
INT-767—A Dual Farnesoid-X Receptor (FXR) and Takeda G Protein-Coupled Receptor-5 (TGR5) Agonist Improves Survival in Rats and Attenuates Intestinal Ischemia Reperfusion Injury

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
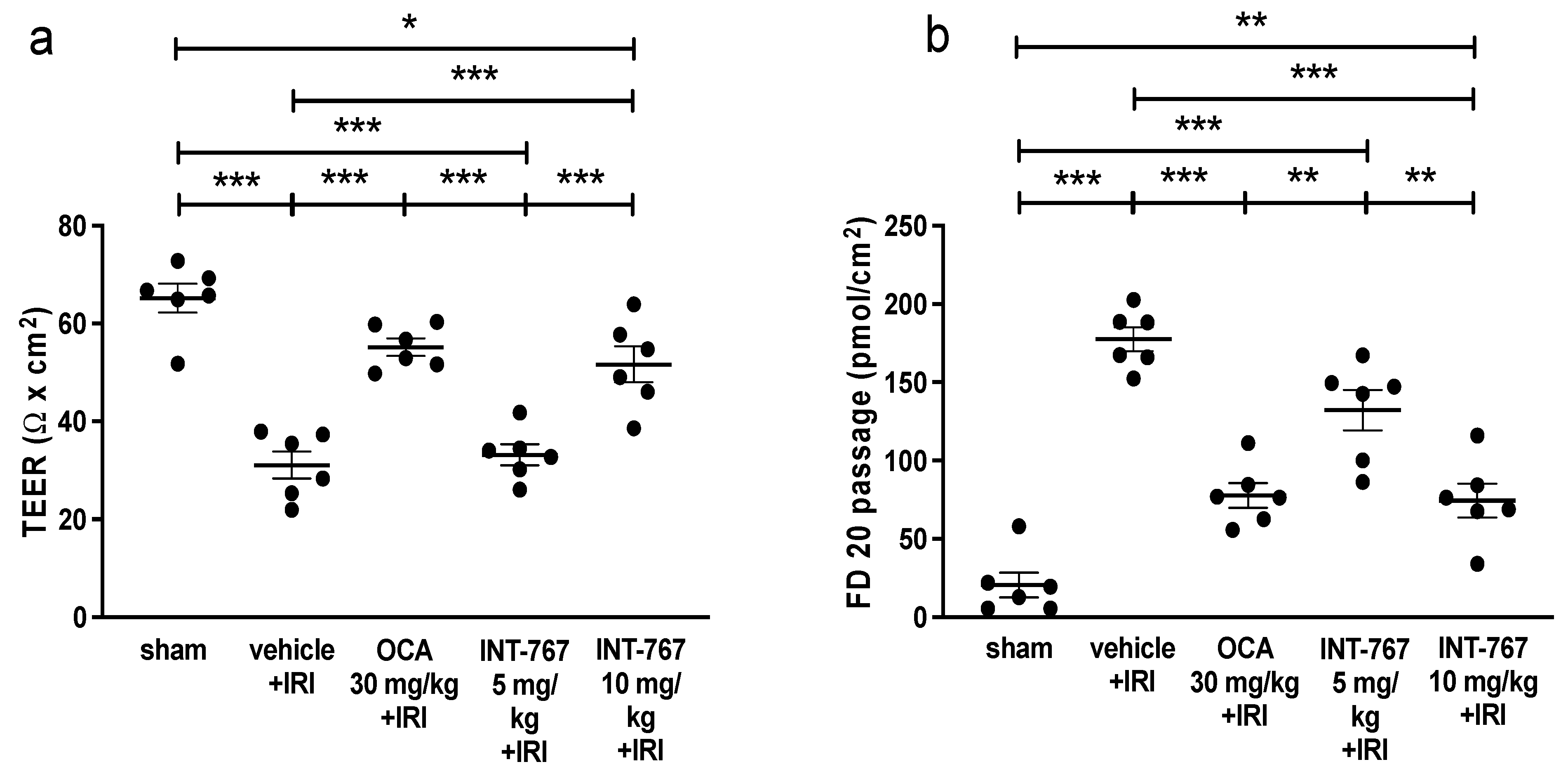
2.1. Experiment 1: Determination of Optimal Dosing of INT-767 Treatment
2.2. Experiment 2: Pharmacological Treatment of Intestinal IRI by INT-767 Administration
2.2.1. INT-767 Treatment Improves 7-Day Survival
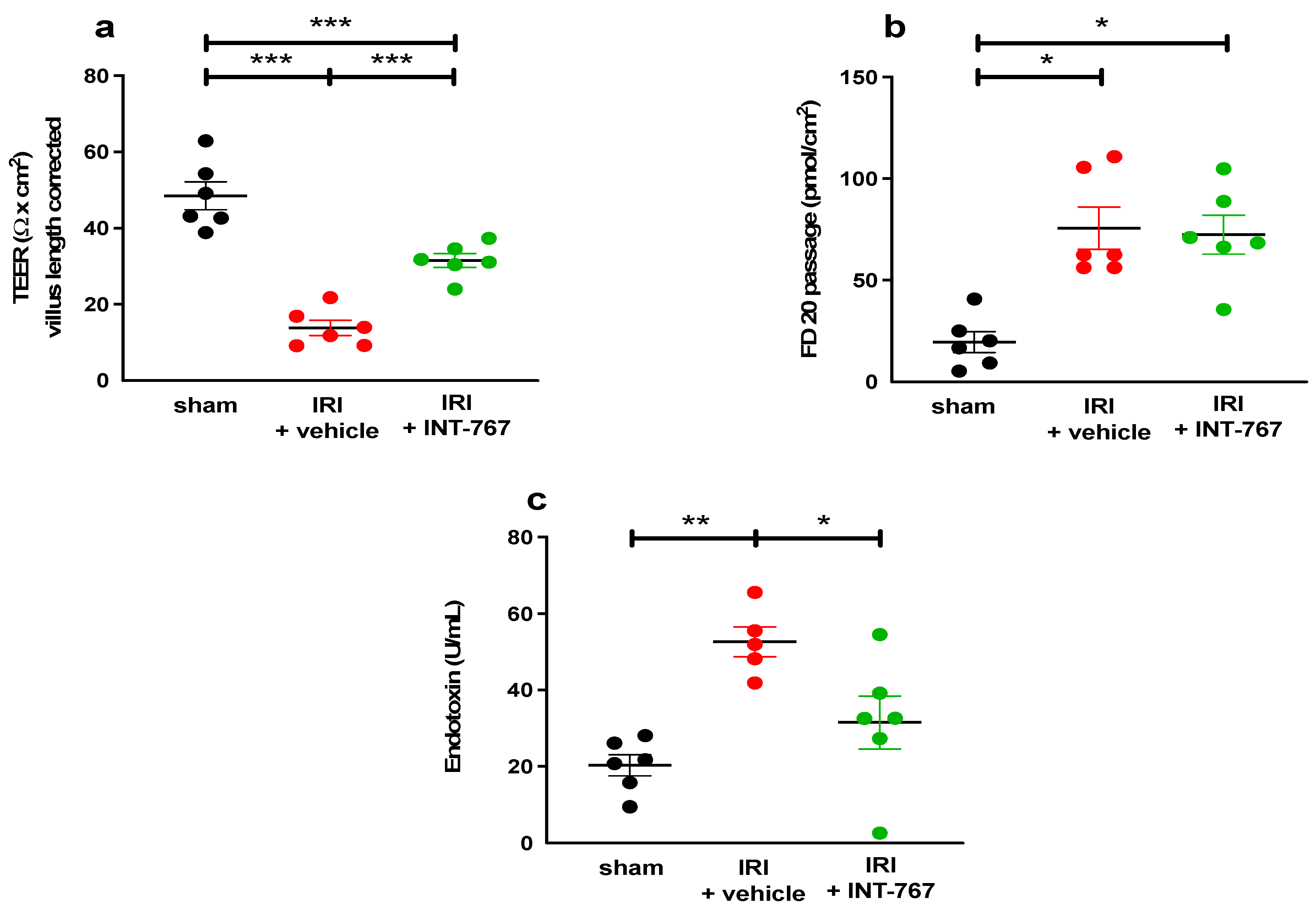
2.2.2. INT-767 Reduces Plasma Intestinal Injury Markers
2.2.3. INT-767 Treatment Reduces IRI-Induced Damage to the Intestinal Wall and Ameliorates the Epithelial Barrier Function
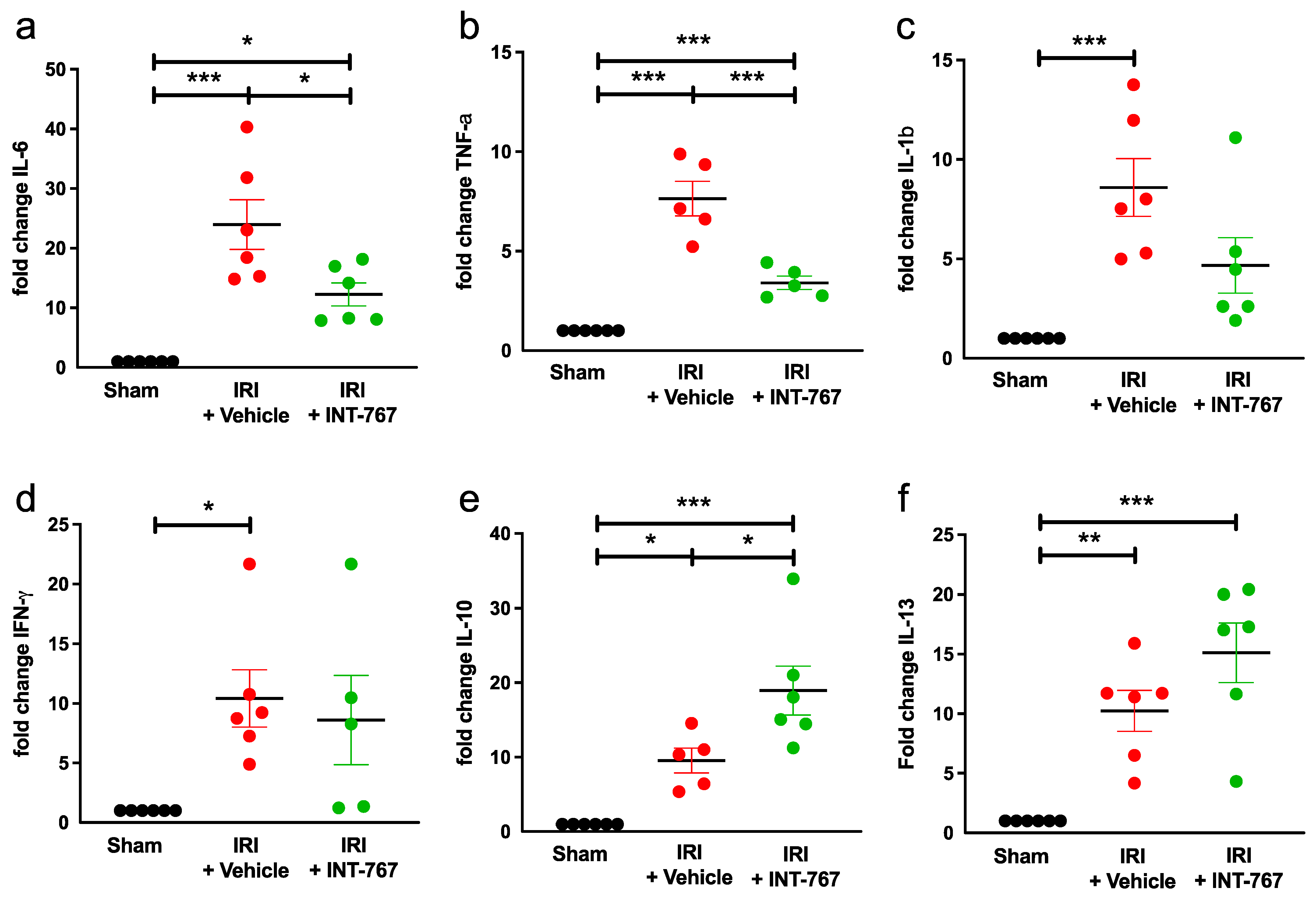
2.2.4. INT-767 Reduces IRI-Induced Pro-Inflammatory Cytokines Expression and Upregulates Anti-Inflammatory Cytokine Expression
2.2.5. INT-767 Induces FXR but Not TGR5 Upregulation
3. Discussion
4. Materials and Methods
4.1. Animal Model
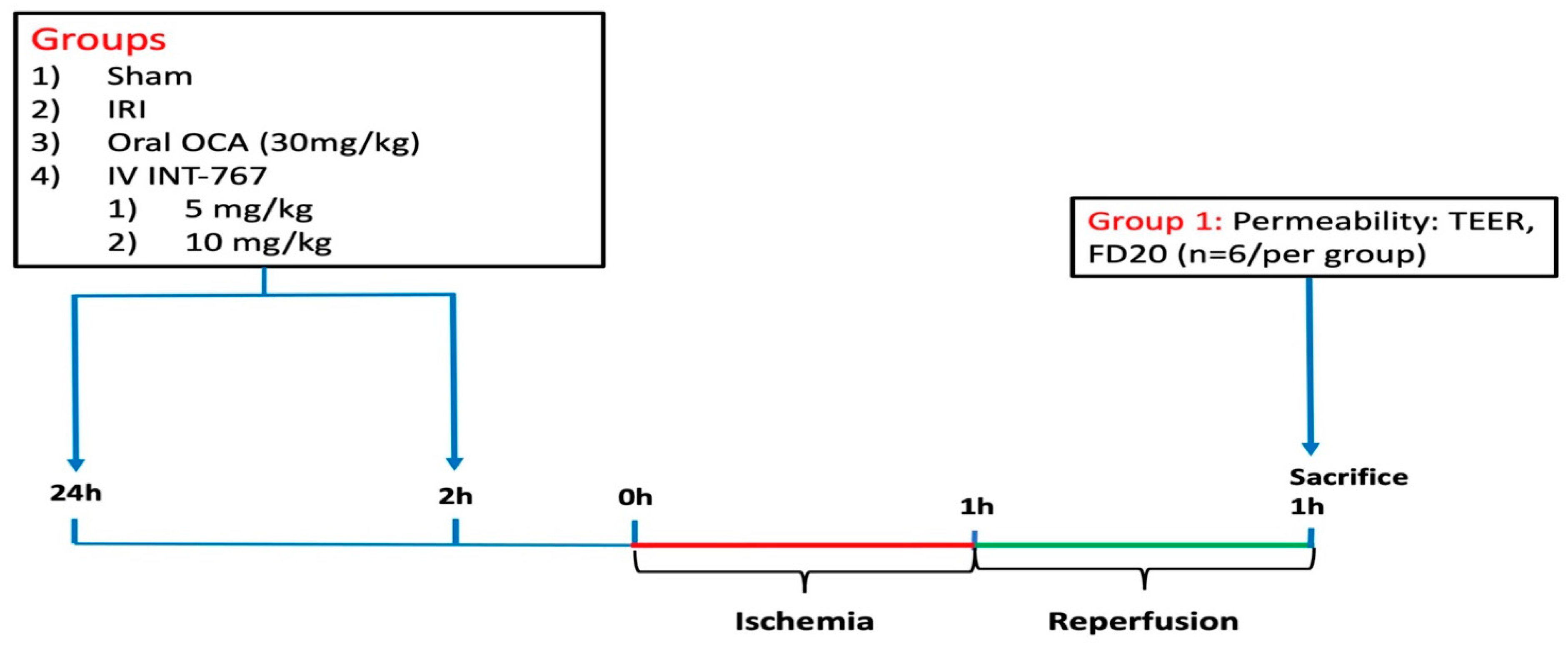
4.2. Experimental Design
4.2.1. Study 1: Determination of Optimal Dosing of INT-767 Treatment
4.2.2. Study 2: Pharmacological Treatment of Intestinal IRI by INT-767 Administration
Effect of INT-767 on Survival
Effect of INT-767 on Intestinal IRI
Damage Biomarkers
Histological Analysis
Evaluation of the Epithelial Barrier Function
Assessment of Bacterial LPS Translocation
Quantitative Reverse-Transcription Polymerase Chain Reaction (qRT-PCR)
Western Blot
Statistical Analysis
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Björck, M.; Koelemay, M.; Acosta, S.; Goncalves, F.B.; Kölbel, T.; Kolkman, J.; Lees, T.; Lefevre, J.; Menyhei, G.; Oderich, G.; et al. Editor’s Choice—Management of the Diseases of Mesenteric Arteries and Veins. Eur. J. Vasc. Endovasc. Surg. 2017, 53, 460–510. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, J.; Lenaerts, K.; Buurman, W.A.; Dejong, C.H.C.; Derikx, J.P.M. Life and death at the mucosal-luminal interface: New perspectives on human intestinal ischemia-reperfusion. World J. Gastroenterol. 2016, 22, 2760–7070. [Google Scholar] [CrossRef] [PubMed]
- Vollmar, B.; Menger, M.D. Intestinal ischemia/reperfusion: Microcirculatory pathology and functional consequences. Langenbeck’s Arch. Surg. 2011, 396, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Mallick, I.H.; Yang, W.; Winslet, M.C.; Seifalian, A.M. Review: Ischemia–Reperfusion Injury of the Intestine and Protective Strategies Against Injury. Dig. Dis. Sci. 2004, 49, 1359–1377. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef]
- Roussel, A.; Castier, Y.; Nuzzo, A.; Pellenc, Q.; Sibert, A.; Panis, Y.; Bouhnik, Y.; Corcos, O. Revascularization of acute mesenteric ischemia after creation of a dedicated multidisciplinary center. J. Vasc. Surg. 2015, 62, 1251–1256. [Google Scholar] [CrossRef]
- Corcos, O.; Castier, Y.; Sibert, A.; Gaujoux, S.; Ronot, M.; Joly, F.; Paugam, C.; Bretagnol, F.; Abdel–Rehim, M.; Francis, F.; et al. Effects of a Multimodal Management Strategy for Acute Mesenteric Ischemia on Survival and Intestinal Failure. Clin. Gastroenterol. Hepatol. 2013, 11, 158–165.e2. [Google Scholar] [CrossRef]
- Vavassori, P.; Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. The Bile Acid Receptor FXR Is a Modulator of Intestinal Innate Immunity. J. Immunol. 2009, 183, 6251–6261. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; Van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.L.; Renooij, W.; Murzilli, S.; Klomp, L.W.J.; Siersema, P.D.; Schipper, M.E.I.; Danese, S.; et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef]
- Ceulemans, L.J.; Verbeke, L.; Decuypere, J.-P.; Farré, R.; De Hertogh, G.; Lenaerts, K.; Jochmans, I.; Monbaliu, D.; Nevens, F.; Tack, J.; et al. Farnesoid X Receptor Activation Attenuates Intestinal Ischemia Reperfusion Injury in Rats. PLoS ONE 2017, 12, e0169331. [Google Scholar] [CrossRef]
- Rizzo, G.; Passeri, D.; De Franco, F.; Ciaccioli, G.; Donadio, L.; Rizzo, G.; Orlandi, S.; Sadeghpour, B.; Wang, X.X.; Jiang, T.; et al. Functional Characterization of the Semisynthetic Bile Acid Derivative INT-767, a Dual Farnesoid X Receptor and TGR5 Agonist. Mol. Pharmacol. 2010, 78, 617–630. [Google Scholar] [CrossRef]
- Roda, A.; Pellicciari, R.; Gioiello, A.; Neri, F.; Camborata, C.; Passeri, D.; De Franco, F.; Spinozzi, S.; Colliva, C.; Adorini, L.; et al. Semisynthetic Bile Acid FXR and TGR5 Agonists: Physicochemical Properties, Pharmacokinetics, and Metabolism in the Rat. J. Pharmacol. Exp. Ther. 2014, 350, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Poole, D.P.; Godfrey, C.; Cattaruzza, F.; Cottrell, G.S.; Kirkland, J.G.; Pelayo, J.C.; Bunnett, N.W.; Corvera, C.U. Expression and function of the bile acid receptor GpBAR1 (TGR5) in the murine enteric nervous system. Neurogastroenterol. Motil. 2010, 22, 814-e228. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-Mediated Bile Acid Sensing Controls Glucose Homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Nakamura, T.; Itadani, H.; Tanaka, K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719. [Google Scholar] [CrossRef]
- Fiorucci, S.; Mencarelli, A.; Palladino, G.; Cipriani, S. Bile-acid-activated receptors: Targeting TGR5 and farnesoid-X-receptor in lipid and glucose disorders. Trends Pharmacol. Sci. 2009, 30, 570–580. [Google Scholar] [CrossRef]
- Pols, T.W.; Noriega, L.G.; Nomura, M.; Auwerx, J.; Schoonjans, K. The bile acid membrane receptor TGR5 as an emerging target in metabolism and inflammation. J. Hepatol. 2011, 54, 1263–1272. [Google Scholar] [CrossRef]
- Baghdasaryan, A.; Claudel, T.; Gumhold, J.; Silbert, D.; Adorini, L.; Roda, A.; Vecchiotti, S.; Gonzalez, F.J.; Schoonjans, K.; Strazzabosco, M.; et al. Dual farnesoid X receptor/TGR5 agonist INT-767 reduces liver injury in the Mdr2 −/− (Abcb4 −/−) mouse cholangiopathy model by promoting biliary HCO3− output. Hepatology 2011, 54, 1303–1312. [Google Scholar] [CrossRef]
- Hu, Y.-B.; Liu, X.-Y.; Zhan, W. Farnesoid X receptor agonist INT-767 attenuates liver steatosis and inflammation in rat model of nonalcoholic steatohepatitis. Drug Des. Dev. Ther. 2018, 12, 2213–2221. [Google Scholar] [CrossRef]
- Kärkkäinen, J.M.; Lehtimäki, T.T.; Manninen, H.; Paajanen, H. Acute Mesenteric Ischemia Is a More Common Cause than Expected of Acute Abdomen in the Elderly. J. Gastrointest. Surg. 2015, 19, 1407–1414. [Google Scholar] [CrossRef]
- Ceulemans, L.J.; Canovai, E.; Verbeke, L.; Pirenne, J.; Farré, R. The expanding role of the bile acid receptor farnesoid X in the intestine and its potential clinical implications. Acta Chir. Belg. 2016, 116, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Nevens, F.; Andreone, P.; Mazzella, G.; Strasser, S.I.; Bowlus, C.; Invernizzi, P.; Drenth, J.P.; Pockros, P.J.; Regula, J.; Beuers, U.; et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N. Engl. J. Med. 2016, 375, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Biagioli, M.; Zampella, A.; Distrutti, E. Bile Acids Activated Receptors Regulate Innate Immunity. Front. Immunol. 2018, 9, 1853. [Google Scholar] [CrossRef] [PubMed]
- McMahan, R.H.; Wang, X.X.; Cheng, L.L.; Krisko, T.; Smith, M.; El Kasmi, K.; Pruzanski, M.; Adorini, L.; Golden-Mason, L.; Levi, M.; et al. Bile Acid Receptor Activation Modulates Hepatic Monocyte Activity and Improves Nonalcoholic Fatty Liver Disease. J. Biol. Chem. 2013, 288, 11761–11770. [Google Scholar] [CrossRef]
- Wang, Y.-D.; Chen, W.-D.; Yu, D.; Forman, B.M.; Huang, W. The G-Protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated B cells (NF-κB) in mice. Hepatology 2011, 54, 1421–1432. [Google Scholar] [CrossRef]
- Perino, A.; Pols, T.W.H.; Nomura, M.; Stein, S.; Pellicciari, R.; Schoonjans, K. TGR5 reduces macrophage migration through mTOR-induced C/EBPβ differential translation. J. Clin. Investig. 2014, 124, 5424–5436. [Google Scholar] [CrossRef]
- Keitel, V.; Stindt, J.; Häussinger, D. Bile Acid-Activated Receptors: GPBAR1 (TGR5) and Other G Protein-Coupled Receptors. In Handbook of Experimental Pharmacology; Springer New York LLC: New York, NY, USA, 2019; Volume 256, pp. 19–49. [Google Scholar] [CrossRef]
- Klag, T.; Stange, E.F.; Wehkamp, J. Defective Antibacterial Barrier in Inflammatory Bowel Disease. Dig. Dis. 2013, 31, 310–316. [Google Scholar] [CrossRef]
- Mastoraki, A.; Mastoraki, S.; Tziava, E.; Touloumi, S.; Krinos, N.; Danias, N.; Lazaris, A.; Arkadopoulos, N. Mesenteric ischemia: Pathogenesis and challenging diagnostic and therapeutic modalities. World J. Gastrointest. Pathophysiol. 2016, 7, 125–130. [Google Scholar] [CrossRef]
- Richter, W.; Vogel, V.; Howe, J.; Steiniger, F.; Brauser, A.; Koch, M.H.; Roessle, M.; Gutsmann, T.; Garidel, P.; Mäntele, W.; et al. Morphology, size distribution, and aggregate structure of lipopolysaccharide and lipid A dispersions from enterobacterial origin. Innate Immun. 2010, 17, 427–438. [Google Scholar] [CrossRef]
- Hollander, D.; Kaunitz, J.D. The “Leaky Gut”: Tight Junctions but Loose Associations? Dig. Dis. Sci. 2020, 65, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Akiba, Y.; Maruta, K.; Takajo, T.; Narimatsu, K.; Said, H.; Kato, I.; Kuwahara, A.; Kaunitz, J.D. Lipopolysaccharides transport during fat absorption in rodent small intestine. Am. J. Gastrointest. Physiol. Liver Physiol. 2020, 318, G1070–G1087. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guo, R.; Zhang, M.; Chen, P.; Li, J.; Sun, Y. Protective effect of emodin on intestinal epithelial tight junction barrier integrity in rats with sepsis induced by cecal ligation and puncture. Exp. Ther. Med. 2020, 19, 3521–3530. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Vuppalanchi, R.; Levy, C.; Floreani, A.; Andreone, P.; LaRusso, N.F.; Shrestha, R.; Trotter, J.; Goldberg, D.; Rushbrook, S.; et al. A randomized, placebo-controlled, phase II study of obeticholic acid for primary sclerosing cholangitis. J. Hepatol. 2020, 73, 94–101. [Google Scholar] [CrossRef]
- Yang, B.; Fung, A.; Pac-Soo, C.; Ma, D. Vascular surgery-related organ injury and protective strategies: Update and future prospects. Br. J. Anaesth. 2016, 117, ii32–ii43. [Google Scholar] [CrossRef] [PubMed]
- Bala, M.; Catena, F.; Kashuk, J.; De Simone, B.; Gomes, C.A.; Weber, D.; Sartelli, M.; Coccolini, F.; Kluger, Y.; Abu-Zidan, F.M.; et al. Acute mesenteric ischemia: Updated guidelines of the World Society of Emergency Surgery. World J. Emerg. Surg. 2022, 17, 54. [Google Scholar] [CrossRef] [PubMed]
- Tilsed, J.V.T.; Casamassima, A.; Kurihara, H.; Mariani, D.; Martinez, I.; Pereira, J.; Ponchietti, L.; Shamiyeh, A.; Al-Ayoubi, F.; Barco, L.A.B.; et al. ESTES guidelines: Acute mesenteric ischaemia. Eur. J. Trauma Emerg. Surg. 2016, 42, 253–270. [Google Scholar] [CrossRef]
- Nuzzo, A.; Maggiori, L.; Paugam-Burtz, C.; Cazals-Hatem, D.; Ronot, M.; Huguet, A.; Becq, A.; Castier, Y.; Weiss, E.; Plessier, A.; et al. Oral Antibiotics Reduce Intestinal Necrosis in Acute Mesenteric Ischemia: A Prospective Cohort Study. Am. J. Gastroenterol. 2019, 114, 348–351. [Google Scholar] [CrossRef]
- Walensi, M.; de Groot, H.; Schulz, R.; Hartmann, M.; Petrat, F. Mesenteric ischemia-reperfusion injury: Clearly improved hemodynamics but only minor protection of the rat small intestine by (sub)therapeutic heparin sodium and enoxaparin doses. J. Surg. Res. 2013, 179, e57–e69. [Google Scholar] [CrossRef]
- Clarysse, M.; Accarie, A.; Farré, R.; Canovai, E.; Monbaliu, D.; Gunst, J.; De Hertogh, G.; Vanuytsel, T.; Pirenne, J.; Ceulemans, L.J. Protective Effect of Oxygen and Isoflurane in Rodent Model of Intestinal Ischemia-Reperfusion Injury. Int. J. Mol. Sci. 2023, 24, 2587. [Google Scholar] [CrossRef]
- Gonzalez, L.M.; Moeser, A.J.; Blikslager, A.T. Animal models of ischemia-reperfusion-induced intestinal injury: Progress and promise for translational research. Am. J. Gastrointest. Physiol. Liver Physiol. 2015, 308, G63–G75. [Google Scholar] [CrossRef] [PubMed]
- Thuijls, G.; van Wijck, K.; Grootjans, J.; Derikx, J.P.M.; van Bijnen, A.A.; Heineman, E.; Dejong, C.H.; Buurman, W.A.; Poeze, M. Early Diagnosis of Intestinal Ischemia Using Urinary and Plasma Fatty Acid Binding Proteins. Ann. Surg. 2011, 253, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Oltean, M.; Olausson, M. The Chiu/Park scale for grading intestinal ischemia–reperfusion: If it ain’t broke don’t fix it! Intensiv. Care Med. 2010, 36, 1095. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.L.; Lim, D.W.; Levesque, C.L.; Vine, D.F.; Muto, M.; Koepke, J.R.; Nation, P.N.; Wizzard, P.R.; Li, J.; Bigam, D.L.; et al. A guide to Ussing chamber studies of mouse intestine. Am. J. Physiol.—Gastrointest. Liver Physiol. 2009, 296, G1151–G1166. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytokine | IRI + Vehicle | IRI + INT-767 | Significance (p-Value) |
|---|---|---|---|
| IL-6 | 20.74 ± 9.31 | 11.2 ± 4.35 | 0.018 |
| TNF-a | 7.14 ± 1.74 | 3.26 ± 0.67 | 0.0002 |
| IL-1b | 7.77 ± 3.25 | 3.54 ± 3.11 | NS (0.0742) |
| INT-γ | 8.99 ± 5.35 | 8.27 ± 7.50 | NS (0.8580) |
| IL-10 | 10.36 ± 3.32 | 16.55 ± 7.34 | 0.0248 |
| IL-13 | 11.57 ± 3.84 | 17.15 ± 5.61 | NS (0.1559) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canovai, E.; Farré, R.; Accarie, A.; Lauriola, M.; De Hertogh, G.; Vanuytsel, T.; Pirenne, J.; Ceulemans, L.J. INT-767—A Dual Farnesoid-X Receptor (FXR) and Takeda G Protein-Coupled Receptor-5 (TGR5) Agonist Improves Survival in Rats and Attenuates Intestinal Ischemia Reperfusion Injury. Int. J. Mol. Sci. 2023, 24, 14881. https://doi.org/10.3390/ijms241914881
Canovai E, Farré R, Accarie A, Lauriola M, De Hertogh G, Vanuytsel T, Pirenne J, Ceulemans LJ. INT-767—A Dual Farnesoid-X Receptor (FXR) and Takeda G Protein-Coupled Receptor-5 (TGR5) Agonist Improves Survival in Rats and Attenuates Intestinal Ischemia Reperfusion Injury. International Journal of Molecular Sciences. 2023; 24(19):14881. https://doi.org/10.3390/ijms241914881
Chicago/Turabian StyleCanovai, Emilio, Ricard Farré, Alison Accarie, Mara Lauriola, Gert De Hertogh, Tim Vanuytsel, Jacques Pirenne, and Laurens J. Ceulemans. 2023. "INT-767—A Dual Farnesoid-X Receptor (FXR) and Takeda G Protein-Coupled Receptor-5 (TGR5) Agonist Improves Survival in Rats and Attenuates Intestinal Ischemia Reperfusion Injury" International Journal of Molecular Sciences 24, no. 19: 14881. https://doi.org/10.3390/ijms241914881
APA StyleCanovai, E., Farré, R., Accarie, A., Lauriola, M., De Hertogh, G., Vanuytsel, T., Pirenne, J., & Ceulemans, L. J. (2023). INT-767—A Dual Farnesoid-X Receptor (FXR) and Takeda G Protein-Coupled Receptor-5 (TGR5) Agonist Improves Survival in Rats and Attenuates Intestinal Ischemia Reperfusion Injury. International Journal of Molecular Sciences, 24(19), 14881. https://doi.org/10.3390/ijms241914881