

Identification of Side Chain Oxidized Sterols as Novel Liver X Receptor Agonists with Therapeutic Potential in the Treatment of Cardiovascular and Neurodegenerative Diseases
 , , ,
, , ,  , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Results
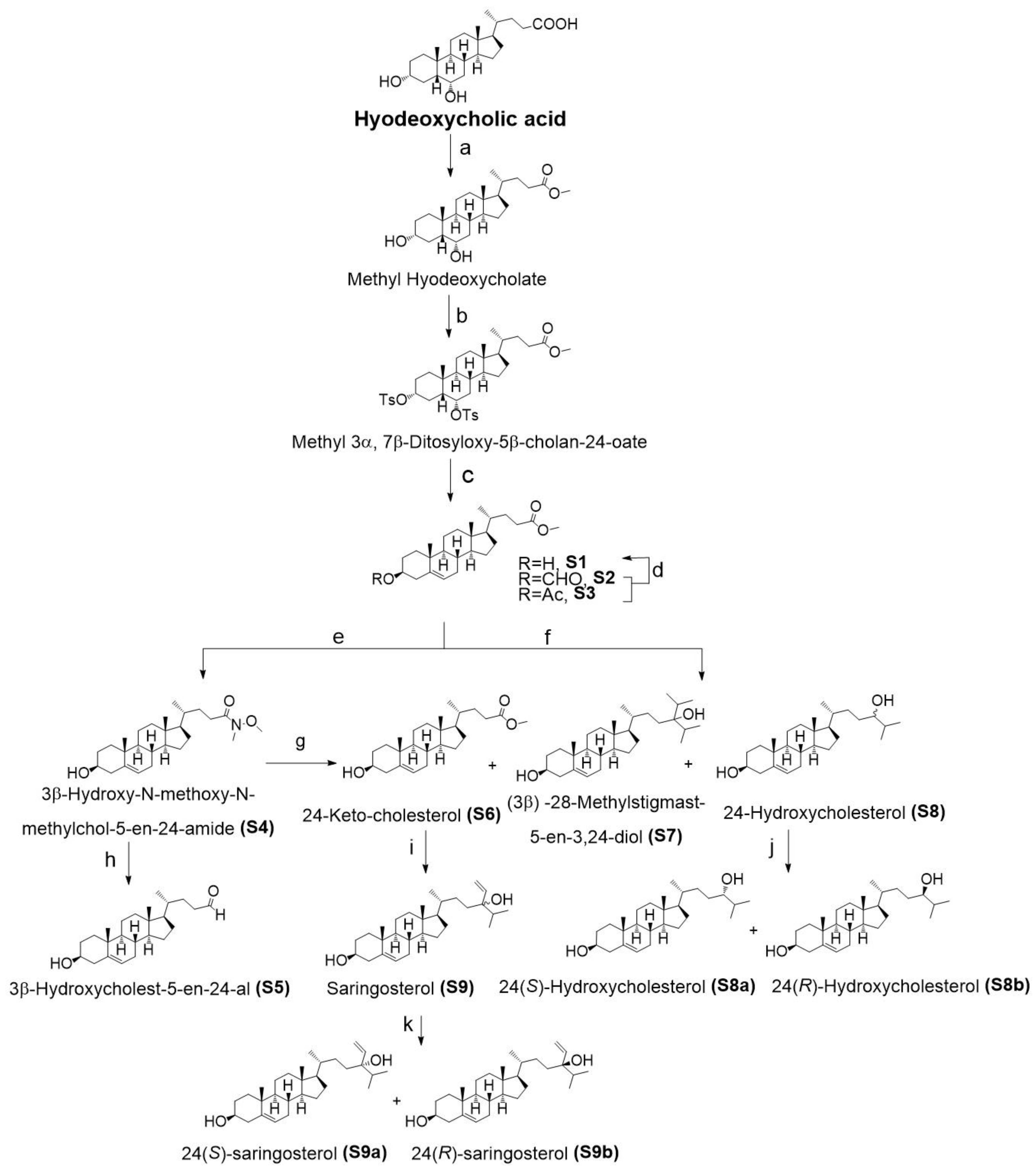
2.1. Chemistry
2.2. LXRα and LXRβ Activation
2.3. LXR-Target Gene Expression in CNS Cell Lines
2.4. Cell-Specific Discrimination of Oxysterols Selectively Regulate SREBF1 and Its Downstream Lipogenic Genes
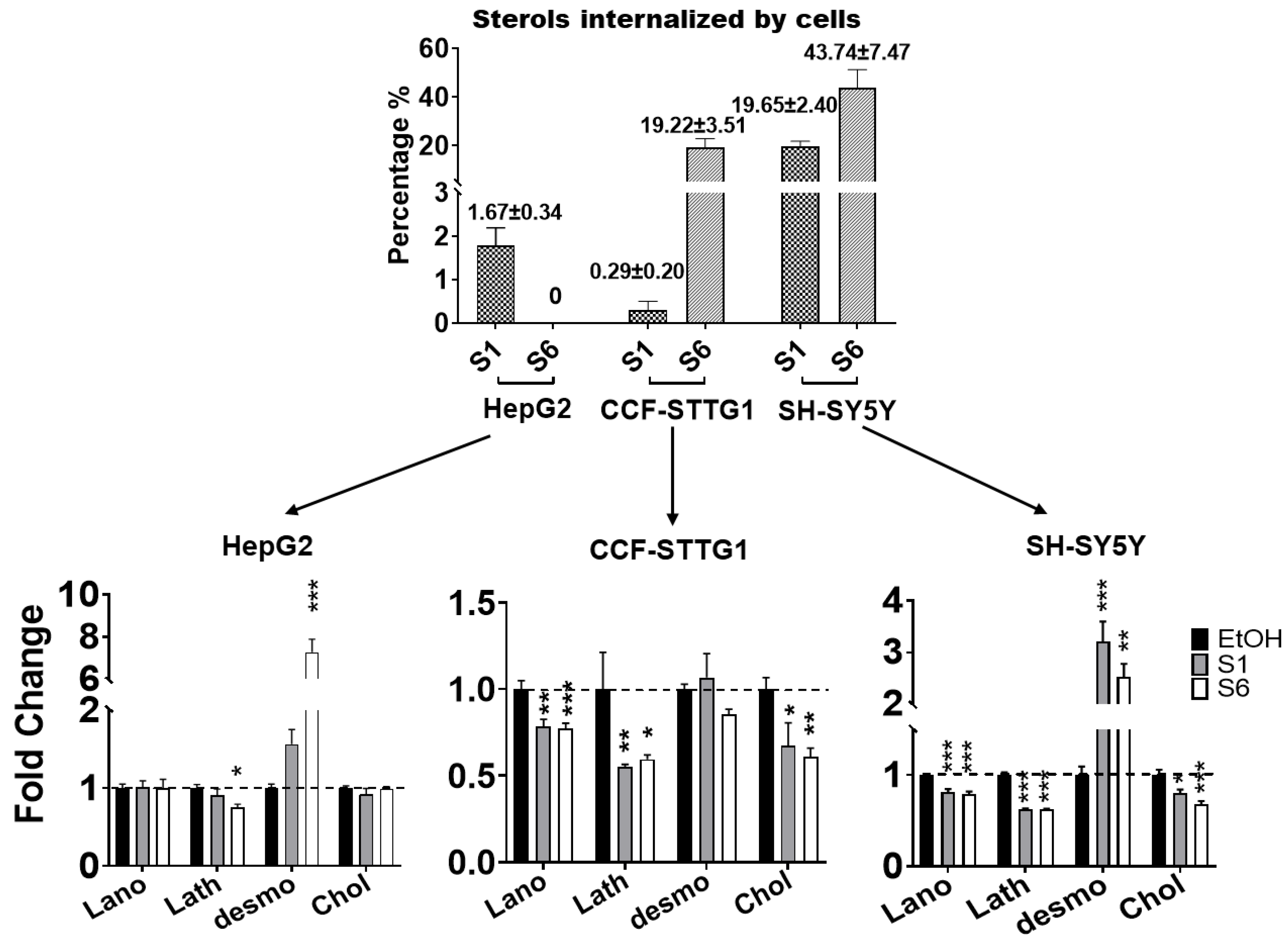
2.5. Effect on Cholesterol Efflux
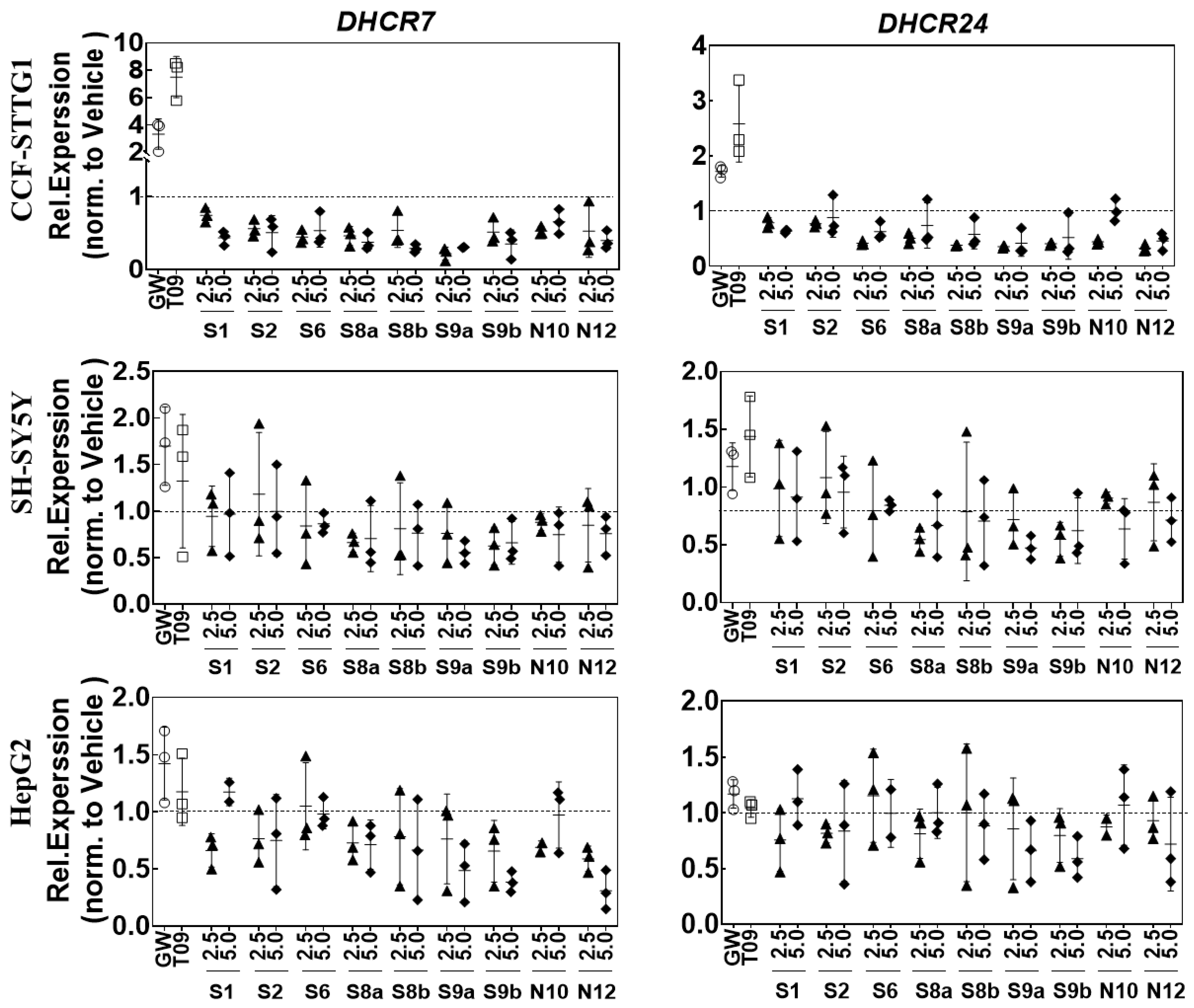
2.6. LXR-Activating Oxidized Sterols Down-Regulate DHCR7 and DHCR24 Gene Expression
3. Discussion
4. Materials and Methods
4.1. Side Chain Oxidized Cholesterol Derivatives
4.2. Phytosterol Separation from Seaweed
4.3. Cell Culture and Transfection
4.4. Reporter Assays
4.5. Quantitative PCR
4.6. Quantitative Analysis of Cholesterol and Cholesterol’s Precursors
4.7. Cholesterol Efflux Studies
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fessler, M.B. The challenges and promise of targeting the Liver X Receptors for treatment of inflammatory disease. Pharmacol. Ther. 2018, 181, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Xiao, X.; Yan, Y.; Zhang, T. Activation of liver X receptors prevents emotional and cognitive dysfunction by suppressing microglial M1-polarization and restoring synaptic plasticity in the hippocampus of mice. Brain Behav. Immun. 2021, 94, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, C.M.; Torrecilla-Parra, M.; Pardo-Marqués, V.; de-Frutos, M.F.; Pérez-García, A.; Tabraue, C.; de la Rosa, J.V.; Martín-Rodriguez, P.; Díaz-Sarmiento, M.; Nuñez, U.; et al. Crosstalk Between LXR and Caveolin-1 Signaling Supports Cholesterol Efflux and Anti-Inflammatory Pathways in Macrophages. Front. Endocrinol. 2021, 12, 635923. [Google Scholar] [CrossRef] [PubMed]
- Zelcer, N.; Tontonoz, P. Liver X receptors as integrators of metabolic and inflammatory signaling. J. Clin. Investig. 2006, 116, 607–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lu, K.; Luo, H.; Liang, D.; Long, X.; Yuan, Y.; Wu, C.; Bao, J. In silico identification of small molecules as novel LXR agonists for the treatment of cardiovascular disease and cancer. J. Mol. Model. 2018, 24, 57. [Google Scholar] [CrossRef]
- Guo, S.; Li, L.; Yin, H. Cholesterol Homeostasis and Liver X Receptor (LXR) in Atherosclerosis. Cardiovasc. Hematol. Disord. Drug Targets 2018, 18, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Geyeregger, R.; Zeyda, M.; Stulnig, T.M. Liver X receptors in cardiovascular and metabolic disease. Cell. Mol. Life Sci. CMLS 2006, 63, 524–539. [Google Scholar] [CrossRef]
- Xu, P.; Li, D.; Tang, X.; Bao, X.; Huang, J.; Tang, Y.; Yang, Y.; Xu, H.; Fan, X. LXR agonists: New potential therapeutic drug for neurodegenerative diseases. Mol. Neurobiol. 2013, 48, 715–728. [Google Scholar] [CrossRef]
- Mouzat, K.; Chudinova, A.; Polge, A.; Kantar, J.; Camu, W.; Raoul, C.; Lumbroso, S. Regulation of Brain Cholesterol: What Role Do Liver X Receptors Play in Neurodegenerative Diseases? Int. J. Mol. Sci. 2019, 20, 3858. [Google Scholar] [CrossRef] [Green Version]
- Janowski, B.A.; Willy, P.J.; Devi, T.R.; Falck, J.R.; Mangelsdorf, D.J. An oxysterol signalling pathway mediated by the nuclear receptor LXRα. Nature 1996, 383, 728–731. [Google Scholar] [CrossRef]
- Song, C.; Hiipakka, R.A.; Liao, S. Selective activation of liver X receptor alpha by 6α-hydroxy bile acids and analogs. Steroids 2000, 65, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; McDonald, J.G.; Patel, A.; Zhang, Y.; Umetani, M.; Xu, F.; Westover, E.J.; Covey, D.F.; Mangelsdorf, D.J.; Cohen, J.C.; et al. Sterol intermediates from cholesterol biosynthetic pathway as liver X receptor ligands. J. Biol. Chem. 2006, 281, 27816–27826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muse, E.D.; Yu, S.; Edillor, C.R.; Tao, J.; Spann, N.J.; Troutman, T.D.; Seidman, J.S.; Henke, A.; Roland, J.T.; Ozeki, K.A. Cell-specific discrimination of desmosterol and desmosterol mimetics confers selective regulation of LXR and SREBP in macrophages. Proc. Natl. Acad. Sci. USA 2018, 115, E4680–E4689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, M.; Wang, W.; Greco, D.; Bellenchi, G.C.; di Porzio, U.; Brown, A.J.; Ikonen, E. What dictates the accumulation of desmosterol in the developing brain? FASEB J. 2013, 27, 865–870. [Google Scholar] [CrossRef] [Green Version]
- Spann, N.J.; Garmire, L.X.; McDonald, J.G.; Myers, D.S.; Milne, S.B.; Shibata, N.; Reichart, D.; Fox, J.N.; Shaked, I.; Heudobler, D.; et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell 2012, 151, 138–152. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Lei, W.; Deng, C.; Wu, Z.; Sun, M.; Jin, Z.; Song, Y.; Yang, Z.; Jiang, S.; Shen, M.J.; et al. The roles of liver X receptor α in inflammation and inflammation-associated diseases. J. Cell. Psysil. 2021, 236, 4807–4828. [Google Scholar] [CrossRef]
- Kidani, Y.; Bensinger, S.J. Liver X receptor and peroxisome proliferator-activated receptor as integrators of lipid homeostasis and immunity. Immunol. Rev. 2012, 249, 72–83. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Qiu, D.K.; Ma, X. Liver X receptors bridge hepatic lipid metabolism and inflammation. J. Dig. Dis. 2012, 13, 69–74. [Google Scholar] [CrossRef]
- Joseph, S.B.; Castrillo, A.; Laffitte, B.A.; Mangelsdorf, D.J.; Tontonoz, P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. J. Nat. Med. 2003, 9, 213–219. [Google Scholar] [CrossRef]
- Carpenter, K.J.; Valfort, A.-C.; Steinauer, N.; Chatterjee, A.; Abuirqeba, S.; Majidi, S.; Sengupta, M.; Di Paolo, R.J.; Shornick, L.P.; Zhang, J. LXR-inverse agonism stimulates immune-mediated tumor destruction by enhancing CD8 T-cell activity in triple negative breast cancer. Sci. Rep. 2019, 9, 1–18. [Google Scholar] [CrossRef]
- Vanmierlo, T.; Weingärtner, O.; van der Pol, S.; Husche, C.; Kerksiek, A.; Friedrichs, S.; Sijbrands, E.; Steinbusch, H.; Grimm, M.; Hartmann, T.; et al. Dietary intake of plant sterols stably increases plant sterol levels in the murine brain. J. Lipid Res. 2012, 53, 726–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djelti, F.; Braudeau, J.; Hudry, E.; Dhenain, M.; Varin, J.; Bièche, I.; Marquer, C.; Chali, F.; Ayciriex, S.; Auzeil, N.; et al. CYP46A1 inhibition, brain cholesterol accumulation and neurodegeneration pave the way for Alzheimer’s disease. Brain A J. Neurol. 2015, 138, 2383–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pikuleva, I.A.; Cartier, N. Cholesterol Hydroxylating Cytochrome P450 46A1: From Mechanisms of Action to Clinical Applications. Front. Aging Neurosci. 2021, 13, 696778. [Google Scholar] [CrossRef] [PubMed]
- Loera-Valencia, R.; Goikolea, J.; Parrado-Fernandez, C.; Merino-Serrais, P.; Maioli, S. Alterations in cholesterol metabolism as a risk factor for developing Alzheimer’s disease: Potential novel targets for treatment. J. Steroid Biochem. Mol. Biol. 2019, 190, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Testa, G.; Staurenghi, E.; Zerbinati, C.; Gargiulo, S.; Iuliano, L.; Giaccone, G.; Fantò, F.; Poli, G.; Leonarduzzi, G.; Gamba, P. Changes in brain oxysterols at different stages of Alzheimer’s disease: Their involvement in neuroinflammation. Redox Biol. 2016, 10, 24–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Lee, C.Y.; Mandrekar, S.; Wilkinson, B.; Cramer, P.; Zelcer, N.; Mann, K.; Lamb, B.; Willson, T.M.; Collins, J.L.; et al. ApoE promotes the proteolytic degradation of Abeta. Neuron 2008, 58, 681–693. [Google Scholar] [CrossRef] [Green Version]
- Vanmierlo, T.; Rutten, K.; Dederen, J.; Bloks, V.W.; van Vark-van der Zee, L.C.; Kuipers, F.; Kiliaan, A.; Blokland, A.; Sijbrands, E.J.; Steinbusch, H.; et al. Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol Aging 2011, 32, 1262–1272. [Google Scholar] [CrossRef]
- Zhu, R.; Ou, Z.; Ruan, X.; Gong, J. Role of liver X receptors in cholesterol efflux and inflammatory signaling (review). Mol. Med. Rep. 2012, 5, 895–900. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.; Tontonoz, P. Coordination of inflammation and metabolism by PPAR and LXR nuclear receptors. Curr. Opin. Genet. Dev. 2008, 18, 461–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radhakrishnan, A.; Sun, L.P.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. Direct binding of cholesterol to the purified membrane region of SCAP: Mechanism for a sterol-sensing domain. Mol. Cell 2004, 15, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. Cholesterol feedback: From Schoenheimer’s bottle to Scap’s MELADL. J. Lipid Res. 2009, 50, S15–S27. [Google Scholar] [CrossRef] [Green Version]
- Repa, J.J.; Liang, G.; Ou, J.; Bashmakov, Y.; Lobaccaro, J.-M.A.; Shimomura, I.; Shan, B.; Brown, M.S.; Goldstein, J.L.; Mangelsdorf, D.J. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRα and LXRβ. Genes Dev. 2000, 14, 2819–2830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Tontonoz, P. Liver X receptors in lipid signalling and membrane homeostasis. Nat. Reviews. Endocrinol. 2018, 14, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Steffensen, K.R.; Neo, S.Y.; Stulnig, T.M.; Vega, V.B.; Rahman, S.S.; Schuster, G.U.; Gustafsson, J.-A.k.; Liu, E.T. Genome-wide expression profiling; a panel of mouse tissues discloses novel biological functions of liver X receptors in adrenals. J. Mol. Endocrinol. 2004, 33, 609–622. [Google Scholar] [CrossRef]
- Annicotte, J.S.; Schoonjans, K.; Auwerx, J. Expression of the liver X receptor α and β in embryonic and adult mice. Anat. Rec. Part A Discov. Mol. Cell. Evol. Biol. Off. Publ. Am. Assoc. Anat. 2004, 277, 312–316. [Google Scholar]
- Ma, C.; Feng, K.; Yang, X.; Yang, Z.; Wang, Z.; Shang, Y.; Fan, G.; Liu, L.; Yang, S.; Li, X.; et al. Targeting macrophage liver X receptors by hydrogel-encapsulated T0901317 reduces atherosclerosis without effect on hepatic lipogenesis. Br. J. Pharmacol. 2021, 178, 1620–1638. [Google Scholar] [CrossRef]
- Navas, F.F.C.; Giorgi, G.; Maggioni, D.; Pacciarini, M.; Russo, V.; Marinozzi, M. C24-hydroxylated stigmastane derivatives as Liver X Receptor agonists. Chem. Phys. Lipids 2018, 212, 44–50. [Google Scholar] [CrossRef]
- Pratiwi, R.; Nantasenamat, C.; Ruankham, W.; Suwanjang, W.; Prachayasittikul, V.; Prachayasittikul, S.; Phopin, K. Mechanisms and Neuroprotective Activities of Stigmasterol Against Oxidative Stress-Induced Neuronal Cell Death via Sirtuin Family. Front. Nutr. 2021, 8, 648995. [Google Scholar] [CrossRef]
- Jansen, P.J.; Lütjohann, D.; Abildayeva, K.; Vanmierlo, T.; Plösch, T.; Plat, J.; von Bergmann, K.; Groen, A.K.; Ramaekers, F.C.; Kuipers, F.; et al. Dietary plant sterols accumulate in the brain. Biochim. Biophys. Acta 2006, 1761, 445–453. [Google Scholar] [CrossRef]
- Vanmierlo, T.; Rutten, K.; van Vark-van der Zee, L.C.; Friedrichs, S.; Bloks, V.W.; Blokland, A.; Ramaekers, F.C.; Sijbrands, E.; Steinbusch, H.; Prickaerts, J.; et al. Cerebral accumulation of dietary derivable plant sterols does not interfere with memory and anxiety related behavior in Abcg5-/- mice. Plant Foods Hum. Nutr. 2011, 66, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Huang, C. Natural modulators of liver X receptors. J. Integr. Med. 2014, 12, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Vanbrabant, K.; Van Meel, D.; Kerksiek, A.; Friedrichs, S.; Dubbeldam, M.; Schepers, M.; Zhan, N.; Gutbrod, K.; Dörmann, P.; Liu, H.-B.; et al. 24 (R, S)-Saringosterol-From artefact to a biological medical agent. J. Steroid Biochem. Mol. Biol. 2021, 212, 105942. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, J.; Fu, Z.; Ye, C.; Zhang, R.; Song, Y.; Zhang, Y.; Li, H.; Ying, H.; Liu, H. 24 (S)-Saringosterol from edible marine seaweed Sargassum fusiforme is a novel selective LXRβ agonist. J. Agric. Food Chem. 2014, 62, 6130–6137. [Google Scholar] [CrossRef] [PubMed]
- Bogie, J.; Hoeks, C.; Schepers, M.; Tiane, A.; Cuypers, A.; Leijten, F.; Chintapakorn, Y.; Suttiyut, T.; Pornpakakul, S.; Struik, D.; et al. Dietary Sargassum fusiforme improves memory and reduces amyloid plaque load in an Alzheimer’s disease mouse model. Sci. Rep. 2019, 9, 4908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martens, N.; Schepers, M.; Zhan, N.; Leijten, F.; Voortman, G.; Tiane, A.; Rombaut, B.; Poisquet, J.; Sande, N.v.d.; Kerksiek, A. 24 (S)-Saringosterol prevents cognitive decline in a mouse model for Alzheimer’s disease. Mar. Drugs 2021, 19, 190. [Google Scholar] [CrossRef]
- Yan, Y.; Niu, Z.; Wang, B.; Zhao, S.; Sun, C.; Wu, Y.; Li, Y.; Ying, H.; Liu, H. Saringosterol from sargassum fusiforme modulates cholesterol metabolism and alleviates atherosclerosis in ApoE-deficient mice. Mar. Drugs 2021, 19, 485. [Google Scholar] [CrossRef]
- Hiebl, V.; Ladurner, A.; Latkolik, S.; Dirsch, V.M. Natural products as modulators of the nuclear receptors and metabolic sensors LXR, FXR and RXR. Biotechnol. Adv. 2018, 36, 1657–1698. [Google Scholar]
- Willy, P.J.; Umesono, K.; Ong, E.S.; Evans, R.M.; Heyman, R.A.; Mangelsdorf, D.J. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev. 1995, 9, 1033–1045. [Google Scholar] [CrossRef] [Green Version]
- Ceroi, A.; Masson, D.; Roggy, A.; Roumier, C.; Chagué, C.; Gauthier, T.; Philippe, L.; Lamarthée, B.; Angelot-Delettre, F.; Bonnefoy, F.J.B. LXR agonist treatment of blastic plasmacytoid dendritic cell neoplasm restores cholesterol efflux and triggers apoptosis. J. Am. Soc. Hematol. 2016, 128, 2694–2707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, J.; Quinn, C.M.; Brown, A.J. SREBP-2 positively regulates transcription of the cholesterol efflux gene, ABCA1, by generating oxysterol ligands for LXR. Biochem. J. 2006, 400, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.-Y.; Linsenbardt, A.J.; Emnett, C.M.; Eisenman, L.N.; Izumi, Y.; Zorumski, C.F.; Mennerick, S. 24 (S)-Hydroxycholesterol as a modulator of neuronal signaling and survival. Neuroscientist 2016, 22, 132–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, T.; Shimano, H.; Amemiya-Kudo, M.; Yahagi, N.; Hasty, A.H.; Matsuzaka, T.; Okazaki, H.; Tamura, Y.; Iizuka, Y.; Ohashi, K.; et al. Identification of liver X receptor-retinoid X receptor as an activator of the sterol regulatory element-binding protein 1c gene promoter. Mol. Cell. Biol. 2001, 21, 2991–3000. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.B.; Laffitte, B.A.; Patel, P.H.; Watson, M.A.; Matsukuma, K.E.; Walczak, R.; Collins, J.L.; Osborne, T.F.; Tontonoz, P. Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors. J. Biol. Chem. 2002, 277, 11019–11025. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.B.; Spiegelman, B.M. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev. 1996, 10, 1096–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, E.; Hylemon, P.; Vlahcevic, Z.; Mallonee, D.; Valerie, K.; Avadhani, N.; Pandak, W. Overexpression of CYP27 in hepatic and extrahepatic cells: Role in the regulation of cholesterol homeostasis. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G293–G301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielska, A.A.; Schlesinger, P.; Covey, D.F.; Ory, D.S. Oxysterols as non-genomic regulators of cholesterol homeostasis. Trends Endocrinol. Metab. TEM 2012, 23, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Janowski, B.A.; Grogan, M.J.; Jones, S.A.; Wisely, G.B.; Kliewer, S.A.; Corey, E.J.; Mangelsdorf, D.J. Structural requirements of ligands for the oxysterol liver X receptors LXRα and LXRβ. Proc. Natl. Acad. Sci. USA 1999, 96, 266–271. [Google Scholar] [CrossRef] [Green Version]
- Lütjohann, D.; Breuer, O.; Ahlborg, G.; Nennesmo, I.; Sidén, A.; Diczfalusy, U.; Björkhem, I. Cholesterol homeostasis in human brain: Evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc. Natl. Acad. Sci. USA 1996, 93, 9799–9804. [Google Scholar] [CrossRef] [Green Version]
- Turley, S.D.; Burns, D.K.; Dietschy, J.M. Preferential utilization of newly synthesized cholesterol for brain growth in neonatal lambs. Am. J. Physiol. 1998, 274, E1099–E1105. [Google Scholar] [CrossRef]
- Turri, M.; Marchi, C.; Adorni, M.P.; Calabresi, L.; Zimetti, F. Emerging role of HDL in brain cholesterol metabolism and neurodegenerative disorders. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2022, 1867, 159123. [Google Scholar] [CrossRef]
- Testa, G.; Gamba, P.; Badilli, U.; Gargiulo, S.; Maina, M.; Guina, T.; Calfapietra, S.; Biasi, F.; Cavalli, R.; Poli, G.J.P.O. Loading into nanoparticles improves quercetin’s efficacy in preventing neuroinflammation induced by oxysterols. PLoS ONE 2014, 9, e96795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kacher, R.; Lamazière, A.; Heck, N.; Kappes, V.; Mounier, C.; Despres, G.; Dembitskaya, Y.; Perrin, E.; Christaller, W.; Sasidharan Nair, S. CYP46A1 gene therapy deciphers the role of brain cholesterol metabolism in Huntington’s disease. Brain 2019, 142, 2432–2450. [Google Scholar] [CrossRef] [PubMed]
- van der Kant, R.; Langness, V.F.; Herrera, C.M.; Williams, D.A.; Fong, L.K.; Leestemaker, Y.; Steenvoorden, E.; Rynearson, K.D.; Brouwers, J.F.; Helms, J.B.; et al. Cholesterol Metabolism Is a Druggable Axis that Independently Regulates Tau and Amyloid-β in iPSC-Derived Alzheimer’s Disease Neurons. Cell Stem Cell 2019, 24, 363–375.e369. [Google Scholar] [CrossRef] [Green Version]
- Doria, M.; Maugest, L.; Moreau, T.; Lizard, G.; Vejux, A. Contribution of cholesterol and oxysterols to the pathophysiology of Parkinson’s disease. Free. Radic. Biol. Med. 2016, 101, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Leoni, V.; Caccia, C. 24S-hydroxycholesterol in plasma: A marker of cholesterol turnover in neurodegenerative diseases. Biochimie 2013, 95, 595–612. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, A.; Brafman, A.; Shafir, M.; Heverin, M.; Gottlieb, H.; Damari, G.; Gozlan-Kelner, S.; Spivak, I.; Moshkin, O.; Fridman, E. Generation of viable cholesterol-free mice. Science 2003, 302, 2087. [Google Scholar] [CrossRef]
- Zhang, X.; McDonald, J.G.; Aryal, B.; Canfrán-Duque, A.; Goldberg, E.L.; Araldi, E.; Ding, W.; Fan, Y.; Thompson, B.M.; Singh, A.K.; et al. Desmosterol suppresses macrophage inflammasome activation and protects against vascular inflammation and atherosclerosis. Proc. Natl. Acad. Sci. USA 2021, 118, e2107682118. [Google Scholar] [CrossRef]
- Huang, L.; Sun, Y.; Zhu, H.; Zhang, Y.; Xu, J.; Shen, Y.M. Synthesis and antimicrobial evaluation of bile acid tridentate conjugates. Steroids 2009, 74, 701–706. [Google Scholar] [CrossRef]
- Huang, L.; Zhu, H.; Xu, X.; Zhang, C.; Shen, Y.-M. Synthesis and characterization of organometallic rhenium (I) and technetium (I) bile acid complexes. J. Organomet. Chem. 2009, 694, 3247–3253. [Google Scholar] [CrossRef]
- Kakiyama, G.; Muto, A.; Shimada, M.; Mano, N.; Goto, J.; Hofmann, A.F.; Iida, T. Chemical synthesis of 3β-sulfooxy-7β-hydroxy-24-nor-5-cholenoic acid: An internal standard for mass spectrometric analysis of the abnormal Δ5-bile acids occurring in Niemann-Pick disease. Steroids 2009, 74, 766–772. [Google Scholar] [CrossRef]
- Iida, T.; Kakiyama, G.; Hibiya, Y.; Miyata, S.; Inoue, T.; Ohno, K.; Goto, T.; Mano, N.; Goto, J.; Nambara, T. Chemical synthesis of the 3-sulfooxy-7-N-acetylglucosaminyl-24-amidated conjugates of 3β, 7β-dihydroxy-5-cholen-24-oic acid, and related compounds: Unusual, major metabolites of bile acid in a patient with Niemann-Pick disease type C1. Steroids 2006, 71, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Tomkinson, N.C.; Willson, T.M.; Russel, J.S.; Spencer, T.A. Efficient, stereoselective synthesis of 24 (S), 25-epoxycholesterol. J. Org. Chem. 1998, 63, 9919–9923. [Google Scholar] [CrossRef]
- Cui, J.G.; Lin, C.W.; Zeng, L.M.; Su, J.Y. Synthesis of polyhydroxysterols (III): Synthesis and structural elucidation of 24-methylenecholest-4-en-3β, 6α-diol. Steroids 2002, 67, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Khripach, V.; Zhabinskii, V.; Antonchik, A. A new synthesis of cerebrosterol and its 24-epimer from lithocholic acid. Russ. J. Bioorganic Chem. 2007, 33, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.H.; Zhou, X.D.; Zhou, W.S. A short and highly stereoselective synthesis of cerebrosterol. Chin. J. Chem. 2002, 20, 1145–1148. [Google Scholar] [CrossRef]
- Wu, Z.-H.; liu, T.; Gu, C.-X.; Shao, C.-L.; Zhou, J.; Wang, C.-Y. Steroids and triterpenoids from the brown alga Kjellmaniella crassifolia. Chem. Nat. Compd. 2012, 48, 158–160. [Google Scholar] [CrossRef]
- Feng, M.T.; Wang, T.; Liu, A.H.; Li, J.; Yao, L.G.; Wang, B.; Guo, Y.W.; Mao, S.C. PTP1B inhibitory and cytotoxic C-24 epimers of Δ(28)-24-hydroxy stigmastane-type steroids from the brown alga Dictyopteris undulata Holmes. Phytochemistry 2018, 146, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Liu, D.-Q.; Liang, T.-J.; Li, J.; Zhang, H.-Y.; Liu, A.-H.; Guo, Y.-W.; Mao, S.-C. Bioactive constituents from the green alga Caulerpa racemosa. Bioorg. Med. Chem. 2015, 23, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Huh, G.-W.; Lee, D.-Y.; In, S.-J.; Lee, D.-G.; Park, S.Y.; Yi, T.-H.; Kang, H.C.; Seo, W.-D.; Baek, N.-I. Fucosterols from Hizikia fusiformis and their proliferation activities on osteosarcoma-derived cell MG63. J. Korean Soc. Appl. Biol. Chem. 2012, 55, 551–555. [Google Scholar] [CrossRef]
- Tang, H.-F.; Yi, Y.-H.; Yao, X.-S.; Xu, Q.-Z.; Zhang, S.-Y.; Lin, H.-W. Bioactive steroids from the brown alga Sargassum carpophyllum. J. Asian Nat. Prod. Res. 2002, 4, 95–101. [Google Scholar] [CrossRef]
- Zwarts, I.; van Zutphen, T.; Kruit, J.K.; Liu, W.; Oosterveer, M.H.; Verkade, H.J.; Uhlenhaut, N.H.; Jonker, J.W. Identification of the fructose transporter GLUT5 (SLC2A5) as a novel target of nuclear receptor LXR. Sci. Rep. 2019, 9, 9299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackay, D.S.; Jones, P.J.; Myrie, S.B.; Plat, J.; Lütjohann, D.J. Methodological considerations for the harmonization of non-cholesterol sterol bio-analysis. J. Chromatogr. B 2014, 957, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Lütjohann, D.; Brzezinka, A.; Barth, E.; Abramowski, D.; Staufenbiel, M.; von Bergmann, K.; Beyreuther, K.; Multhaup, G.; Bayer, T.A. Profile of cholesterol-related sterols in aged amyloid precursor protein transgenic mouse brain. J. Lipid Res. 2002, 43, 1078–1085. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell line | HEK293 | CCF-STTG1 | SH-SY5Y | CHME3 | HepG2 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LXR | α | β | α | β | α | β | α | β | α | β | ||
| Compouds | 5.0μM | |||||||||||
| S1 | 2.69 | 3.29 | 1.73 | 1.37 | 7.85 | 6.30 | 1.89 | 2.26 | 2.56 | 2.33 | ||
| S2 | 1.90 | 1.80 | 0.76 | 1.32 | 5.44 | 5.09 | 1.75 | 1.24 | 2.40 | 2.55 | ||
| S3 | 2.72 | 1.74 | 1.39 | 1.51 | 1.97 | 1.74 | 1.46 | 1.28 | 1.05 | 1.39 | Fold change | |
| S4 | 1.62 | 1.25 | 1.34 | 1.45 | 0.19 | 0.33 | 1.51 | 1.98 | 0.82 | 0.80 | 3.5 < X | |
| S5 | 1.33 | 0.99 | 1.07 | 1.16 | 0.24 | 0.45 | 1.13 | 1.05 | 0.67 | 0.71 | 3.0 < X ≤ 3.5 | |
| S6 | 2.34 | 2.82 | 2.28 | 2.87 | 9.90 | 7.26 | 1.87 | 2.67 | 3.22 | 2.73 | 2.5 < X ≤ 3.0 | |
| S7 | 1.67 | 1.53 | 1.30 | 1.12 | 1.08 | 1.33 | 1.24 | 1.39 | 0.83 | 1.45 | 2.0 < X ≤ 2.5 | |
| S8a | 2.51 | 2.61 | 1.72 | 2.25 | 7.79 | 6.90 | 1.73 | 2.40 | 2.64 | 3.70 | 1.5 < X ≤ 2.0 | |
| S8b | 2.07 | 2.12 | 1.93 | 2.45 | 3.56 | 2.72 | 1.51 | 1.39 | 1.54 | 2.01 | 1.0 < X ≤ 1.5 | |
| S9a | 1.55 | 1.96 | 1.26 | 2.12 | 4.40 | 3.77 | 1.70 | 2.01 | 2.34 | 2.75 | ≤1.0 | |
| S9b | 1.17 | 1.22 | 0.62 | 0.49 | 0.96 | 1.87 | 1.14 | 1.07 | 1.37 | 1.14 | ||
| N10 | 2.86 | 3.67 | 0.86 | 0.91 | 12.56 | 8.82 | 2.20 | 2.75 | 2.88 | 4.15 | ||
| N11 | 1.55 | 1.45 | 1.02 | 1.05 | 2.56 | 2.36 | 1.40 | 1.77 | 1.87 | 2.30 | ||
| N12 | 2.17 | 2.57 | 0.91 | 1.30 | 7.89 | 7.71 | 2.47 | 3.05 | 1.38 | 4.23 | ||
| N13 | 1.21 | 0.86 | 1.00 | 1.32 | 0.09 | 0.20 | 1.33 | 1.66 | 0.37 | 0.59 | ||
| Gene (Human) | Forward | Reverse | Order/Design Date |
|---|---|---|---|
| APOE | ACCCAGGAACTGAGGGC | CTCCTTGGACAGCCGTG | 13 November 2017 |
| ABCA1 | TCTCTGTTCGGCTGAGCTAC | TGCAGAGGGCATGGCTTTAT | 26 June 2017 |
| ABCG1 | GGTCGCTCCATCATTTGCAC | GCAGACTTTTCCCCGGTACA | 26 June 2017 |
| SREBF1 | ACAGCCATGAAGACAGACGG | CAAGATGGTTCCGCCACTCA | 15 September 2020 |
| ACACA | GGGTCAAGTCCTTCCTGCTC | GGACTGTCGAGTCACCTTAAGTA | 30 August 2022 |
| FASN | CACAGACGAGAGCACCTTTGA | CAGGTCTATGAGGCCTATCTGG | 22 October 2019 |
| SCD1 | GCTGTCAAAGAGAAGGGGAGT | AGCCAGGTTTGTAGTACCTCCT | 10 May 2021 |
| NR1H3 (LXRA) | GTTATAACCGGGAAGACTTTGC | AAACTCGGCATCATTGAGTTG | 29 August 2018 |
| NR1H2 (LXRB) | AAGCAAGTGCCTGGTTTCCT | GCAGCATGATCTCGATAGTGGA | 26 June 2017 |
| DHCR7 | TGGGCCAAGACTCCACCTAT | ACGTGTACAGAAGCACCTGG | 12 July 2021 |
| DHCR24 | GTCTCACTACGTGTCGGGAA | CTCCACACGGACAATCTGTTTC | 10 May 2021 |
| CYP27A1 | GGGCAAGTACCCAGTACGGA | TGGTGTCCTTCCGTGGTGAA | 8 June 2021 |
| CYP46A1 | TGTGTTTGGTGAGAGACTCTTCG | GCCAGGTCTATGACTCTCCG | 14 October 2020 |
| HPRT1 | TGACACTGGCAAAACAATGCA | GGTCCTTTTCACCAGCAAGCT | 10 February 2017 |
| B2M | CTCCGTGGCCTTAGCTGTG | TTTGGAGTACGCTGGATAGCCT | 10 February 2017 |
| SDHA | TGGGAACAAGAGGGCATCTG | CCACCACTGCATCAAATTCATG | 12 May 2011 |
| ACTB | CTCCCTGGAGAAGAGCTACG | GAAGGAAGGCTGGAAGAGTG | 12 May 2011 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhan, N.; Wang, B.; Martens, N.; Liu, Y.; Zhao, S.; Voortman, G.; van Rooij, J.; Leijten, F.; Vanmierlo, T.; Kuipers, F.; et al. Identification of Side Chain Oxidized Sterols as Novel Liver X Receptor Agonists with Therapeutic Potential in the Treatment of Cardiovascular and Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 1290. https://doi.org/10.3390/ijms24021290
Zhan N, Wang B, Martens N, Liu Y, Zhao S, Voortman G, van Rooij J, Leijten F, Vanmierlo T, Kuipers F, et al. Identification of Side Chain Oxidized Sterols as Novel Liver X Receptor Agonists with Therapeutic Potential in the Treatment of Cardiovascular and Neurodegenerative Diseases. International Journal of Molecular Sciences. 2023; 24(2):1290. https://doi.org/10.3390/ijms24021290
Chicago/Turabian StyleZhan, Na, Boyang Wang, Nikita Martens, Yankai Liu, Shangge Zhao, Gardi Voortman, Jeroen van Rooij, Frank Leijten, Tim Vanmierlo, Folkert Kuipers, and et al. 2023. "Identification of Side Chain Oxidized Sterols as Novel Liver X Receptor Agonists with Therapeutic Potential in the Treatment of Cardiovascular and Neurodegenerative Diseases" International Journal of Molecular Sciences 24, no. 2: 1290. https://doi.org/10.3390/ijms24021290
APA StyleZhan, N., Wang, B., Martens, N., Liu, Y., Zhao, S., Voortman, G., van Rooij, J., Leijten, F., Vanmierlo, T., Kuipers, F., Jonker, J. W., Bloks, V. W., Lütjohann, D., Palumbo, M., Zimetti, F., Adorni, M. P., Liu, H., & Mulder, M. T. (2023). Identification of Side Chain Oxidized Sterols as Novel Liver X Receptor Agonists with Therapeutic Potential in the Treatment of Cardiovascular and Neurodegenerative Diseases. International Journal of Molecular Sciences, 24(2), 1290. https://doi.org/10.3390/ijms24021290