
The Pitfall of White Blood Cell Cystine Measurement to Diagnose Juvenile Cystinosis

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Case
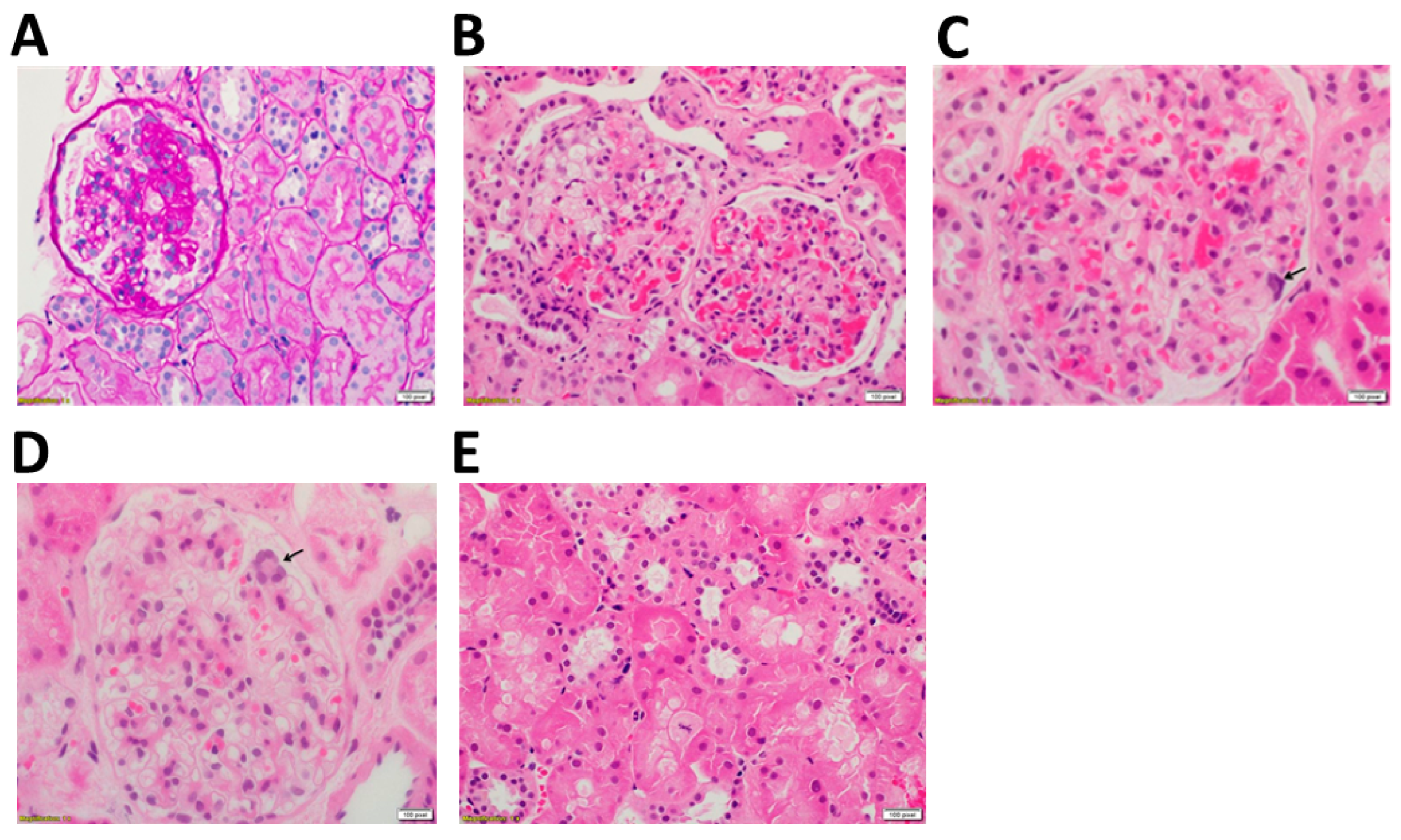
2.1. Clincal and Technical Examination
2.2. Cystine Measurement
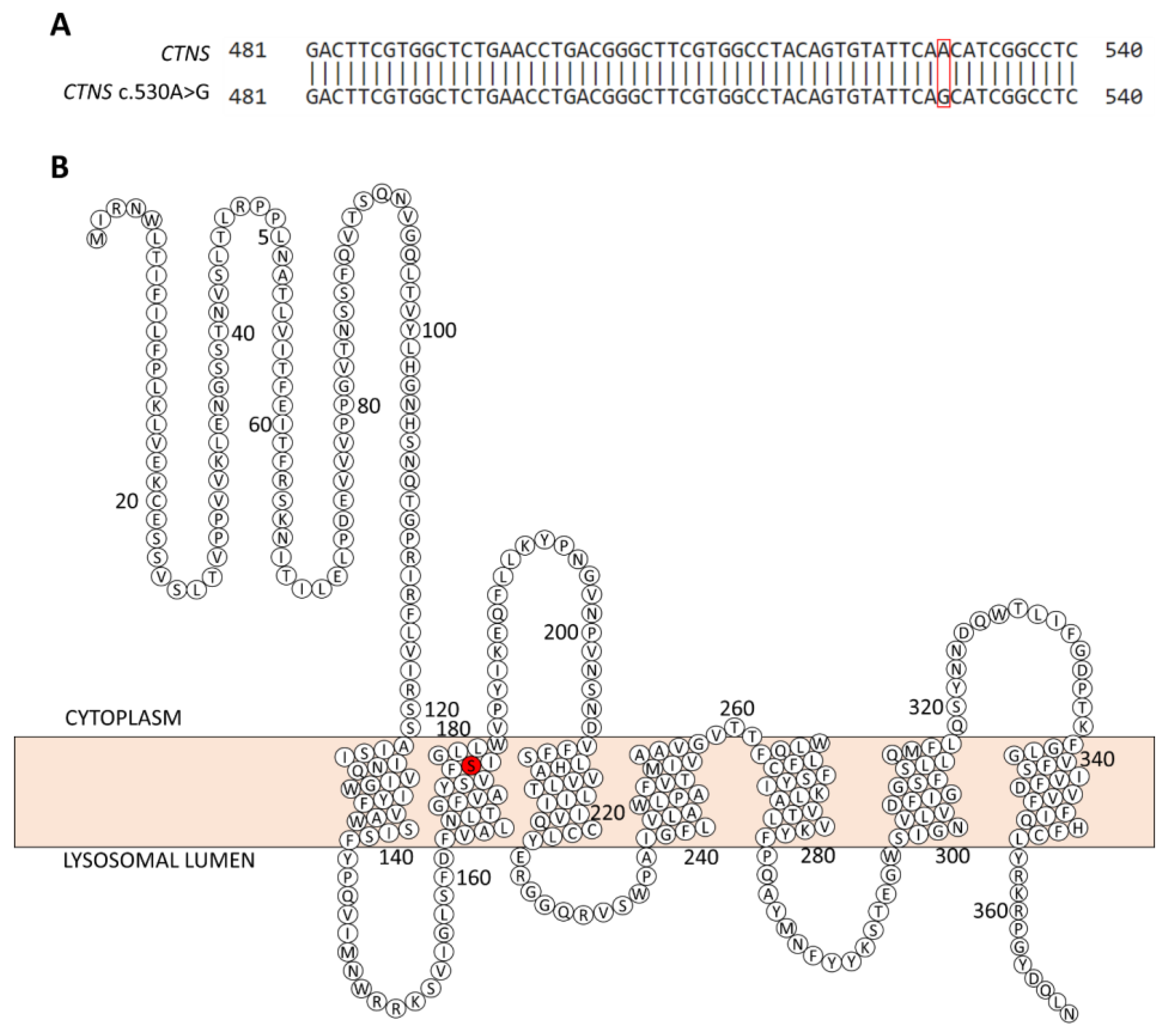
2.3. Genetic Analysis
3. Discussion
4. Materials and Methods
4.1. Patient
4.2. Generation of Conditionally Immortalized Cell Lines
4.3. gDNA Isolation from Blood
4.4. gDNA Isolation from ciPTECs and ciPODOs
4.5. Allele-Specific PCR
4.6. Sanger Sequencing of gDNA
4.7. Measurement of Intracellular Cystine in Cultured Kidney Cells
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Town, M.M.; Jean, G.; Cherqui, S.; Attard, M.; Forestier, L.; Whitmore, S.A.; Callen, D.F.; Gribouval, O.; Broyer, M.; Bates, G.; et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat. Genet. 1998, 18, 319–324. [Google Scholar] [CrossRef] [PubMed]
- David, D.; Berlingerio, S.P.; Elmonem, M.A.; Arcolino, F.O.; Soliman, N.; Heuvel, B.V.D.; Gijsbers, R.; Levtchenko, E. Molecular Basis of Cystinosis: Geographic Distribution, Functional Consequences of Mutations in the CTNS Gene, and Potential for Repair. Nephron 2019, 141, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Gahl, W.A.; Thoene, J.G.; Schneider, J.A. Cystinosis. N. Engl. J. Med. 2002, 347, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Elmonem, M.A.; Veys, K.R.; Soliman, N.A.; van Dyck, M.; Heuvel, L.P.V.D.; Levtchenko, E. Cystinosis: A review. Orphanet J. Rare Dis. 2016, 11, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özkan, B.; Çayır, A.; Koşan, C.; Alp, H. Cystinosis presenting with findings of Bartter syndrome. J. Clin. Res. Pediatr. Endocrinol. 2011, 3, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Cherqui, S.; Courtoy, P.J. The renal Fanconi syndrome in cystinosis: Pathogenic insights and therapeutic perspectives. Nat. Rev. Nephrol. 2017, 13, 115–131. [Google Scholar] [CrossRef]
- Ivanova, E.A.; De Leo, M.G.; van den Heuvel, L.; Pastore, A.; Dijkman, H.; De Matteis, M.A.; Levtchenko, E.N. Endo-lysosomal dysfunction in human proximal tubular epithelial cells deficient for lysosomal cystine transporter cystinosin. PLoS ONE 2015, 10, e0120998. [Google Scholar] [CrossRef] [Green Version]
- Servais, A.; Morinière, V.; Grünfeld, J.-P.; Noël, L.-H.; Goujon, J.-M.; Chadefaux-Vekemans, B.; Antignac, C. Late-onset nephropathic cystinosis: Clinical presentation, outcome, and genotyping. Clin. J. Am. Soc. Nephrol. 2008, 3, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Veys, K.R.; Elmonem, M.; Arcolino, F.O.; Heuvel, L.V.D.; Levtchenko, E. Nephropathic cystinosis: An update. Curr. Opin. Pediatr. 2017, 29, 168–178. [Google Scholar] [CrossRef]
- Anikster, Y.; Lucero, C.; Guo, J.; Huizing, M.; Shotelersuk, V.; Bernardini, I.; McDowell, G.; Iwata, F.; I Kaiser-Kupfer, M.; Jaffe, R.; et al. Ocular non-nephropathic cystinosis: Clinical, biochemical, and molecular correlations. Pediatr. Res. 2000, 47, 17. [Google Scholar] [CrossRef]
- Emma, F.; Hoff, W.V.; Hohenfellner, K.; Topaloglu, R.; Greco, M.; Ariceta, G.; Bettini, C.; Bockenhauer, D.; Veys, K.; Pape, L.; et al. An international cohort study spanning five decades assessed outcomes of nephropathic cystinosis. Kidney Int. 2021, 100, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Veys, K.; Zadora, W.; Hohenfellner, K.; Bockenhauer, D.; Janssen, M.C.H.; Niaudet, P.; Servais, A.; Topaloglu, R.; Besouw, M.; Novo, R.; et al. Outcome of infantile nephropathic cystinosis depends on early intervention, not genotype: A multicenter sibling cohort study. J. Inherit. Metab. Dis. 2022. [Google Scholar] [CrossRef] [PubMed]
- Shams, F.; Livingstone, I.; Oladiwura, D.; Ramaesh, K. Treatment of corneal cystine crystal accumulation in patients with cystinosis. Clin. Ophthalmol. 2014, 8, 2077–2084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilmer, M.J.; Schoeber, J.P.; van den Heuvel, L.P.; Levtchenko, E.N. Cystinosis: Practical tools for diagnosis and treatment. Pediatr. Nephrol. 2011, 26, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Veys, K.R.; Elmonem, M.A.; Van Dyck, M.; Janssen, M.C.; Cornelissen, E.A.; Hohenfellner, K.; Prencipe, G.; Heuvel, L.P.V.D.; Levtchenko, E. Chitotriosidase as a Novel Biomarker for Therapeutic Monitoring of Nephropathic Cystinosis. J. Am. Soc. Nephrol. 2020, 31, 1092–1106. [Google Scholar] [CrossRef]
- Tak, T.; Tesselaar, K.; Pillay, J.; Borghans, J.A.M.; Koenderman, L. What’s your age again? Determination of human neutrophil half-lives revisited. J. Leukoc. Biol. 2013, 94, 595–601. [Google Scholar] [CrossRef]
- Levtchenko, E.; de Graaf-Hess, A.; Wilmer, M.; Heuvel, L.V.D.; Monnens, L.; Blom, H. Comparison of Cystine Determination in Mixed Leukocytes vs Polymorphonuclear Leukocytes for Diagnosis of Cystinosis and Monitoring of Cysteamine Therapy. Clin. Chem. 2004, 50, 1686–1688. [Google Scholar] [CrossRef] [Green Version]
- Aldahmesh, M.A.; Humeidan, A.; Almojalli, H.A.; Khan, A.O.; Rajab, M.; Al-Abbad, A.A.; Meyer, B.F.; Alkuraya, F.S. Characterization of CTNS mutations in Arab patients with cystinosis. Ophthalmic Genet. 2009, 30, 185–189. [Google Scholar] [CrossRef]
- Wilmer, M.J.; Saleem, M.A.; Masereeuw, R.; Ni, L.; van der Velden, T.J.; Russel, F.G.; Mathieson, P.W.; Monnens, L.A.; Heuvel, L.P.V.D.; Levtchenko, E.N. Novel conditionally immortalized human proximal tubule cell line expressing functional influx and efflux transporters. Cell Tissue Res. 2010, 339, 449–457. [Google Scholar] [CrossRef] [Green Version]
- De Graaf-Hess, A.; Trijbels, F.; Blom, H. New method for determining cystine in leukocytes and fibroblasts. Clin. Chem. 1999, 45, 2224–2228. [Google Scholar] [CrossRef]
- Tietze, F.; Butler, J.D. Elevated cystine levels in cultured skin fibroblasts from patients with I-Cell disease. Pediatr. Res. 1979, 13, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, J.A.; Rosenbloom, F.M.; Bradley, K.H.; Seegmiller, J.E. Increased free-cystine content of fibroblasts cultured from patients with cystinosis. Biochem. Biophys. Res. Commun. 1967, 29, 527–531. [Google Scholar] [CrossRef]
- Sumayao, R.; Newsholme, P.; McMorrow, T. The Role of Cystinosin in the Intermediary Thiol Metabolism and Redox Homeostasis in Kidney Proximal Tubular Cells. Antioxidants 2018, 7, 179. [Google Scholar] [CrossRef] [Green Version]
- Betts, M.J.; Russell, R.B. Amino Acid Properties and Consequences of Substitutions. In Bioinformatics for Geneticists; John Wiley & Sons, Ltd.: Chichester, UK, 2003; pp. 289–316. [Google Scholar]
- Kalatzis, V.; Cohen-Solal, L.; Cordier, B.; Frishberg, Y.; Kemper, M.; Nuutinen, E.M.; Legrand, E.; Cochat, P.; Antignac, C. Identification of 14 novel CTNS mutations and characterization of seven splice site mutations associated with cystinosis. Hum. Mutat. 2002, 20, 439–446. [Google Scholar] [CrossRef]
- Kalatzis, V.; Nevo, N.; Cherqui, S.; Gasnier, B.; Antignac, C. Molecular pathogenesis of cystinosis: Effect of CTNS mutations on the transport activity and subcellular localization of cystinosin. Hum. Mol. Genet. 2004, 13, 1361–1371. [Google Scholar] [CrossRef]
- What Are White Blood Cells? Available online: https://www.stanfordchildrens.org/en/topic/default?id=what-are-white-blood-cells-160-35 (accessed on 25 August 2022).
- Lasagni, L.; Lazzeri, E.; Shankland, S.J.; Anders, H.-J.; Romagnani, P. Podocyte mitosis—A catastrophe. Curr. Mol. Med. 2013, 13, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D.; Czerniak, S.; DiRocco, D.P.; Hasnain, W.; Cheema, R.; Bonventre, J.V. Repair of injured proximal tubule does not involve specialized progenitors. Proc. Natl. Acad. Sci. USA 2011, 108, 9226–9231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogetseder, A.; Palan, T.; Bacic, D.; Kaissling, B.; Le Hir, M. Proximal tubular epithelial cells are generated by division of differentiated cells in the healthy kidney. Am. J. Physiol. Cell Physiol. 2007, 292, C807–C813. [Google Scholar] [CrossRef] [Green Version]
- Itahana, K.; Zou, Y.; Itahana, Y.; Martinez, J.-L.; Beausejour, C.; Jacobs, J.J.L.; van Lohuizen, M.; Band, V.; Campisi, J.; Dimri, G.P. Control of the replicative life span of human fibroblasts by p16 and the polycomb Protein Bmi-1. Mol. Cell. Biol. 2003, 23, 389. [Google Scholar] [CrossRef] [Green Version]
- Langman, C.B.; Barshop, B.A.; Deschênes, G.; Emma, F.; Goodyer, P.; Lipkin, G.; Midgley, J.P.; Ottolenghi, C.; Servais, A.; Soliman, N.A.; et al. Controversies and research agenda in nephropathic cystinosis: Conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2016, 89, 1192–1203. [Google Scholar] [CrossRef]
- Levtchenko, E.; Blom, H.; Wilmer, M.; Heuvel, L.V.D.; Monnens, L. ACE inhibitor enalapril diminishes albuminuria in patients with cystinosis. Clin. Nephrol. 2003, 60, 386–389. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Measurement at First Visit | Unit | Normal Range | |
|---|---|---|---|
| Clinical examination | |||
| Weight | 83.1 | kg | |
| Height | 167.8 | cm | |
| Blood Pressure | 127/75 | mmHg | |
| Blood | |||
| Hemoglobin | 14.0 | g/L | 14.0–18.0 |
| Calcium | 2.4 | mmol/L | 2.2–2.6 |
| Phosphate | 1.0 | mmol/L | 0.8–1.5 |
| AP | 137.0 | U/L | 40.0–130.0 |
| PTH | 35.0 | ng/L | 14.9–56.9 |
| 25-OH Vitamin D | 23.9 | µg/L | 30.0–60.0 |
| TSH | 2.5 | mIU/L | 0.3–4.2 |
| free T4 | 15.6 | pmol/L | 11.6–21.9 |
| LH | 4.7 | IU/L | 1.7–8.6 |
| FSH | 2.0 | IU/L | 1.2–7.7 |
| Testosterone | 696.0 | ng/dL | 300.0–1000.0 |
| Urine | |||
| Protein/creatinine | 0.7 | g/g | <0.2 |
| Albumin/creatinine | 602.0 | mg/g | <30.0 |
| Alpha-1 microglobulin/creatinine | 17.9 | mg/g | <11.7 |
| TmP/GFR | 1.0 | mmol/L | 0.8–1.2 |
| CKD-EPI eGFR | 120 | ml/min/1.73 m2 | >90.0 |
| Cystatin C-eGFR | 88 | ml/min/1.73 m2 | >90.0 |
| Case (nmol ½ Cystine/mg Protein) | Reference Values (nmol ½ Cystine/mg Protein) 1 | |
|---|---|---|
| PMN leukocytes | 1.31 | Normal: <0.20 [4] Heterozygotes: <1.00 [4] (Nephropathic) cystinosis: >3.00 [4] |
| Skin fibroblasts | 3.40 | Normal: <0.07 (mean) [22] Heterozygotes: <0.34 (mean) [22] (Nephropathic) cystinosis: >1.00 [21] |
| Podocytes (urine-derived) | 8.59 ± 0.62 | Normal: 5.01 ± 1.65 ** |
| Proximal tubular epithelial cells (urine-derived) | 13.39 ± 0.69 | Normal: 2.04 ± 0.68 ** |
| Exon 1 | Sequence |
|---|---|
| CTNS EX 3 FOR | AGC TGA TTC AAC ATT CCC CTG |
| CTNS EX 3 REV | TAG CCA CCA TTT CCC TCT TTA C |
| CTNS EX 4 FOR | TGT CAT TGA TTT GGG TCC TTC C |
| CTNS EX 4 REV | TAG GGC TTG TCT TAC AGG TA |
| CTNS EX 5 FOR | GAT CTC ACT GTC CAG CTT CT |
| CTNS EX 5 REV | TCC CTA CCC ATC CGT TAA G |
| CTNS EX 6 FOR | GCG GGG TCC TCG GTA ACT G |
| CTNS EX 6 REV | GGC CCC CTT CTT GTC ACG |
| CTNS EX 7 FOR | CTT CAG AAG CCC AGC CTC AGC |
| CTNS EX 7 REV | CGA GAG AGC CTG CAC ATA CG |
| CTNS EX 8 FOR | CCC TGC CCT GTC TTG TCC |
| CTNS EX 8 REV | CAG AGA TGT AGG GCA GGC AA |
| CTNS EX 9 FOR | CCT CAC CAC CCA GCT TCT CC |
| CTNS EX 9 REV | GTG GCG GGT GTT GGC TG |
| CTNS EX 10 FOR | GGC CTC TGT GTG GGT CC |
| CTNS EX 10 REV | GGC CAT GTA GCT CTC ACC TC |
| CTNS EX 11 FOR | GCC CTC CGT CTG TCT GTC CG |
| CTNS EX 11 REV | GCC CGA TGC CCC AGC CGC |
| CTNS EX 12 FOR | GCC AAC CTA ACA CCA GCT TC |
| CTNS EX 12 REV | AGA GGC TGG GTA CAC TGG GT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bondue, T.; Kouraich, A.; Berlingerio, S.P.; Veys, K.; Marie, S.; Alsaad, K.O.; Al-Sabban, E.; Levtchenko, E.; van den Heuvel, L. The Pitfall of White Blood Cell Cystine Measurement to Diagnose Juvenile Cystinosis. Int. J. Mol. Sci. 2023, 24, 1253. https://doi.org/10.3390/ijms24021253
Bondue T, Kouraich A, Berlingerio SP, Veys K, Marie S, Alsaad KO, Al-Sabban E, Levtchenko E, van den Heuvel L. The Pitfall of White Blood Cell Cystine Measurement to Diagnose Juvenile Cystinosis. International Journal of Molecular Sciences. 2023; 24(2):1253. https://doi.org/10.3390/ijms24021253
Chicago/Turabian StyleBondue, Tjessa, Anas Kouraich, Sante Princiero Berlingerio, Koenraad Veys, Sandrine Marie, Khaled O. Alsaad, Essam Al-Sabban, Elena Levtchenko, and Lambertus van den Heuvel. 2023. "The Pitfall of White Blood Cell Cystine Measurement to Diagnose Juvenile Cystinosis" International Journal of Molecular Sciences 24, no. 2: 1253. https://doi.org/10.3390/ijms24021253
APA StyleBondue, T., Kouraich, A., Berlingerio, S. P., Veys, K., Marie, S., Alsaad, K. O., Al-Sabban, E., Levtchenko, E., & van den Heuvel, L. (2023). The Pitfall of White Blood Cell Cystine Measurement to Diagnose Juvenile Cystinosis. International Journal of Molecular Sciences, 24(2), 1253. https://doi.org/10.3390/ijms24021253