; Relton, C.L.; Gaunt, T.R.; Slagboom, P.E.; Heijmans, B.T. Age-related DNA methylation changes are tissue-specific with ELOVL2 promoter methylation as exception. Epigenetics Chromatin 2018, 11, 25. [Google Scholar] [CrossRef]
- Hannon, E.; Lunnon, K.; Schalkwyk, L.; Mill, J. Interindividual methylomic variation across blood, cortex, and cerebellum: Implications for epigenetic studies of neurological and neuropsychiatric phenotypes. Epigenetics 2015, 10, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Braun, P.R.; Han, S.; Hing, B.; Nagahama, Y.; Gaul, L.N.; Heinzman, J.T.; Grossbach, A.J.; Close, L.; Dlouhy, B.J.; Howard, M.A., 3rd; et al. Genome-wide DNA methylation comparison between live human brain and peripheral tissues within individuals. Transl. Psychiatry 2019, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Illumina, Infinium HumanMethylation450 BeadChip. Data Sheet: Epigenetics. 2012. Available online: https://www.illumina.com/content/dam/illumina-marketing/documents/products/datasheets/datasheet_humanmethylation450.pdf (accessed on 16 February 2023).
- Morris, T.J.; Beck, S. Analysis pipelines and packages for Infinium HumanMethylation450 BeadChip (450k) data. Methods 2015, 72, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Jimenez, N.; Allard, C.; Bouchard, L.; Perron, P.; Bustamante, M.; Bilbao, J.R.; Hivert, M.F. Comparison of Illumina 450K and EPIC arrays in placental DNA methylation. Epigenetics 2019, 14, 1177–1182. [Google Scholar] [CrossRef]
- Infinium MethylationEPIC v2.0 Kit. Available online: https://www.illumina.com/products/by-type/microarray-kits/infinium-methylation-epic.html (accessed on 16 February 2023).
- van Dongen, J.; Ehli, E.A.; Jansen, R.; van Beijsterveldt, C.E.M.; Willemsen, G.; Hottenga, J.J.; Kallsen, N.A.; Peyton, S.A.; Breeze, C.E.; Kluft, C.; et al. Genome-wide analysis of DNA methylation in buccal cells: A study of monozygotic twins and mQTLs. Epigenetics Chromatin 2018, 11, 54. [Google Scholar] [CrossRef]
- Sugden, K.; Hannon, E.J.; Arseneault, L.; Belsky, D.W.; Corcoran, D.L.; Fisher, H.L.; Houts, R.M.; Kandaswamy, R.; Moffitt, T.E.; Poulton, R.; et al. Patterns of Reliability: Assessing the Reproducibility and Integrity of DNA Methylation Measurement. Patterns 2020, 1. [Google Scholar] [CrossRef]
- Olstad, E.W.; Nordeng, H.M.E.; Sandve, G.K.; Lyle, R.; Gervin, K. Low reliability of DNA methylation across Illumina Infinium platforms in cord blood: Implications for replication studies and meta-analyses of prenatal exposures. Clin. Epigenet. 2022, 14, 80. [Google Scholar] [CrossRef]
- Li, M.; Zou, D.; Li, Z.; Gao, R.; Sang, J.; Zhang, Y.; Li, R.; Xia, L.; Zhang, T.; Niu, G.; et al. EWAS Atlas: A curated knowledgebase of epigenome-wide association studies. Nucleic Acids Res. 2019, 47, D983–D988. [Google Scholar] [CrossRef]
- Bonder, M.J.; Luijk, R.; Zhernakova, D.V.; Moed, M.; Deelen, P.; Vermaat, M.; van Iterson, M.; van Dijk, F.; van Galen, M.; Bot, J.; et al. Disease variants alter transcription factor levels and methylation of their binding sites. Nat. Genet. 2017, 49, 131–138. [Google Scholar] [CrossRef]
- Seale, K.; Horvath, S.; Teschendorff, A.; Eynon, N.; Voisin, S. Making sense of the ageing methylome. Nat. Rev. Genet. 2022, 23, 585–605. [Google Scholar] [CrossRef]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Kananen, L.; Marttila, S.; Nevalainen, T.; Kummola, L.; Junttila, I.; Mononen, N.; Kähönen, M.; Raitakari, O.T.; Hervonen, A.; Jylhä, M.; et al. The trajectory of the blood DNA methylome ageing rate is largely set before adulthood: Evidence from two longitudinal studies. Age 2016, 38, 65. [Google Scholar] [CrossRef] [PubMed]
- Voisin, S.; Seale, K.; Jacques, M.; Landen, S.; Harvey, N.R.; Haupt, L.M.; Griffiths, L.R.; Ashton, K.J.; Coffey, V.G.; Thompson, J.M.; et al. Exercise is associated with younger methylome and transcriptome profiles in human skeletal muscle. Aging Cell 2023, e13859. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Liu, J.; Beck, S.; Pan, S.; Capper, D.; Lechner, M.; Thirlwell, C.; Breeze, C.E.; Teschendorff, A.E. A pan-tissue DNA methylation atlas enables in silico decomposition of human tissue methylomes at cell-type resolution. Nat. Methods 2022, 19, 296–306. [Google Scholar] [CrossRef]
- Titus, A.J.; Gallimore, R.M.; Salas, L.A.; Christensen, B.C. Cell-type deconvolution from DNA methylation: A review of recent applications. Hum. Mol. Genet. 2017, 26, R216–R224. [Google Scholar] [CrossRef]
- Logue, M.W.; Smith, A.K.; Wolf, E.J.; Maniates, H.; Stone, A.; Schichman, S.A.; McGlinchey, R.E.; Milberg, W.; Miller, M.W. The correlation of methylation levels measured using Illumina 450K and EPIC BeadChips in blood samples. Epigenomics 2017, 9, 1363–1371. [Google Scholar] [CrossRef]
- Cheung, K.; Burgers, M.J.; Young, D.A.; Cockell, S.; Reynard, L.N. Correlation of Infinium HumanMethylation450K and MethylationEPIC BeadChip arrays in cartilage. Epigenetics 2020, 15, 594–603. [Google Scholar] [CrossRef]
- Zhang, B.; Zhou, Y.; Lin, N.; Lowdon, R.F.; Hong, C.; Nagarajan, R.P.; Cheng, J.B.; Li, D.; Stevens, M.; Lee, H.J.; et al. Functional DNA methylation differences between tissues, cell types, and across individuals discovered using the M&M algorithm. Genome Res. 2013, 23, 1522–1540. [Google Scholar]
- Johnson, A.A.; Akman, K.; Calimport, S.R.; Wuttke, D.; Stolzing, A.; de Magalhães, J.P. The role of DNA methylation in aging, rejuvenation, and age-related disease. Rejuvenation Res. 2012, 15, 483–494. [Google Scholar] [CrossRef]
- Tantoh, D.M.; Wu, M.-C.; Chuang, C.-C.; Chen, P.-H.; Tyan, Y.S.; Nfor, O.N.; Lu, W.-Y.; Liaw, Y.-P. AHRR cg05575921 methylation in relation to smoking and PM2.5 exposure among Taiwanese men and women. Clin. Epigenetics 2020, 12, 117. [Google Scholar] [CrossRef]
- Johansson, A.; Enroth, S.; Gyllensten, U. Continuous Aging of the Human DNA Methylome Throughout the Human Lifespan. PLoS ONE 2013, 8, e67378. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Boezen, H.M.; Postma, D.S.; Los, H.; Postmus, P.E.; Snieder, H.; Boomsma, D.I. Genetic and environmental influences on objective intermediate asthma phenotypes in Dutch twins. Eur. Respir. J. 2010, 36, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, G.; de Geus, E.J.C.; Bartels, M.; van Beijsterveldt, C.E.M.T.; Brooks, A.I.; Estourgie-van Burk, G.F.; Fugman, D.A.; Hoekstra, C.; Hottenga, J.-J.; Kluft, K.; et al. The Netherlands Twin Register Biobank: A Resource for Genetic Epidemiological Studies. Twin Res. Human. Genet. 2010, 13, 231–245. [Google Scholar] [CrossRef] [PubMed]
- van Dongen, J.; Nivard, M.G.; Willemsen, G.; Hottenga, J.J.; Helmer, Q.; Dolan, C.V.; Ehli, E.A.; Davies, G.E.; van Iterson, M.; Breeze, C.E.; et al. Genetic and environmental influences interact with age and sex in shaping the human methylome. Nat. Commun. 2016, 7, 11115. [Google Scholar] [CrossRef] [PubMed]
- Illumina, Infinium HTS Assay Protocol Guide. In Illumina. 2019. Available online: https://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/infinium_assays/infinium-hts/infinium-hts-assay-reference-guide-15045738-04.pdf (accessed on 16 February 2023).
- Illumina, Analyzing Standards and Custom Infinium Genotyping Products Training Guide. In Illumina. Available online: https://support.illumina.com/content/dam/illumina-support/courses/eval-inf-controls/story_html5.html (accessed on 16 February 2023).
- Infinium® Genotyping Data Analysis. In Illumina®; Illumina, I. (Ed.) 2014; Vol. 970-2007-005, Available online: https://www.illumina.com/Documents/products/technotes/technote_infinium_genotyping_data_analysis.pdf (accessed on 16 February 2023).
- Infinium HD Assay Methylation Protocol Guide. Available online: https://support-docs.illumina.com/ARR/Inf_HD_Methylation/Content/ARR/FrontPages/inf_hd_methylation_pg.htm (accessed on 16 February 2023).
- van Iterson, M.; Tobi, E.W.; Slieker, R.C.; den Hollander, W.; Luijk, R.; Slagboom, P.E.; Heijmans, B.T. MethylAid: Visual and interactive quality control of large Illumina 450k datasets. Bioinformatics 2014, 30, 3435–3437. [Google Scholar] [CrossRef]
- Fortin, J.-P.; Labbe, A.; Lemire, M.; Zanke, B.W.; Hudson, T.J.; Fertig, E.J.; Greenwood, C.M.T.; Hansen, K.D. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014, 15, 503. [Google Scholar] [CrossRef]
- van Iterson, M.; Cats, D.; Hop, P.; Heijmans, B.T. omicsPrint: Detection of data linkage errors in multiple omics studies. Bioinformatics 2018, 34, 2142–2143. [Google Scholar] [CrossRef]
- Min, J.L.; Hemani, G.; Davey Smith, G.; Relton, C.; Suderman, M. Meffil: Efficient normalization and analysis of very large DNA methylation datasets. Bioinformatics 2018, 34, 3983–3989. [Google Scholar] [CrossRef]
- Zhou, W.; Laird, P.W.; Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 2017, 45, e22. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Individuals | Males | Females | CpGs |
|---|---|---|---|---|
| Longitudinal | 197 | 95 | 102 | 759,263 |
| Tissue | 58 | 21 | 37 | 759,263 |
| Platform | 83 | 36 | 47 | 386,805 |
| Experiment | Significant and Strongly Correlated CpGs |
|---|---|
| Longitudinal | 136,833 |
| Tissue | 7674 |
| Platform | 96,891 |
| Overlapping | 3674 |
| Trait | Odds Ratio | p-Value | CpGs Identified | Background |
|---|---|---|---|---|
| Ancestry | 8.887 | 0 | 328 | 10,618 |
| Kabuki syndrome (KS) | 25.596 | 3.18 × 10−306 | 162 | 1891 |
| Respiratory allergies (RAs) | 78.282 | 3.43 × 10−296 | 109 | 485 |
| Alzheimer’s disease (AD) | 19.434 | 4.39 × 10−160 | 94 | 1392 |
| Gestational diabetes mellitus | 6.541 | 1.50 × 10−135 | 156 | 6599 |
| Ankylosing spondylitis | 60.141 | 1.25 × 10−105 | 41 | 222 |
| Childhood stress | 26.544 | 2.17 × 10−98 | 50 | 550 |
| Primary Sjögren’s syndrome (pSS) | 7.565 | 3.88 × 10−96 | 97 | 3526 |
| Klinefelter syndrome | 64.282 | 3.79 × 10−92 | 35 | 179 |
| Leukoaraiosis (LA) | 25.757 | 1.50 × 10−91 | 47 | 531 |
| CpGs | mQTL Associations | Cis/Trans-mQTL Breakdown | CpGs with At Least 1 Association | CpGs Cis/Trans-mQTL Breakdown | |
|---|---|---|---|---|---|
| Bonder et al. [21] | 405,709 | 299,853 | 272,037/27,816 | 145,792 (35.9%) | 142,126 (35.0%)/10,141 (2.5%) |
| Longitudinal | 69,570 | 121,253 | 107,601/13,652 | 42,881 (61.6%) | 41,609 (59.8%)/4662 (6.7%) |
| Tissue | 4408 | 7402 | 6613/789 | 2181 (49.5%) | 2119 (48.1%)/168 (3.8%) |
| Platform | 96,891 | 165,484 | 146,330/19,154 | 61,503 (63.5%) | 59,561 (61.5%)/6849 (7.1%) |
| Overlapping | 3674 | 7016 | 6235/781 | 1989 (54.1%) | 1932 (52.6%)/163 (4.4%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Asselt, A.J.; Beck, J.J.; Finnicum, C.T.; Johnson, B.N.; Kallsen, N.; Hottenga, J.J.; de Geus, E.J.C.; BIOS Consortium; Boomsma, D.I.; Ehli, E.A.; et al. Genome-Wide DNA Methylation Profiles in Whole-Blood and Buccal Samples—Cross-Sectional, Longitudinal, and across Platforms. Int. J. Mol. Sci. 2023, 24, 14640. https://doi.org/10.3390/ijms241914640
Van Asselt AJ, Beck JJ, Finnicum CT, Johnson BN, Kallsen N, Hottenga JJ, de Geus EJC, BIOS Consortium, Boomsma DI, Ehli EA, et al. Genome-Wide DNA Methylation Profiles in Whole-Blood and Buccal Samples—Cross-Sectional, Longitudinal, and across Platforms. International Journal of Molecular Sciences. 2023; 24(19):14640. https://doi.org/10.3390/ijms241914640
Chicago/Turabian StyleVan Asselt, Austin J., Jeffrey J. Beck, Casey T. Finnicum, Brandon N. Johnson, Noah Kallsen, Jouke Jan Hottenga, Eco J. C. de Geus, BIOS Consortium, Dorret I. Boomsma, Erik A. Ehli, and et al. 2023. "Genome-Wide DNA Methylation Profiles in Whole-Blood and Buccal Samples—Cross-Sectional, Longitudinal, and across Platforms" International Journal of Molecular Sciences 24, no. 19: 14640. https://doi.org/10.3390/ijms241914640
APA StyleVan Asselt, A. J., Beck, J. J., Finnicum, C. T., Johnson, B. N., Kallsen, N., Hottenga, J. J., de Geus, E. J. C., BIOS Consortium, Boomsma, D. I., Ehli, E. A., & van Dongen, J. (2023). Genome-Wide DNA Methylation Profiles in Whole-Blood and Buccal Samples—Cross-Sectional, Longitudinal, and across Platforms. International Journal of Molecular Sciences, 24(19), 14640. https://doi.org/10.3390/ijms241914640