
Mechanistic Investigation of the Androgen Receptor DNA-Binding Domain and Modulation via Direct Interactions with DNA Abasic Sites: Understanding the Mechanisms Involved in Castration-Resistant Prostate Cancer

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
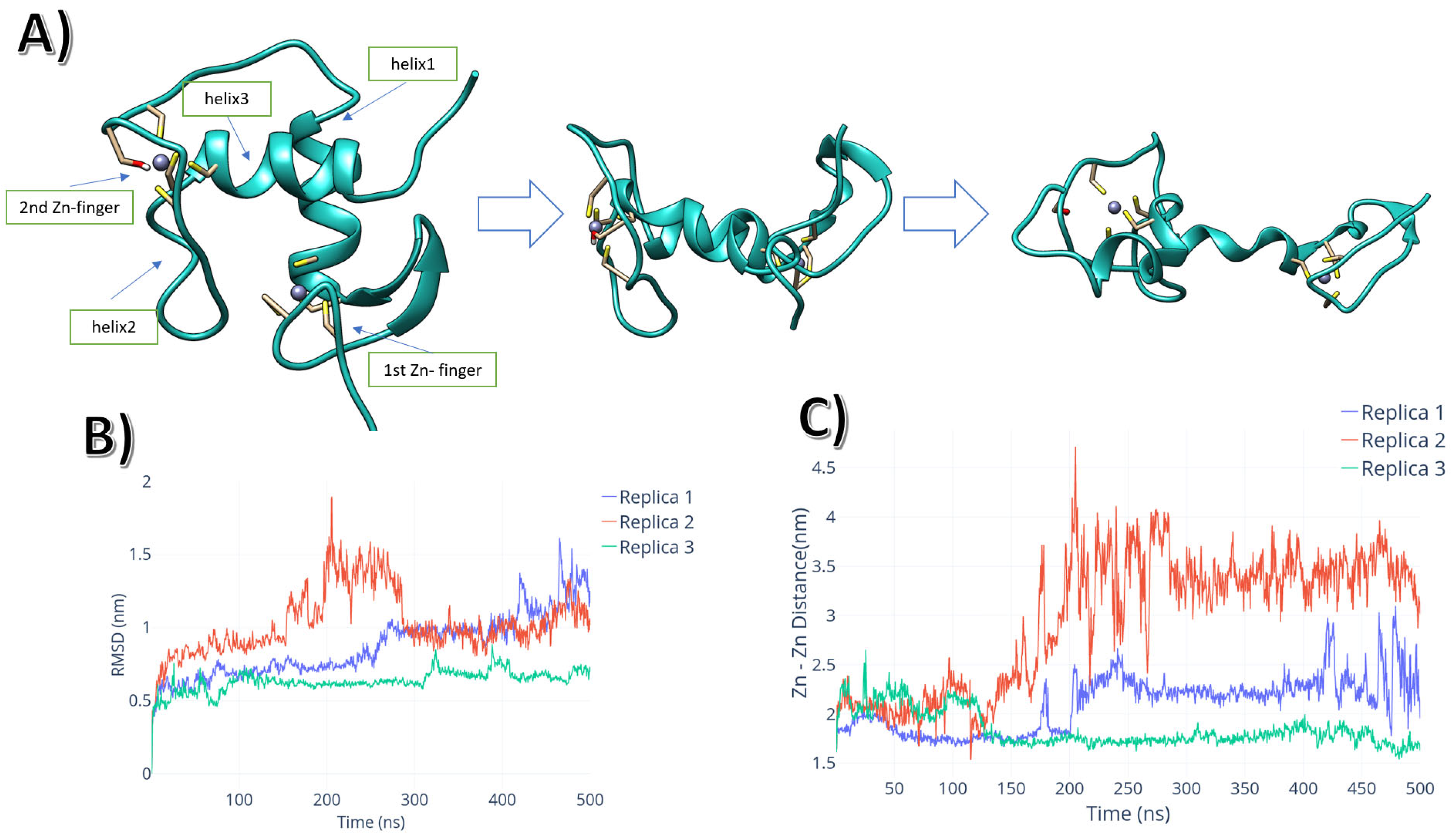
2.1. Intrinsic Molecular Dynamics of AR-DBD Homodimers in the Presence and Absence of DNA
2.2. Impact of Cancer-Linked Mutations within the AR-DBD
2.3. Impact of Abasic Lesions within ARE on Interactions with AR-DBD Domain
3. Discussion
4. Materials and Methods
4.1. Molecular Dynamics Simulations
4.2. Quantum Mechanical Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviation | Definition |
| APE1 | Apurinic/apyrimidinic endonuclease 1 |
| AR | Androgen receptor |
| ARE | Androgen-responsive element |
| BER | Base excision repair |
| CRPC | Castration-resistant prostate cancer |
| DBD | DNA-binding domain |
| DFT | Density functional theory |
| HOMO | Highest-occupied molecular orbital |
| LBD | Ligand-binding domain |
| LUMO | Lowest-unoccupied molecular orbital |
| MD | Molecular dynamics |
| NTD | N-terminal domain |
| PBC | Periodic boundary conditions |
| PME | Particle Mesh-Ewald |
| PPI | Protein-protein interaction |
| QM | Quantum mechanical |
| RMSD | Root mean-squared deviation |
| RMSF | Root-mean-square fluctuation |
| WT | Wild-type |
References
- Gatta, G.; Mallone, S.; Van der Zwan, J.M.; Trama, A.; Siesling, S.; Capocaccia, R.; Hackl, M.; Van Eycken, E.; Henau, K.; Hedelin, G.; et al. Cancer survival in Europe 1999–2007 by country and age: Results of EUROCARE-5—A population-based study. Lancet Oncol. 2014, 15, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.H.; Parsi, M.; Potdar, R.R. Triple-arm androgen blockade for advanced prostate cancer: A review. Med. Oncol. 2021, 38, 75. [Google Scholar] [CrossRef]
- Center, M.M.; Jemal, A.; Lortet-Tieulent, J.; Ward, E.; Ferlay, J.; Brawley, O.; Bray, F. International variation in prostate cancer incidence and mortality rates. Eur. Urol. 2012, 61, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, H.U.; El-Shater Bosaily, A.; Brown, L.C.; Gabe, R.; Kaplan, R.; Parmar, M.K.; Collaco-Moraes, Y.; Ward, K.; Hindley, R.G.; Freeman, A.; et al. Diagnostic accuracy of multi-parametric MRI and TRUS biopsy in prostate cancer (PROMIS): A paired validating confirmatory study. Lancet 2017, 389, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Gelmann, E.P. Molecular biology of the androgen receptor. J. Clin. Oncol. 2002, 20, 3001–3015. [Google Scholar] [CrossRef]
- Rizzo, A.; Mollica, V.; Rosellini, M.; Marchetti, A.; Ricci, A.D.; Fiorentino, M.; Battelli, N.; Santoni, M.; Massari, F. Exploring the association between metastatic sites and androgen receptor splice variant 7 (AR-V7) in castration-resistant prostate cancer patients: A meta-analysis of prospective clinical trials. Pathol. Res. Pract. 2021, 222, 153440. [Google Scholar] [CrossRef]
- Nagandla, H.; Robertson, M.J.; Putluri, V.; Putluri, N.; Coarfa, C.; Weigel, N.L. Isoform-specific Activities of Androgen Receptor and its Splice Variants in Prostate Cancer Cells. Endocrinology 2021, 162, bqaa227. [Google Scholar] [CrossRef]
- Cao, H.; Wang, D.; Gao, R.; Chen, L.; Feng, Y. Down regulation of U2AF1 promotes ARV7 splicing and prostate cancer progression. Biochem. Biophys. Res. Commun. 2021, 541, 56–62. [Google Scholar] [CrossRef]
- Thadani-Mulero, M.; Portella, L.; Sun, S.; Sung, M.; Matov, A.; Vessella, R.L.; Corey, E.; Nanus, D.M.; Plymate, S.R.; Giannakakou, P. Androgen receptor splice variants determine taxane sensitivity in prostate cancer. Cancer Res. 2014, 74, 2270–2282. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasnov, G.S.; Dmitriev, A.A.; Sadritdinova, A.F.; Volchenko, N.N.; Slavnova, E.N.; Danilova, T.V.; Snezhkina, A.V.; Melnikova, N.V.; Fedorova, M.S.; Lakunina, V.A.; et al. Molecular genetic mechanisms of drug resistance in prostate cancer. Mol. Biol. 2015, 49, 638–648. [Google Scholar] [CrossRef]
- Tan, M.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [Green Version]
- Wade, C.A.; Kyprianou, N. Profiling Prostate Cancer Therapeutic Resistance. Int. J. Mol. Sci. 2018, 19, 904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Mahony, O.A.; Steinkamp, M.P.; Albertelli, M.A.; Brogley, M.; Rehman, H.; Robins, D.M. Profiling human androgen receptor mutations reveals treatment effects in a mouse model of prostate cancer. Mol. Cancer Res. 2008, 6, 1691–1701. [Google Scholar] [CrossRef]
- Messner, E.A.; Steele, T.M.; Tsamouri, M.M.; Hejazi, N.; Gao, A.C.; Mudryj, M.; Ghosh, P.M. The androgen receptor in prostate cancer: Effect of structure, ligands and spliced variants on therapy. Biomedicines 2020, 8, 422. [Google Scholar] [CrossRef]
- Komori, S.; Sakata, K.; Kasumi, H.; Tsuji, Y.; Hamada, K.; Koyama, K. A substitutional mutation in the DNA binding domain of the androgen receptor causes complete androgen insensitivity syndrome. Gynecol. Endocrinol. 1999, 13, 327–332. [Google Scholar] [CrossRef]
- Morova, T.; McNeill, D.R.; Lallous, N.; Gönen, M.; Dalal, K.; Wilson, D.M.; Gürsoy, A.; Keskin, Ö.; Lack, N.A. Androgen receptor-binding sites are highly mutated in prostate cancer. Nat. Commun. 2020, 11, 832. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Yao, G.; Guan, X.; Ni, Z.; Ma, W.; Wilson, E.M.; French, F.S.; Liu, Q.; Zhang, Y. Research resource: Genome-wide mapping of in vivo androgen receptor binding sites in mouse epididymis. Mol. Endocrinol. 2010, 24, 2392–2405. [Google Scholar] [CrossRef]
- Denayer, S.; Helsen, C.; Thorrez, L.; Haelens, A.; Claessens, F. The Rules of DNA Recognition by the Androgen Receptor. Mol. Endocrinol. 2010, 24, 898. [Google Scholar] [CrossRef]
- Bagherpoor Helabad, M.; Volkenandt, S.; Imhof, P. Molecular Dynamics Simulations of a Chimeric Androgen Receptor Protein (SPARKI) Confirm the Importance of the Dimerization Domain on DNA Binding Specificity. Front. Mol. Biosci. 2020, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, S.K.; Tew, B.Y.; Lim, M.; Stankavich, B.; He, M.; Pufall, M.; Hu, W.; Chen, Y.; Jones, J.O. Mechanistic Investigation of the Androgen Receptor DNA-Binding Domain Inhibitor Pyrvinium. ACS Omega 2019, 4, 2472–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.O.; Bolton, E.C.; Huang, Y.; Feau, C.; Guy, R.K.; Yamamoto, K.R.; Hann, B.; Diamond, M.I. Non-competitive androgen receptor inhibition in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 7233–7238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabanés Zariquiey, F.; de Souza, J.V.; Bronowska, A.K. Cosolvent Analysis Toolkit (CAT): A robust hotspot identification platform for cosolvent simulations of proteins to expand the druggable proteome. Sci. Rep. 2019, 9, 19118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadal, M.; Prekovic, S.; Gallastegui, N.; Helsen, C.; Abella, M.; Zielinska, K.; Gay, M.; Vilaseca, M.; Taulès, M.; Houtsmuller, A.B.; et al. Structure of the homodimeric androgen receptor ligand-binding domain. Nat. Commun. 2017, 8, 14388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serçinoğlu, O.; Bereketoglu, C.; Olsson, P.E.; Pradhan, A. In silico and in vitro assessment of androgen receptor antagonists. Comput. Biol. Chem. 2021, 92, 107490. [Google Scholar] [CrossRef] [PubMed]
- Shao, G.; Bao, J.; Pan, X.; He, X.; Qi, Y.; Zhang, J.Z.H. Computational Analysis of Residue-Specific Binding Free Energies of Androgen Receptor to Ligands. Front. Mol. Biosci. 2021, 8, 646524. [Google Scholar] [CrossRef]
- Chen, G.; Wang, X.; Zhang, S.; Lu, Y.; Sun, Y.; Zhang, J.; Li, Z.; Lu, J. Androgen receptor mutants detected in recurrent prostate cancer exhibit diverse functional characteristics. Prostate 2005, 63, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Tilley, W.D.; Buchanan, G.; Hickey, T.E.; Bentel, J.M. Mutations in the androgen receptor gene are associated with progression of human prostate cancer to androgen independence. Clin. Cancer Res. 1996, 2, 277–285. [Google Scholar]
- Lallous, N.; Snow, O.; Sanchez, C.; Parra Nuñez, A.K.; Sun, B.; Hussain, A.; Lee, J.; Morin, H.; Leblanc, E.; Gleave, M.E.; et al. Evaluation of Darolutamide (ODM201) Efficiency on Androgen Receptor Mutants Reported to Date in Prostate Cancer Patients. Cancers 2021, 13, 2939. [Google Scholar] [CrossRef]
- Marcelli, M.; Ittmann, M.; Mariani, S.; Sutherland, R.; Nigam, R.; Murthy, L.; Zhao, Y.; Diconcini, D.; Puxeddu, E.; Esen, A.; et al. Androgen Receptor Mutations in Prostate Cancer. Cancer Res. 2000, 60, 944–949. [Google Scholar]
- Ta, H.Q.; Gioeli, D. The convergence of DNA damage checkpoint pathways and androgen receptor signaling in prostate cancer. Endocr. Relat. Cancer 2014, 21, R395–R407. [Google Scholar] [CrossRef] [PubMed]
- Thompson, T.C.; Li, L. Connecting androgen receptor signaling and the DNA damage response: Development of new therapies for advanced prostate cancer. Mol. Cell. Oncol. 2017, 4, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loizzo, D.; Pandolfo, S.D.; Rogers, D.; Cerrato, C.; Di Meo, N.A.; Autorino, R.; Mirone, V.; Ferro, M.; Porta, C.; Stella, A.; et al. Novel Insights into Autophagy and Prostate Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2022, 23, 3826. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, M.M. Abasic and oxidized abasic site reactivity in DNA: Enzyme inhibition, cross-linking, and nucleosome catalyzed reactions. Acc. Chem. Res. 2014, 47, 646–655. [Google Scholar] [CrossRef] [Green Version]
- Rozelle, A.L.; Cheun, Y.; Vilas, C.K.; Koag, M.C.; Lee, S. DNA interstrand cross-links induced by the major oxidative adenine lesion 7,8-dihydro-8-oxoadenine. Nat. Commun. 2021, 12, 1897. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjørås, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Raper, A.T.; Maxwell, B.A.; Suo, Z. Dynamic Processing of a Common Oxidative DNA Lesion by the First Two Enzymes of the Base Excision Repair Pathway. J. Mol. Biol. 2021, 433, 166811. [Google Scholar] [CrossRef]
- Sassa, A.; Odagiri, M. Understanding the sequence and structural context effects in oxidative DNA damage repair. DNA Repair 2020, 93, 102906. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.L.; Zubair, M.; John, K.; Poirier, M.C.; Martin, F.L. Carcinogens and DNA damage. Biochem. Soc. Trans. 2018, 46, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Foloppe, N.; MacKerell, A.D. Intrinsic conformational properties of deoxyribonucleosides: Implicated role for cytosine in the equilibrium among the A, B, and Z forms of DNA. Biophys. J. 1999, 76, 3206–3218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppel, Y.; Berthet, N.; Coulombeau, C.; Coulombeau, C.; Garcia, J.; Lhomme, J. Solution conformation of an abasic DNA undecamer duplex d(CGCACXCACGC)·d(GCGTGTGTGCG): The unpaired trymine stacks inside the helix. Biochemistry 1997, 36, 4817–4830. [Google Scholar] [CrossRef]
- Berman, H.M.; Olson, W.K.; Beveridge, D.L.; Westbrook, J.; Gelbin, A.; Demeny, T.; Hsieh, S.H.; Srinivasan, A.R.; Schneider, B. The nucleic acid database. A comprehensive relational database of three-dimensional structures of nucleic acids. Biophys. J. 1992, 63, 751–759. [Google Scholar] [CrossRef] [Green Version]
- Davletgildeeva, A.T.; Kuznetsova, A.A.; Fedorova, O.S.; Kuznetsov, N.A. Activity of Human Apurinic/Apyrimidinic Endonuclease APE1 Toward Damaged DNA and Native RNA With Non-canonical Structures. Front. Cell Dev. Biol. 2020, 8, 590848. [Google Scholar] [CrossRef]
- Azam, S.; Jouvet, N.; Jilani, A.; Vongsamphanh, R.; Yang, X.; Yang, S.; Ramotar, D. Human glyceraldehyde-3-phosphate dehydrogenase plays a direct role in reactivating oxidized forms of the DNA repair enzyme APE1. J. Biol. Chem. 2008, 283, 30632–30641. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, E.; Giménez, R.; Alexandra Cañas, M.; Aguilera, L.; Aguilar, J.; Badia, J.; Baldomà, L. Glyceraldehyde-3-phosphate dehydrogenase is required for efficient repair of cytotoxic DNA lesions in Escherichia coli. Int. J. Biochem. Cell Biol. 2015, 60, 202–212. [Google Scholar] [CrossRef] [Green Version]
- Ilina, E.S.; Khodyreva, S.N.; Lavrik, O.I. Unusual interaction of human apurinic/apyrimidinic endonuclease 1 (APE1) with abasic sites via the Schiff-base-dependent mechanism. Biochimie 2018, 150, 88–99. [Google Scholar] [CrossRef]
- Shaffer, P.L.; Jivan, A.; Dollins, D.E.; Claessens, F.; Gewirth, D.T.; Richardson, J.S. Structural Basis of Androgen Receptor Binding to Selective Androgen Response Elements. Proc. Natl. Acad. Sci. USA 2004, 101, 4758–4763. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera? A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Sousa Da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Crystal structure and pair potentials: A molecular-dynamics study. Phys. Rev. Lett. 1980, 45, 1196–1199. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Frisch, J.M. GAUSSIAN09. Available online: https://gaussian.com/ (accessed on 31 October 2022).
- Dennington, R.; Keith, T.; Millam, J. Gauss View, Version 5; Semichem Inc.: Shawnee, KS, USA, 2009.








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, S.; Kondal, M.D.; Ahmad, A.; Zhu, R.; Fan, L.; Zaborniak, P.; Madden, K.S.; de Souza, J.V.; Bronowska, A.K. Mechanistic Investigation of the Androgen Receptor DNA-Binding Domain and Modulation via Direct Interactions with DNA Abasic Sites: Understanding the Mechanisms Involved in Castration-Resistant Prostate Cancer. Int. J. Mol. Sci. 2023, 24, 1270. https://doi.org/10.3390/ijms24021270
Xu S, Kondal MD, Ahmad A, Zhu R, Fan L, Zaborniak P, Madden KS, de Souza JV, Bronowska AK. Mechanistic Investigation of the Androgen Receptor DNA-Binding Domain and Modulation via Direct Interactions with DNA Abasic Sites: Understanding the Mechanisms Involved in Castration-Resistant Prostate Cancer. International Journal of Molecular Sciences. 2023; 24(2):1270. https://doi.org/10.3390/ijms24021270
Chicago/Turabian StyleXu, Shangze, Matthew D. Kondal, Ayaz Ahmad, Ruidi Zhu, Lanyu Fan, Piotr Zaborniak, Katrina S. Madden, João V. de Souza, and Agnieszka K. Bronowska. 2023. "Mechanistic Investigation of the Androgen Receptor DNA-Binding Domain and Modulation via Direct Interactions with DNA Abasic Sites: Understanding the Mechanisms Involved in Castration-Resistant Prostate Cancer" International Journal of Molecular Sciences 24, no. 2: 1270. https://doi.org/10.3390/ijms24021270
APA StyleXu, S., Kondal, M. D., Ahmad, A., Zhu, R., Fan, L., Zaborniak, P., Madden, K. S., de Souza, J. V., & Bronowska, A. K. (2023). Mechanistic Investigation of the Androgen Receptor DNA-Binding Domain and Modulation via Direct Interactions with DNA Abasic Sites: Understanding the Mechanisms Involved in Castration-Resistant Prostate Cancer. International Journal of Molecular Sciences, 24(2), 1270. https://doi.org/10.3390/ijms24021270