

T Cell Receptor Sequences Amplified during Severe COVID-19 and Multisystem Inflammatory Syndrome in Children Mimic SARS-CoV-2, Its Bacterial Co-Infections and Host Autoantigens


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
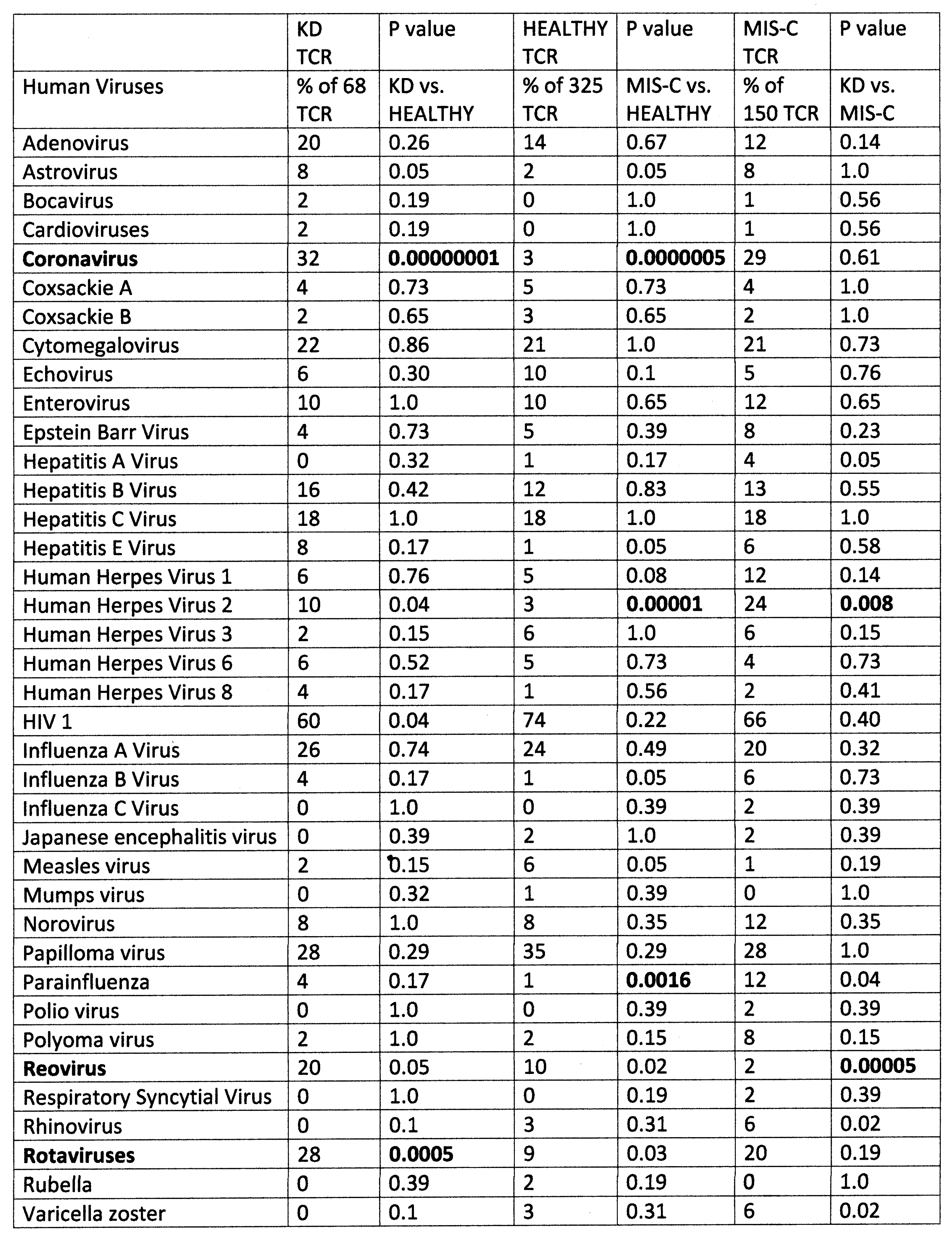
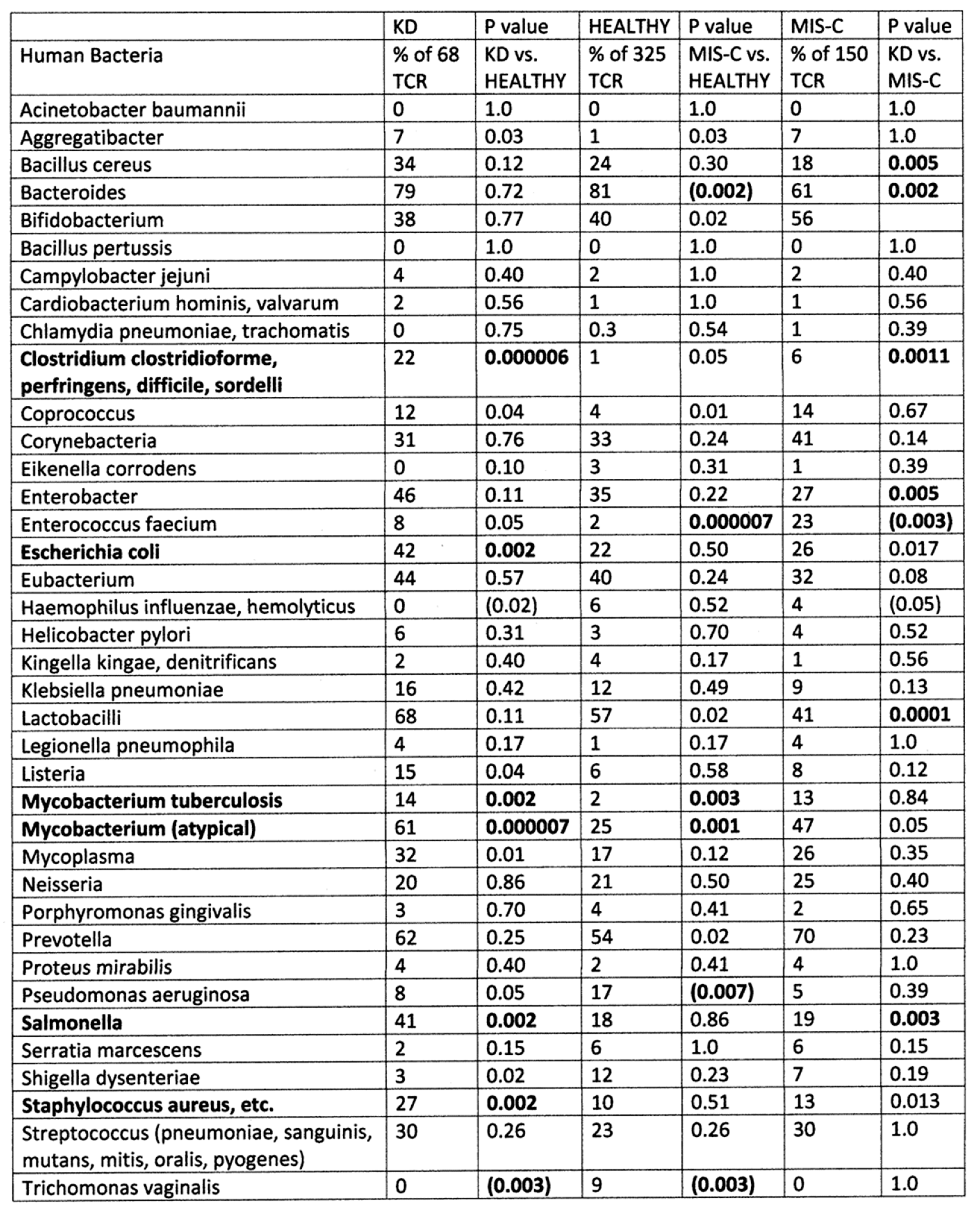
2. Results
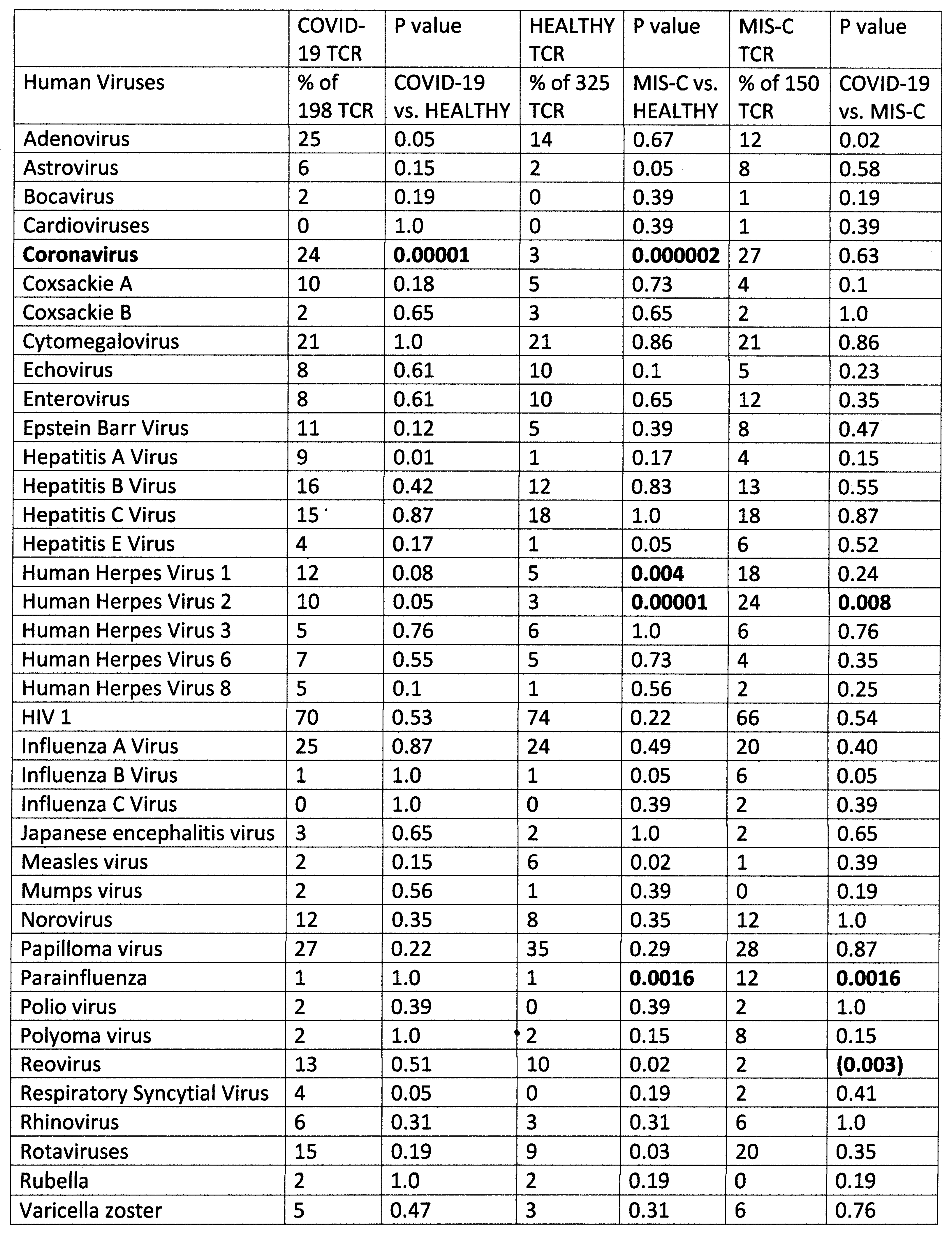
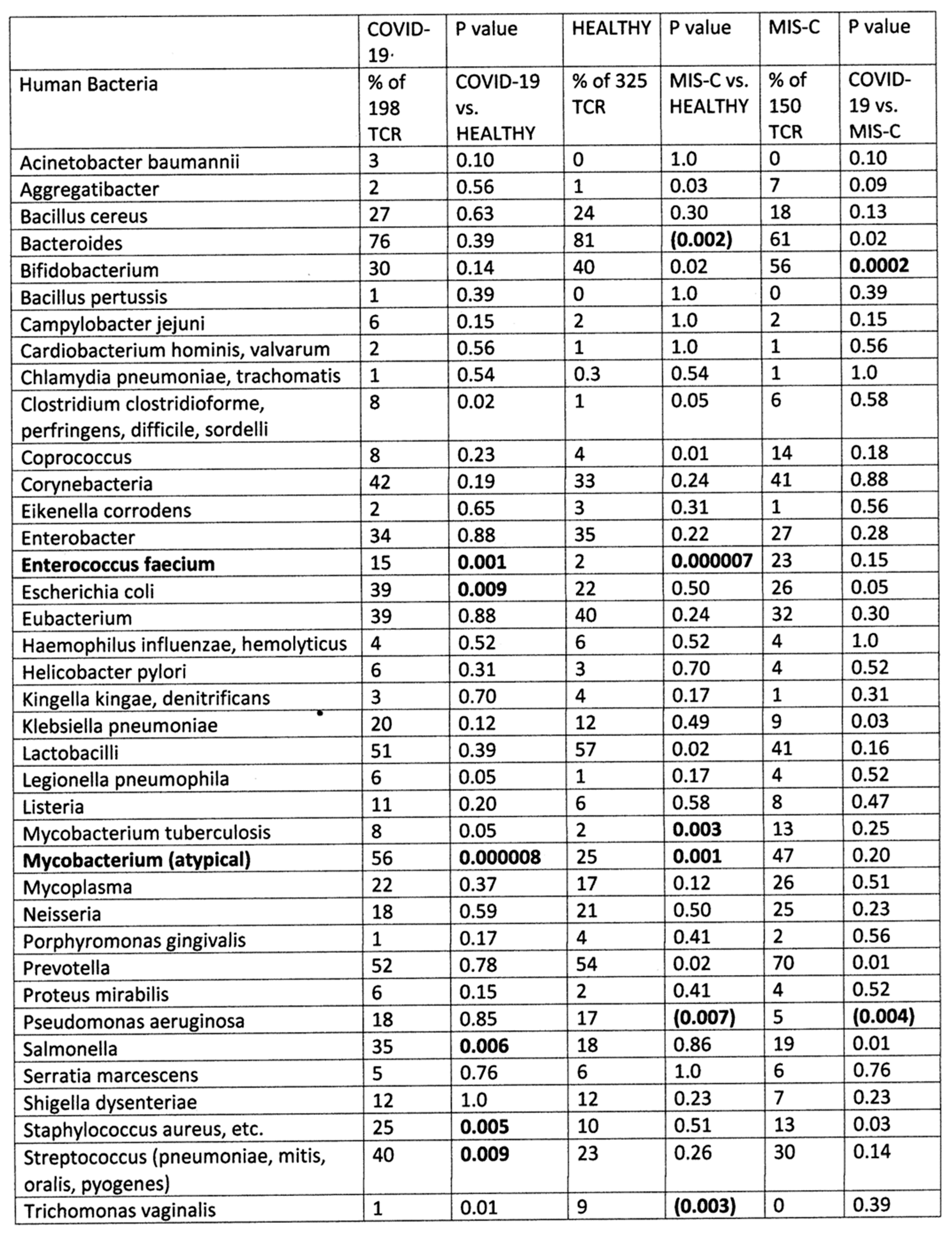
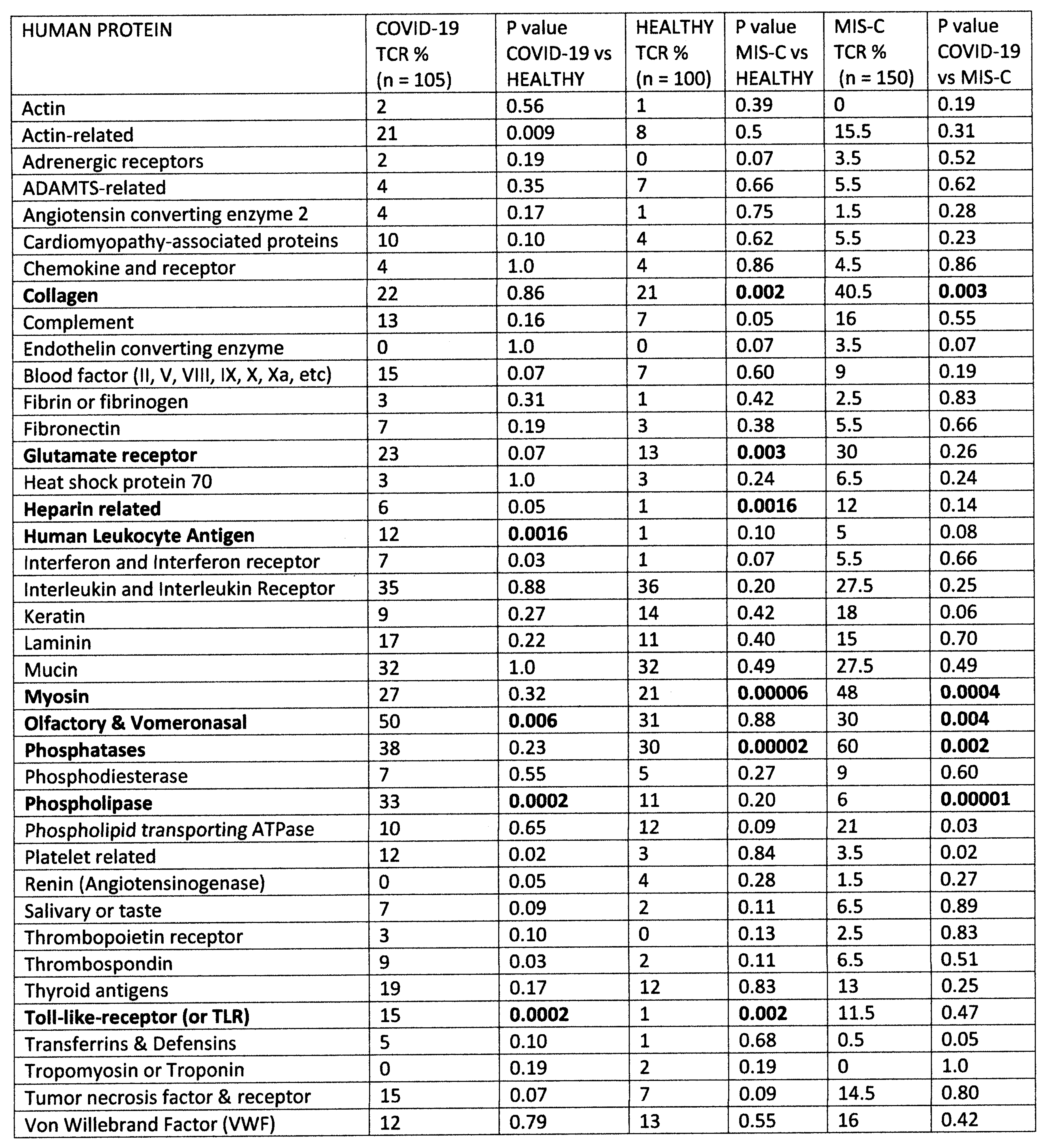
2.1. Statistical Analysis of COVID-19 TCR Sequence Similarity to Microbial Sequences
2.2. Analyzing TCR Sets from Individual Patients
2.3. Comparing TCR Mimicry Distributions in MIS-C and KD Patients
3. Discussion
3.1. Summary of Results
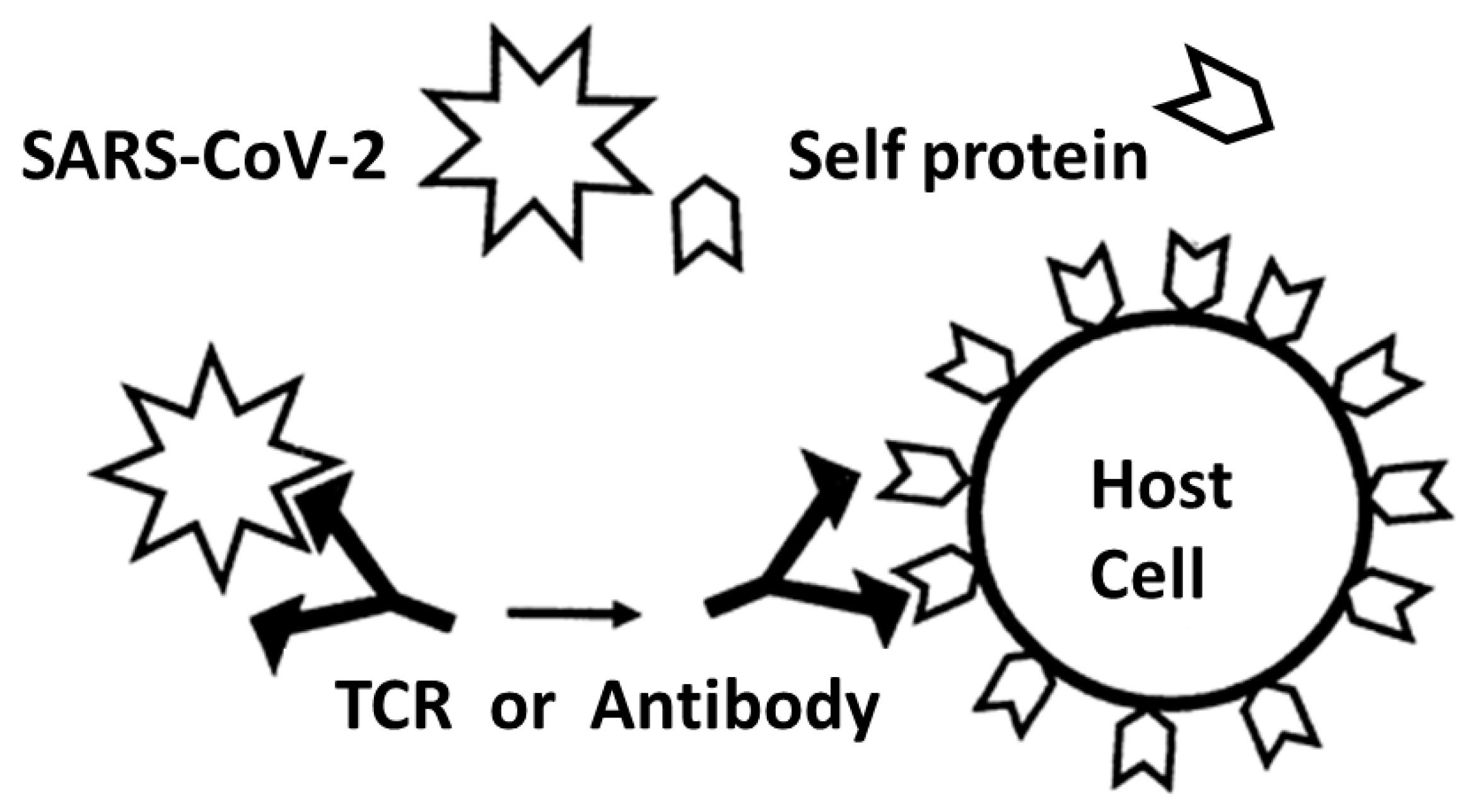
3.2. Explaining the TCR Mimicry of Pathogen and Host Antigens
3.3. Further Tests of the Theories
3.4. TCR Sequences as Clues to the Causes of Autoimmune Diseases and Their Specific Treatment
3.5. Implications for the Prevention of COVID-19-Associated Autoimmune Syndromes
3.6. Limitations of the Study
4. Materials and Methods
4.1. Similarity Searches
4.2. TCR Sources
4.3. Statistics
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
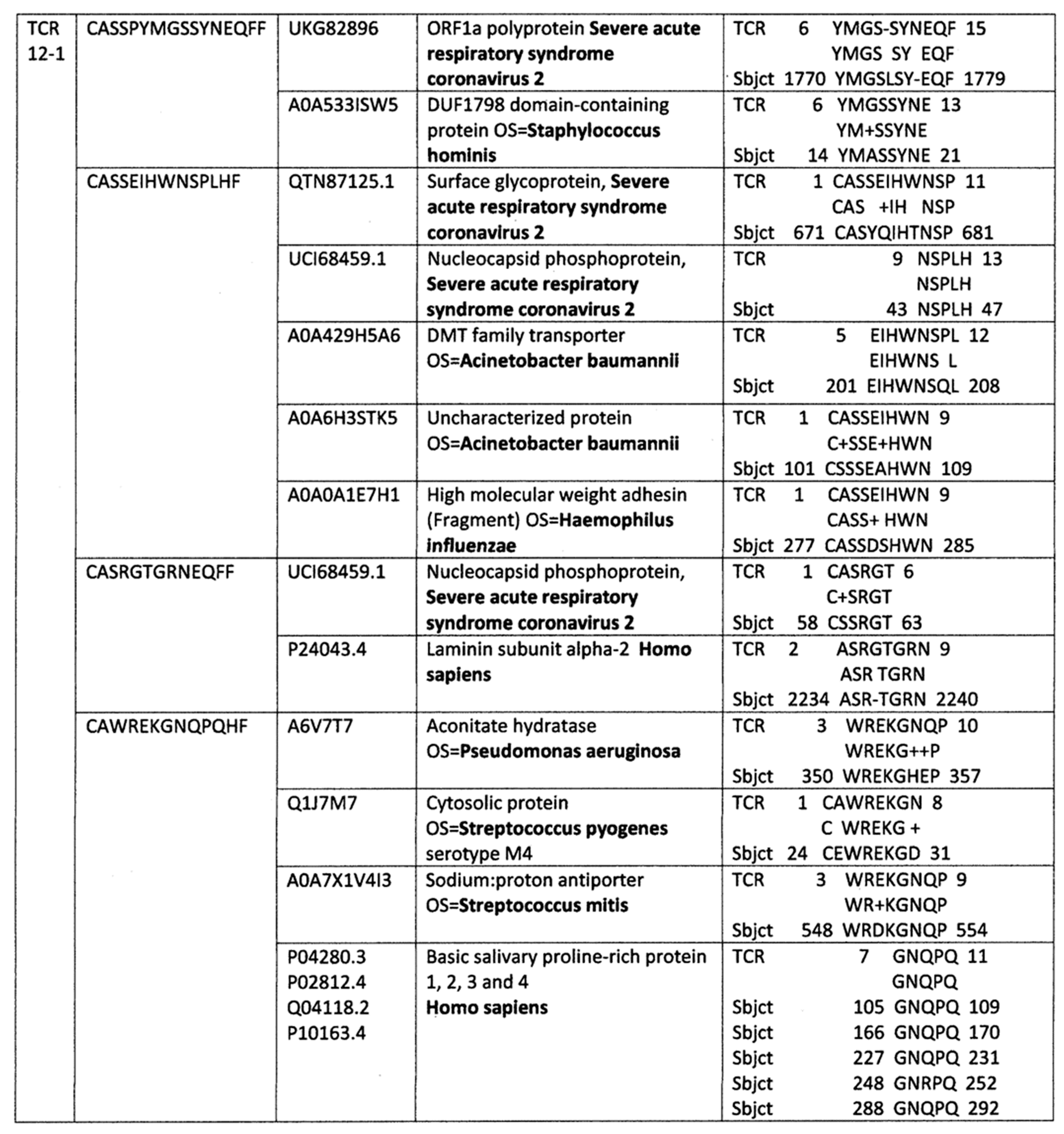
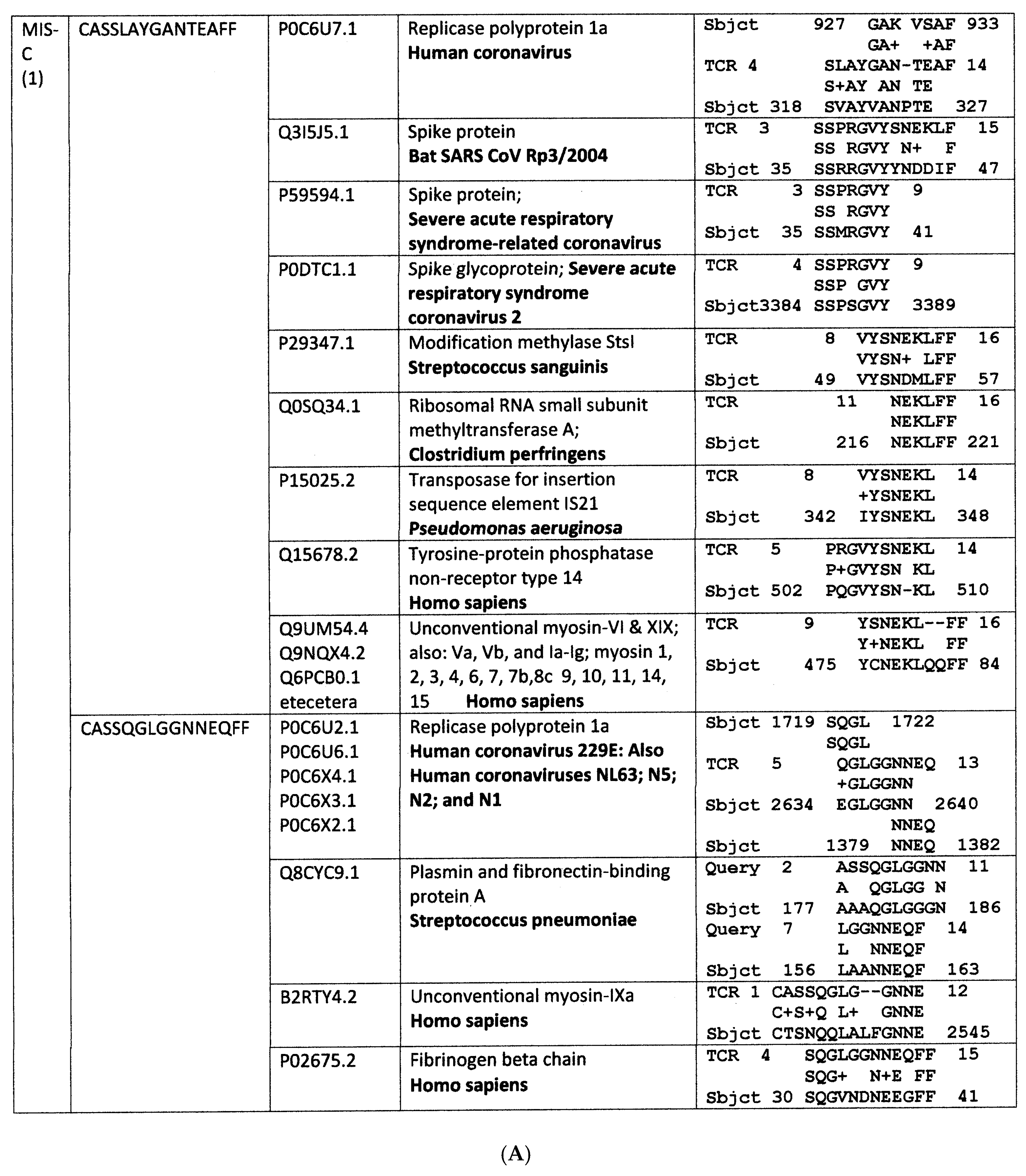
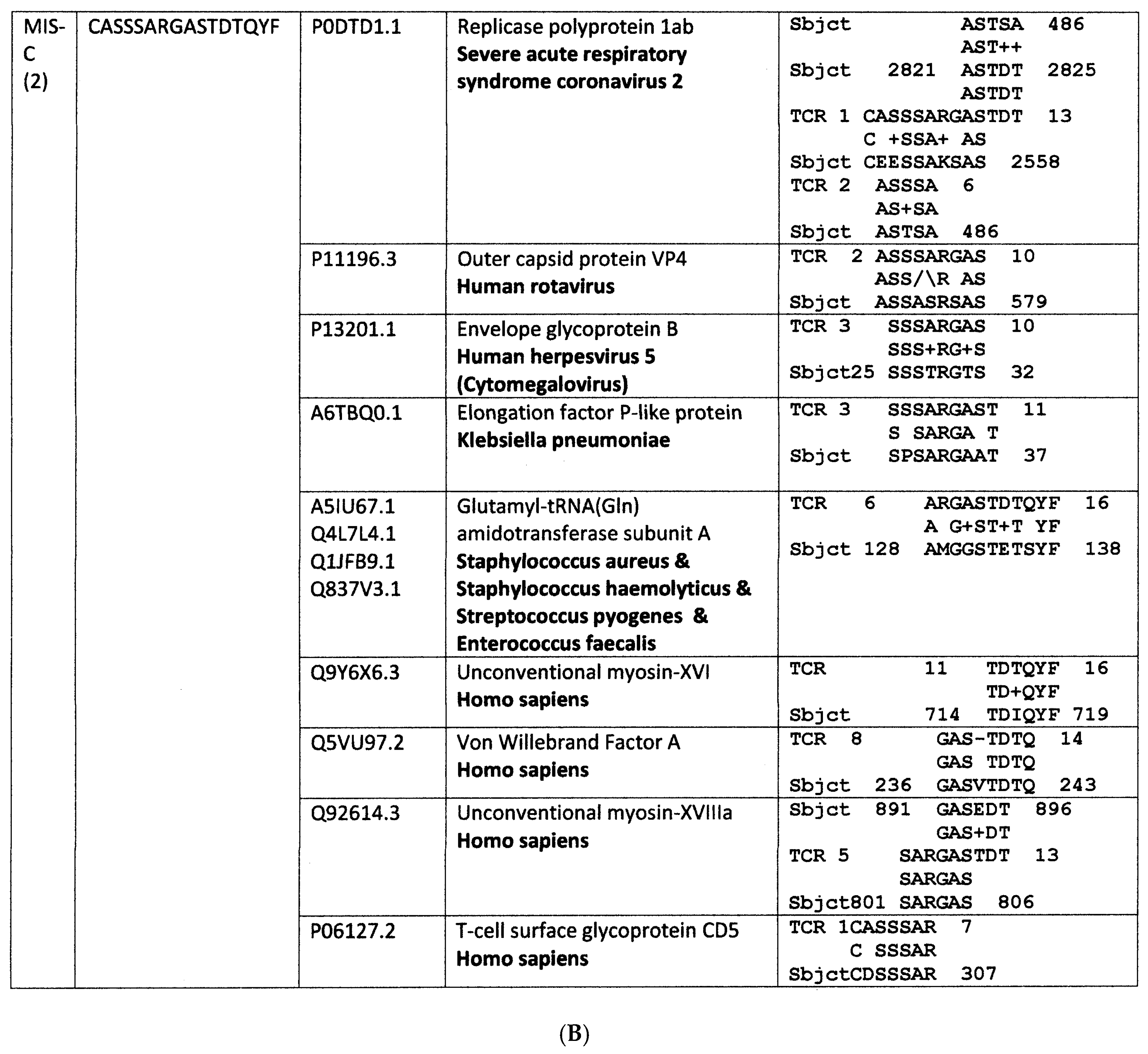
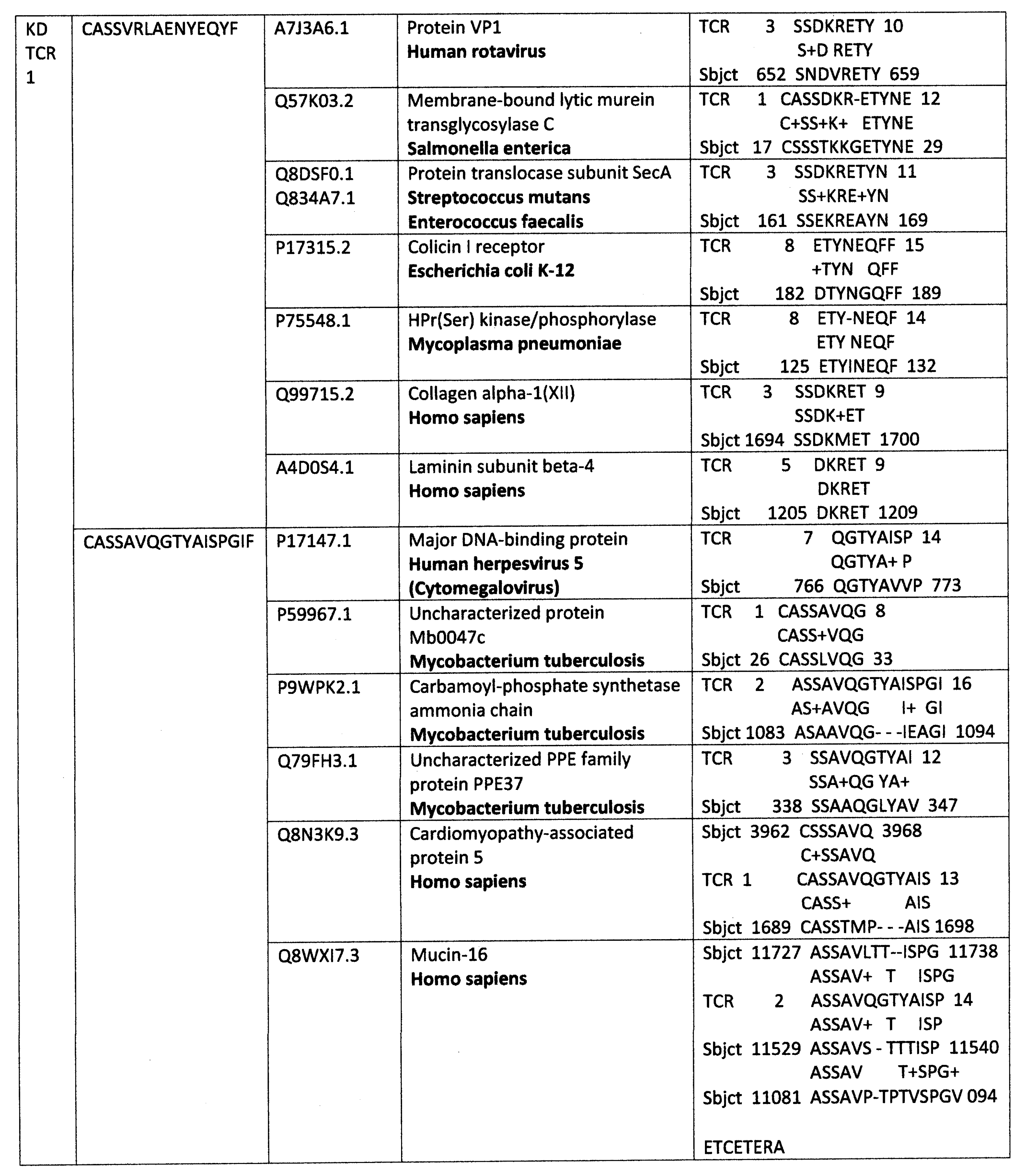
Appendix A. Detailed Analyses Demonstrating Mimicry between TCR Sequences from Three Individual Surviving COVID-19 Patients, 14-1, 29-1 and 32-1 from [74], and Viral, Bacterial and Human Proteins








Appendix B. Detailed Analyses Demonstrating Mimicry between TCR Sequences from Two Individuals Surviving Kawasaki Disease from [79], and Viral, Bacterial and Human Proteins








References
- Wucherpfennig, K.W.; Hafler, D.A. A review of T-cell receptors in multiple sclerosis: Clonal expansion and persistence of human T-cells specific for an immunodominant myelin basic protein peptide. Ann. N. Y. Acad. Sci. 1995, 756, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Ernst, N.; Iglesias, A.; Dornmair, K.; Wekerle, H.; Hohlfeld, R. T cell receptor repertoire in polymyositis: Clonal expansion of autoaggressive CD8+ T cells. J. Exp. Med. 1995, 181, 1863–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Offner, H.; Vandenbark, A.A. T cell receptor V genes in multiple sclerosis: Increased use of TCRAV8 and TCRBV5 in MBP-specific clones. Int. Rev. Immunol. 1999, 18, 9–36. [Google Scholar] [CrossRef] [PubMed]
- Frimpong, A.; Ofori, M.F.; Degoot, A.M.; Kusi, K.A.; Gershom, B.; Quartey, J.; Kyei-Baafour, E.; Nguyen, N.; Ndifon, W. Perturbations in the T cell receptor β repertoire during malaria infection in children: A preliminary study. Front. Immunol. 2022, 13, 971392. [Google Scholar] [CrossRef] [PubMed]
- Plasilova, M.; Risitano, A.; Maciejewski, J.P. Application of the molecular analysis of the T-cell receptor repertoire in the study of immune-mediated hematologic diseases. Hematology 2003, 8, 173–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codina-Busqueta, E.; Scholz, E.; Muñoz-Torres, P.M.; Roura-Mir, C.; Costa, M.; Xufré, C.; Planas, R.; Vives-Pi, M.; Jaraquemada, D.; Martí, M. TCR bias of in vivo expanded T cells in pancreatic islets and spleen at the onset in human type 1 diabetes. J. Immunol. 2011, 186, 3787–3797. [Google Scholar] [CrossRef] [Green Version]
- Risitano, A.M.; Maciejewski, J.P.; Green, S.; Plasilova, M.; Zeng, W.; Young, N.S. In-vivo dominant immune responses in aplastic anaemia: Molecular tracking of putatively pathogenetic T-cell clones by TCR beta-CDR3 sequencing. Lancet 2004, 364, 355–364. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S. Autoimmunity and the microbiome: T-cell receptor mimicry of "self" and microbial antigens mediates self tolerance in holobionts. BioEssays 2016, 38, 1068–1083. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Autoreactive T-cell receptor (Vbeta/D/Jbeta) sequences in diabetes are homologous to insulin, glucagon, the insulin receptor, and the glucagon receptor. J. Mol. Recognit. 2009, 22, 177–187. [Google Scholar] [CrossRef]
- Zhang, P.; Minardi, L.M.; Kuenstner, J.T.; Zekan, S.M.; Kruzelock, R. Anti-microbial Antibodies, Host Immunity, and Autoimmune Disease. Front. Med. 2018, 5, 153. [Google Scholar] [CrossRef]
- Koutsoumpas, A.; Polymeros, D.; Tsiamoulos, Z.; Smyk, D.; Karamanolis, G.; Triantafyllou, K.; Rigopoulou, E.I.; Forbes, A.; Vergani, D.; Bogdanos, D.P.; et al. Peculiar antibody reactivity to human connexin 37 and its microbial mimics in patients with Crohn’s disease. J. Crohns Colitis 2011, 5, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polymeros, D.; Bogdanos, D.P.; Day, R.; Arioli, D.; Vergani, D.; Forbes, A. Does cross-reactivity between mycobacterium avium paratuberculosis and human intestinal antigens characterize Crohn’s disease? Gastroenterology 2006, 131, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Lake, D.F.; Schluter, S.F.; Wang, E.; Bernstein, R.M.; Edmundson, A.B.; Marchalonis, J.J. Autoantibodies to the alpha/beta T-cell receptors in human immunodeficiency virus infection: Dysregulation and mimicry. Proc. Natl. Acad. Sci. USA 1994, 91, 10849–10853. [Google Scholar] [CrossRef] [Green Version]
- Marchalonis, J.J.; Kaymaz, H.; Schluter, S.F.; Yocum, D.E. Naturally occurring human autoantibodies to defined T-cell receptor and light chain peptides. Adv. Exp. Med. Biol. 1994, 347, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Silvestris, F.; Williams, R.C., Jr.; Dammacco, F. Autoreactivity in HIV-1 infection: The role of molecular mimicry. Clin. Immunol. Immunopathol. 1995, 75, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.S.; DeWitt, S.H. Semen alloantigens and lymphocytotoxic antibodies in AIDS and ICL. Genetica 1995, 95, 133–156. [Google Scholar] [CrossRef]
- Talotta, R.; Robertson, E. Autoimmunity as the comet tail of COVID-19 pandemic. World J. Clin. Cases 2020, 8, 3621–3644. [Google Scholar] [CrossRef]
- Moise, L.; Beseme, S.; Tassone, R.; Liu, R.; Kibria, F.; Terry, F.; Martin, W.; De Groot, A.S. T cell epitope redundancy: Cross-conservation of the TCR face between pathogens and self and its implications for vaccines and autoimmunity. Expert Rev. Vaccines 2016, 15, 607–617. [Google Scholar] [CrossRef] [Green Version]
- Moise, L.; Terry, F.; Gutierrez, A.H.; Tassone, R.; Losikoff, P.; Gregory, S.H.; Bailey-Kellogg, C.; Martin, W.D.; De Groot, A.S. Smarter vaccine design will circumvent regulatory T cell-mediated evasion in chronic HIV and HCV infection. Front. Microbiol. 2016, 5, 502. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S. Antigenic complementarity among AIDS-associated infectious agents and molecular mimicry of lymphocyte proteins as inducers of lymphocytotoxic antibodies and circulating immune complexes. J. Clin. Virol. 2004, 31 (Suppl. 1), S16–S25. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Human Immunodeficiency Virus Proteins Mimic Human T Cell Receptors Inducing Cross-Reactive Antibodies. Int. J. Mol. Sci. 2017, 18, 2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotzny, F.; Filgueiras, I.S.; Kedor, C.; Freitag, H.; Wittke, K.; Bauer, S.; Sepúlveda, N.; Mathias da Fonseca, D.L.; Baiocchi, G.C.; Marques, A.H.C.; et al. Dysregulated autoantibodies targeting vaso- and immunoregulatory receptors in Post COVID Syndrome correlate with symptom severity. Front. Immunol. 2022, 13, 981532. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Ampudia, Y.; Monsalve, D.M.; Rojas, M.; Rodríguez, Y.; Zapata, E.; Ramírez-Santana, C.; Anaya, J.M. Persistent Autoimmune Activation and Proinflammatory State in Post-Coronavirus Disease 2019 Syndrome. J. Infect. Dis. 2022, 225, 2155–2162. [Google Scholar] [CrossRef]
- Rojas, M.; Rodríguez, Y.; Acosta-Ampudia, Y.; Monsalve, D.M.; Zhu, C.; Li, Q.Z.; Ramírez-Santana, C.; Anaya, J.M. Autoimmunity is a hallmark of post-COVID syndrome. J. Transl. Med. 2022, 20, 129. [Google Scholar] [CrossRef]
- Sacchi, M.C.; Tamiazzo, S.; Stobbione, P.; Agatea, L.; De Gaspari, P.; Stecca, A.; Lauritano, E.C.; Roveta, A.; Tozzoli, R.; Guaschino, R.; et al. SARS-CoV-2 infection as a trigger of autoimmune response. Clin. Transl. Sci. 2021, 14, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Yong, S.J. Long COVID or post-COVID-19 syndrome: Putative pathophysiology, risk factors, and treatments. Infect. Dis. 2021, 53, 737–754. [Google Scholar] [CrossRef]
- Castanares-Zapatero, D.; Chalon, P.; Kohn, L.; Dauvrin, M.; Detollenaere, J.; de Noordhout, C.M.; Primus-de Jong, C.; Cleemput, I.; Van den Heede, K. Pathophysiology and mechanism of long COVID: A comprehensive review. Ann. Med. 2022, 54, 1473–1487. [Google Scholar] [CrossRef]
- Global Burden of Disease Long COVID Collaborators; Hanson, S.W.; Abbafati, C.; Aerts, J.G.; Al-Aly, Z.; Ashbaugh, C.; Ballouz, T.; Blyuss, O.; Bobkova, P.; Bonsel, G.; et al. Estimated Global Proportions of Individuals With Persistent Fatigue, Cognitive, and Respiratory Symptom Clusters Following Symptomatic COVID-19 in 2020 and 2021. JAMA 2022, 328, 1604–1615. [Google Scholar] [CrossRef]
- Kruger, A.; Vlok, M.; Turner, S.; Venter, C.; Laubscher, G.J.; Kell, D.B.; Pretorius, E. Proteomics of fibrin amyloid microclots in long COVID/post-acute sequelae of COVID-19 (PASC) shows many entrapped pro-inflammatory molecules that may also contribute to a failed fibrinolytic system. Cardiovasc. Diabetol. 2022, 21, 190. [Google Scholar] [CrossRef]
- Kell, D.B.; Laubscher, G.J.; Pretorius, E. A central role for amyloid fibrin microclots in long COVID/PASC: Origins and therapeutic implications. Biochem. J. 2022, 479, 537–559. [Google Scholar] [CrossRef]
- Pretorius, E.; Vlok, M.; Venter, C.; Bezuidenhout, J.A.; Laubscher, G.J.; Steenkamp, J.; Kell, D.B. Persistent clotting protein pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc. Diabetol. 2021, 20, 172. [Google Scholar] [CrossRef] [PubMed]
- Di Gennaro, L.; Valentini, P.; Sorrentino, S.; Ferretti, M.A.; De Candia, E.; Basso, M.; Lancellotti, S.; De Cristofaro, R.; De Rose, C.; Mariani, F.; et al. Extended coagulation profile of children with Long Covid: A prospective study. Sci. Rep. 2022, 12, 18392. [Google Scholar] [CrossRef] [PubMed]
- Connors, J.M.; Levy, J.H. COVID-19 and its implications for thrombosis and anticoagulation. Blood 2020, 135, 2033–2040. [Google Scholar] [CrossRef] [PubMed]
- Lund, L.C.; Hallas, J.; Nielsen, H.; Koch, A.; Mogensen, S.H.; Brun, N.C.; Christiansen, C.F.; Thomsen, R.W.; Pottegård, A. Post-acute effects of SARS-CoV-2 infection in individuals not requiring hospital admission: A Danish population-based cohort study. Lancet Infect. Dis. 2021, 21, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Taquet, M.; Husain, M.; Geddes, J.R.; Luciano, S.; Harrison, P.J. Cerebral venous thrombosis and portal vein thrombosis: A retrospective cohort study of 537,913 COVID-19 cases. EClinicalMedicine 2021, 39, 101061. [Google Scholar] [CrossRef] [PubMed]
- Abou-Ismail, M.Y.; Diamond, A.; Kapoor, S.; Arafah, Y.; Nayak, L. The hypercoagulable state in COVID-19: Incidence, pathophysiology, and management. Thromb. Res. 2020, 194, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Taha, M.; Samavati, L. Antiphospholipid antibodies in COVID-19: A meta-analysis and systematic review. RMD Open 2021, 7, e001580. [Google Scholar] [CrossRef]
- Najim, M.; Rahhal, A.; Khir, F.; Aljundi, A.H.; Yousef, S.A.; Ibrahim, F.; Amer, A.; Mohamed, A.S.; Saleh, S.; Alfaridi, D.; et al. Prevalence and clinical significance of antiphospholipid antibodies in patients with coronavirus disease 2019 admitted to intensive care units: A prospective observational study. Rheumatol. Int. 2021, 41, 1243–1252. [Google Scholar] [CrossRef]
- Hendrickson, K.W.; Knox, D.B.; Bledsoe, J.R.; Peltan, I.D.; Jacobs, J.R.; Lloyd, J.F.; Dean, N.C.; Woller, S.C.; Brown, S.M. Comparative frequency of venous thromboembolism in patients admitted to the hospital with SARS-CoV-2 infection vs. community-acquired pneumonia. Ann. Am. Thorac. Soc. 2022, 19, 1233–1235. [Google Scholar] [CrossRef]
- Llitjos, J.F.; Leclerc, M.; Chochois, C.; Monsallier, J.M.; Ramakers, M.; Auvray, M.; Merouani, K. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J. Thromb. Haemost. 2020, 18, 1743–1746. [Google Scholar] [CrossRef]
- Bisaccia, G.; Ricci, F.; Recce, V.; Serio, A.; Iannetti, G.; Chahal, A.A.; Ståhlberg, M.; Khanji, M.Y.; Fedorowski, A.; Gallina, S. Post-Acute Sequelae of COVID-19 and Cardiovascular Autonomic Dysfunction: What Do We Know? J. Cardiovasc. Dev. Dis. 2021, 8, 156. [Google Scholar] [CrossRef] [PubMed]
- Lui, D.T.W.; Lee, C.H.; Chow, W.S.; Lee, A.C.H.; Tam, A.R.; Fong, C.H.Y.; Law, C.Y.; Leung, E.K.H.; To, K.K.W.; Tan, K.C.B.; et al. Insights from a Prospective Follow-up of Thyroid Function and Autoimmunity among COVID-19 Survivors. Endocrinol. Metab. 2021, 36, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Bertin, D.; Kaphan, E.; Weber, S.; Babacci, B.; Arcani, R.; Faucher, B.; Ménard, A.; Brodovitch, A.; Mege, J.L.; Bardin, N. Persistent IgG anticardiolipin autoantibodies are associated with post-COVID syndrome. Int. J. Infect. Dis. 2021, 113, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Hassani, N.S.; Talakoob, H.; Karim, H.; Bazargany, M.H.M.; Rastad, H. Cardiac Magnetic Resonance Imaging Findings in 2954 COVID-19 Adult Survivors: A Comprehensive Systematic Review. J. Magn. Reson. Imaging 2022, 55, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Lee, G.M.; Razzaghi, H.; Lorman, V.; Mejias, A.; Pajor, N.M.; Thacker, D.; Webb, R.; Dickinson, K.; Bailey, L.C.; et al. Clinical Features and Burden of Postacute Sequelae of SARS-CoV-2 Infection in Children and Adolescents. JAMA Pediatr. 2022, 176, 1000–1009. [Google Scholar] [CrossRef]
- Porritt, R.A.; Binek, A.; Paschold, L.; Rivas, M.N.; McArdle, A.; Yonker, L.M.; Alter, G.; Chandnani, H.K.; Lopez, M.; Fasano, A.; et al. The autoimmune signature of hyperinflammatory multisystem inflammatory syndrome in children. J. Clin. Investig. 2021, 131, e151520. [Google Scholar] [CrossRef]
- Ramaswamy, A.; Brodsky, N.N.; Sumida, T.S.; Comi, M.; Asashima, H.; Hoehn, K.B.; Li, N.; Liu, Y.; Shah, A.; Ravindra, N.G.; et al. Immune dysregulation and autoreactivity correlate with disease severity in SARS-CoV-2-associated multisystem inflammatory syndrome in children. Immunity 2021, 54, 1083–1095.e7. [Google Scholar] [CrossRef]
- Vella, L.A.; Giles, J.R.; Baxter, A.E.; Oldridge, D.A.; Diorio, C.; Kuri-Cervantes, L.; Alanio, C.; Pampena, M.B.; Wu, J.E.; Chen, Z.; et al. Deep immune profiling of MIS-C demonstrates marked but transient immune activation compared to adult and pediatric COVID-19. Sci. Immunol. 2021, 6, eabf7570. [Google Scholar] [CrossRef]
- Bizjak, M.; Emeršič, N.; Zajc Avramovič, M.; Barbone, F.; Ronchese, F.; Della Paolera, S.; Conversano, E.; Amoroso, S.; Vidoni, M.; Vesel Tajnšek, T.; et al. High incidence of multisystem inflammatory syndrome and other autoimmune diseases after SARS-CoV-2 infection compared to COVID-19 vaccination in children and adolescents in south central Europe. Clin. Exp. Rheumatol. 2022. [Google Scholar] [CrossRef]
- Chen, M.R.; Kuo, H.C.; Lee, Y.J.; Chi, H.; Li, S.C.; Lee, H.C.; Yang, K.D. Phenotype, Susceptibility, Autoimmunity, and Immunotherapy Between Kawasaki Disease and Coronavirus Disease-19 Associated Multisystem Inflammatory Syndrome in Children. Front. Immunol. 2021, 12, 632890. [Google Scholar] [CrossRef]
- Wang, Y.; Li, T. Advances in understanding Kawasaki disease-related immuno-inflammatory response and vascular endothelial dysfunction. Pediatr. Investig. 2022, 6, 271–279. [Google Scholar] [CrossRef]
- Mahajan, A.; Yadav, S.; Maheshwari, A.; Mahto, D.; Divya, K.; Ackshaya, R.; Meena, H.; Shakya, S.; Kumar, V. Profile of children with Kawasaki disease associated with tropical infections. Indian J. Pediatr. 2022, 89, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Ae, R.; Shibata, Y.; Kosami, K.; Nakamura, Y.; Hamada, H. Kawasaki Disease and Pediatric Infectious Diseases During the Coronavirus Disease 2019 Pandemic. J. Pediatr. 2021, 239, 50–58.e2. [Google Scholar] [CrossRef] [PubMed]
- Kamidani, S.; Panagiotakopoulos, L.; Licata, C.; Daley, M.F.; Yih, W.K.; Zerbo, O.; Tseng, H.F.; DeSilva, M.B.; Nelson, J.C.; Groom, H.C.; et al. Kawasaki Disease Following the 13-valent Pneumococcal Conjugate Vaccine and Rotavirus Vaccines. Pediatrics 2022, 150, e2022058789. [Google Scholar] [CrossRef] [PubMed]
- Yung, C.F.; Ma, X.; Cheung, Y.B.; Oh, B.K.; Soh, S.; Thoon, K.C. Kawasaki Disease following administration of 13-valent pneumococcal conjugate vaccine in young children. Sci. Rep. 2019, 9, 14705. [Google Scholar] [CrossRef] [Green Version]
- Newhouse, C.N.; Finn, L.; Gragnani, C.M.; Hathaway, S.; Nunez, D.; Malenfant, J.; Fernandes, P.; Kim, M.; Terashita, D.; Balter, S. Epidemiology of Exposures, Preceding Illness and Testing History in Children With Multisystem Inflammatory Syndrome in Children in the First 18 Months of the COVID-19 Pandemic, Los Angeles County, California. Pediatr. Infect. Dis. J. 2022, 41, e453–e455. [Google Scholar] [CrossRef]
- Dufort, E.M.; Koumans, E.H.; Chow, E.J.; Rosenthal, E.M.; Muse, A.; Rowlands, J.; Barranco, M.A.; Maxted, A.M.; Rosenberg, E.S.; Easton, D.; et al. Multisystem Inflammatory Syndrome in Children in New York State. N. Engl. J. Med. 2020, 383, 347–358. [Google Scholar] [CrossRef]
- Fattorini, L.; Creti, R.; Palma, C.; Pantosti, A.; Unit of Antibiotic Resistance and Special Pathogens; Unit of Antibiotic Resistance and Special Pathogens of the Department of Infectious Diseases, Istituto Superiore di Sanità, Rome. Bacterial coinfections in COVID-19: An underestimated adversary. Ann. Ist. Super Sanita 2020, 56, 359–364. [Google Scholar] [CrossRef]
- Rawson, T.M.; Moore, L.S.P.; Zhu, N.; Ranganathan, N.; Skolimowska, K.; Gilchrist, M.; Satta, G.; Cooke, G.; Holmes, A. Bacterial and Fungal Coinfection in Individuals With Coronavirus: A Rapid Review To Support COVID-19 Antimicrobial Prescribing. Clin. Infect. Dis. 2020, 71, 2459–2468. [Google Scholar] [CrossRef]
- Sreenath, K.; Batra, P.; Vinayaraj, E.V.; Bhatia, R.; SaiKiran, K.; Singh, V.; Singh, S.; Verma, N.; Singh, U.B.; Mohan, A.; et al. Coinfections with Other Respiratory Pathogens among Patients with COVID-19. Microbiol. Spectr. 2021, 9, e0016321. [Google Scholar] [CrossRef]
- Lai, C.C.; Wang, C.Y.; Hsueh, P.R. Co-infections among patients with COVID-19: The need for combination therapy with non-anti-SARS-CoV-2 agents? J. Microbiol. Immunol. Infect 2020, 53, 505–512. [Google Scholar] [CrossRef]
- Iversen, K.; Ihlemann, N.; Gill, S.U.; Madsen, T.; Elming, H.; Jensen, K.T.; Bruun, N.E.; Høfsten, D.E.; Fursted, K.; Christensen, J.J.; et al. Partial Oral versus Intravenous Antibiotic Treatment of Endocarditis. N. Engl. J. Med. 2019, 380, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Martínez, A.; Fernández-Cruz, A.; Domínguez, F.; Forteza, A.; Cobo, M.; Sánchez-Romero, I.; Asensio, A. Hospital-acquired infective endocarditis during Covid-19 pandemic. Infect. Prev. Pract. 2020, 2, 100080. [Google Scholar] [CrossRef] [PubMed]
- Quintero-Martinez, J.A.; Hindy, J.R.; Mahmood, M.; Gerberi, D.J.; DeSimone, D.C.; Baddour, L.M. A clinical profile of infective endocarditis in patients with recent COVID-19: A systematic review. Am. J. Med. Sci. 2022, 364, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Righi, E.; Lambertenghi, L.; Gorska, A.; Sciammarella, C.; Ivaldi, F.; Mirandola, M.; Sartor, A.; Tacconelli, E. Impact of COVID-19 and Antibiotic Treatments on Gut Microbiome: A Role for Enterococcus spp. Biomedicines 2022, 10, 2786. [Google Scholar] [CrossRef]
- Giacobbe, D.R.; Labate, L.; Tutino, S.; Baldi, F.; Russo, C.; Robba, C.; Ball, L.; Dettori, S.; Marchese, A.; Dentone, C.; et al. Enterococcal bloodstream infections in critically ill patients with COVID-19: A case series. Ann. Med. 2021, 53, 1779–1786. [Google Scholar] [CrossRef]
- Gaibani, P.; D’Amico, F.; Bartoletti, M.; Lombardo, D.; Rampelli, S.; Fornaro, G.; Coladonato, S.; Siniscalchi, A.; Re, M.C.; Viale, P.; et al. The Gut Microbiota of Critically Ill Patients With COVID-19. Front. Cell. Infect. Microbiol. 2021, 11, 670424. [Google Scholar] [CrossRef]
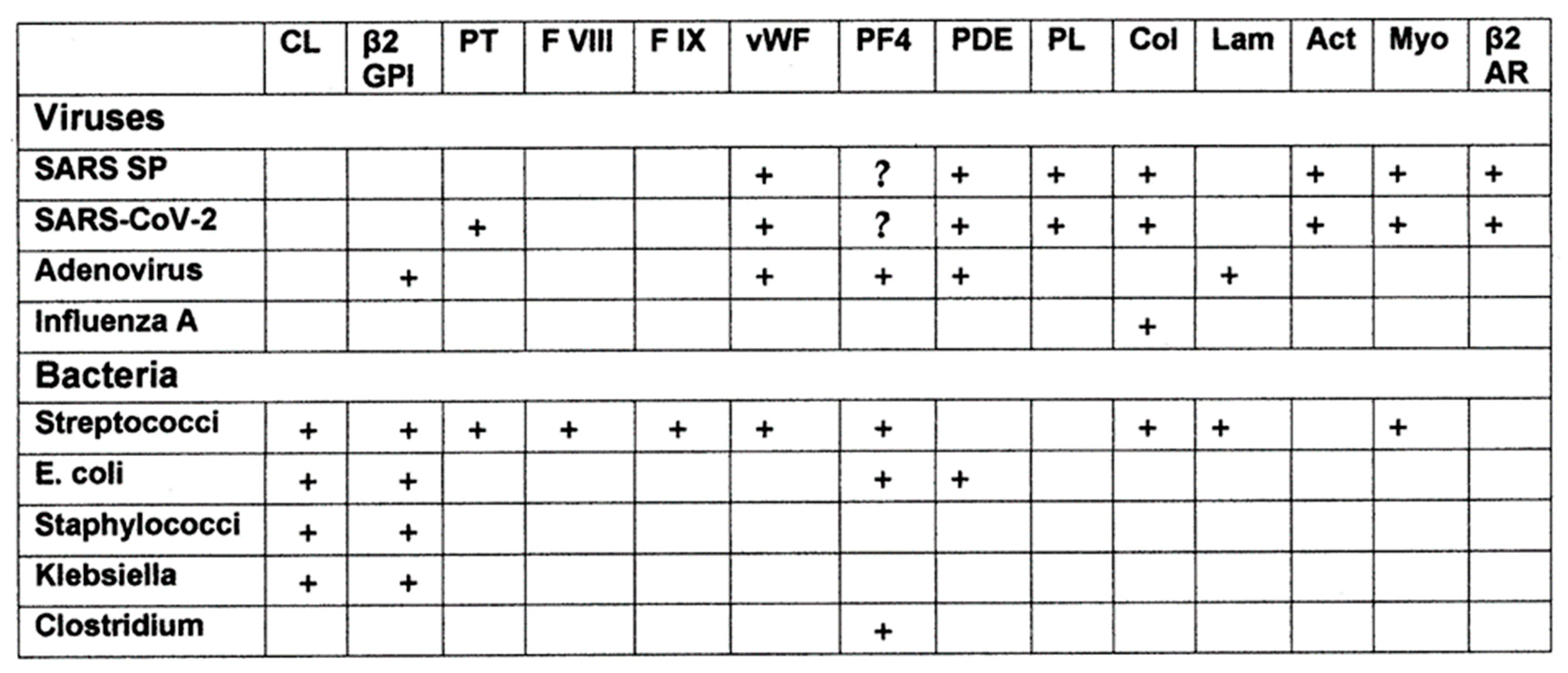
- Kanduc, D. Thromboses and hemostasis disorders associated with COVID-19: The possible causal role of cross-reactivity and immunological imprinting. Glob. Med. Genet. 2021, 8, 162–170. [Google Scholar] [CrossRef]
- Root-Bernstein, R. COVID-19 coagulopathies: Human blood proteins mimic SARS-CoV-2 virus, vaccine proteins and bacterial co-infections inducing autoimmunity: Combinations of bacteria and SARS-CoV-2 synergize to induce autoantibodies targeting cardiolipin, cardiolipin-binding proteins, platelet factor 4, prothrombin, and coagulation factors. BioEssays 2021, 43, e2100158. [Google Scholar] [CrossRef]
- Root-Bernstein, R.; Huber, J.; Ziehl, A. Complementary Sets of Autoantibodies Induced by SARS-CoV-2, Adenovirus and Bacterial Antigens Cross-React with Human Blood Protein Antigens in COVID-19 Coagulopathies. Int. J. Mol. Sci. 2022, 23, 11500. [Google Scholar] [CrossRef]
- Cunningham, M.W.; McCormack, J.M.; Fenderson, P.G.; Ho, M.K. Human and murine antibodies cross-reactive with streptococcal M protein and myosin recognize the sequence GLN-LYS-SER-LYS-GLN in M protein. J. Immunol. 1989, 143, 2677–2683. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.S.; Podufaly, A.; Aimone, F. Antigenic Complementarity between Influenza A Virus and Haemophilus Influenzae may Drive Lethal Co-Infection Such As That Seen In 1918-19. J. Virol. Antivir. Res 2013, 2, 1. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S. Rethinking molecular mimicry in rheumatic heart disease and autoimmune myocarditis: Laminin, collagen IV, CAR, and B1AR as initial targets of disease. Front. Pediatr. 2014, 2, 85. [Google Scholar] [CrossRef] [Green Version]
- Schultheiß, C.; Paschold, L.; Simnica, D.; Mohme, M.; Willscher, E.; von Wenserski, L.; Scholz, R.; Wieters, I.; Dahlke, C.; Tolosa, E.; et al. Next-Generation Sequencing of T and B Cell Receptor Repertoires from COVID-19 Pa-tients Showed Signatures Associated with Severity of Disease. Immunity 2020, 53, 442–455.e4. [Google Scholar] [CrossRef] [PubMed]
- Folga, B.A.; Karpenko, C.J.; Grygiel-Górniak, B. SARS-CoV-2 infection in the context of Kawasaki disease and multisystem inflammatory syndrome in children. Med. Microbiol. Immunol. 2022, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sobh, A.; Mosa, D.M.; Khaled, N.; Korkor, M.S.; Noureldin, M.A.; Eita, A.M.; Elnagdy, M.H.; El-Bayoumi, M.A. How multisystem inflammatory syndrome in children discriminated from Kawasaki disease: A differentiating score based on an inception cohort study. Clin. Rheumatol. 2022, 21, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Y.; Xu, B.W.; Du, J.B. Similarities and differences between multiple inflammatory syndrome in children associated with COVID-19 and Kawasaki disease: Clinical presentations, diagnosis, and treatment. World J. Pediatr. 2021, 17, 335–340. [Google Scholar] [CrossRef]
- Gámez-González, L.B.; Escrcega-Jurez, A.S.; Aguilar-Soto, D.E.; Rascón, M.C.; Espinosa, A.C.G.; Yamazaki-Nakashimada, M.A. Multisystem inflammatory syndrome in neonates associated with SARS-CoV-2 infection, a different entity? J. Neonatal-Perinat. Med. 2022, 1–9. [Google Scholar] [CrossRef]
- Ramaswamy, A.; Brodsky, N.N.; Sumida, T.S.; Comi, M.; Asashima, H.; Hoehn, K.B.; Li, N.; Liu, Y.; Shah, A.; Ravindra, N.G.; et al. Post-infectious inflammatory disease in MIS-C features elevated cytotoxicity signatures and autoreactivity that correlates with severity. medRxiv 2021. [Google Scholar] [CrossRef]
- Yoshioka, T.; Matsutani, T.; Iwagami, S.; Toyosaki-Maeda, T.; Yutsudo, T.; Tsuruta, Y.; Suzuki, H.; Uemura, S.; Takeuchi, T.; Koike, M.; et al. Polyclonal expansion of TCRBV2- and TCRBV6-bearing T cells in patients with Kawasaki disease. Immunology 1999, 96, 465–472. [Google Scholar] [CrossRef]
- Shanshal, M.; Ahmed, H.S. COVID-19 and Herpes Simplex Virus Infection: A Cross-Sectional Study. Cureus 2021, 13, e18022. [Google Scholar] [CrossRef]
- Katz, J.; Yue, S.; Xue, W. Herpes simplex and herpes zoster viruses in COVID-19 patients. Ir. J. Med. Sci. 2022, 191, 1093–1097. [Google Scholar] [CrossRef] [PubMed]
- Menger, J.; Apostolidou, S.; Edler, C.; Kniep, I.; Kobbe, R.; Singer, D.; Sperhake, J.P. Fatal outcome of SARS-CoV-2 infection (B1.1.7) in a 4-year-old child. Int. J. Leg. Med. 2022, 136, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Yuksel, S.; Demirkan, N.C.; Comut, E.; Yilmaz, M.; Gurses, D. Histopathological and Clinical Analysis of Skin Rashes in Children With Multisystem Inflammatory Syndrome Associated With COVID-19. Am. J. Dermatopathol. 2022, 44, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Giray, T.; Biçer, S.; Küçük, Ö.; Çöl, D.; Yalvaç, Z.; Gürol, Y.; Yilmaz, G.; Saç, A.; Mogol, Y. Four cases with Kawasaki disease and viral infection: Aetiology or association. Infez. Med. 2016, 24, 340–344. [Google Scholar] [PubMed]
- Johnson, D.; Azimi, P. Kawasaki disease associated with Klebsiella pneumoniae bacteremia and parainfluenza type 3 virus infection. Pediatr. Infect. Dis. 1985, 4, 100. [Google Scholar] [CrossRef] [PubMed]
- Schnaar, D.A.; Bell, D.M. Kawasaki syndrome in two cousins with parainfluenza virus infection. Am. J. Dis. Child. 1982, 136, 554–555. [Google Scholar] [CrossRef]
- Embil, J.A.; McFarlane, E.S.; Murphy, D.M.; Krause, V.W.; Stwart, H.B. Adenovirus type 2 isolated from a patient with fatal Kawasaki disease. Can. Med. Assoc. J. 1985, 132, 1400. [Google Scholar]
- Jaggi, P.; Kajon, A.E.; Mejias, A.; Ramilo, O.; Leber, A. Human adenovirus infection in Kawasaki disease: A confounding bystander? Clin. Infect. Dis. 2013, 56, 58–64. [Google Scholar] [CrossRef] [Green Version]
- Song, E.; Kajon, A.E.; Wang, H.; Salamon, D.; Texter, K.; Ramilo, O.; Leber, A.; Jaggi, P. Clinical and virologic characteristics may aid distinction of acute adenovirus disease from Kawasaki disease with incidental adenovirus detection. J. Pediatr. 2016, 170, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, S.R.; Anderson, M.S.; Glodé, M.P.; Robinson, C.C.; Holmes, K.V. Blinded case-control study of the relationship between human coronavirus NL63 and Kawasaki syndrome. J. Infect. Dis. 2006, 194, 1697–1701. [Google Scholar] [CrossRef]
- Shirato, K.; Imada, Y.; Kawase, M.; Nakagaki, K.; Matsuyama, S.; Taguchi, F. Possible involvement of infection with human coronavirus 229E, but not NL63, in Kawasaki disease. J. Med. Virol. 2014, 86, 2146–2153. [Google Scholar] [CrossRef]
- Esper, F.; Shapiro, E.D.; Weibel, C.; Ferguson, D.; Landry, M.L.; Kahn, J.S. Association between a novel human coronavirus and Kawasaki disease. J. Infect. Dis. 2005, 191, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Thiele, G.M.; Sakiyama, Y.; Matsumoto, S.; Purtilo, D.T. Adenovirus infection in patients with Kawasaki disease. J. Med. Virol. 1990, 32, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Lee, K.Y.; Han, J.W.; Lee, J.S.; Whang, K.T. Epstein-Barr virus antibodies in Kawasaki disease. Yonsei Med. J. 2006, 47, 475–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchette, N.J.; Melish, M.E.; Hicks, R.; Kihara, S.; Sam, E.; Ching, D. Epstein-Barr virus and other herpesvirus infections in Kawasaki syndrome. J. Infect. Dis. 1990, 161, 680–684. [Google Scholar] [CrossRef]
- Lim, J.H.; Kim, Y.K.; Min, S.H.; Kim, S.W.; Lee, Y.H.; Lee, J.M. Seasonal Trends of Viral Prevalence and Incidence of Kawasaki Disease: A Korea Public Health Data Analysis. J. Clin. Med. 2021, 10, 3301. [Google Scholar] [CrossRef]
- Kim, G.B.; Park, S.; Kwon, B.S.; Han, J.W.; Park, Y.W.; Hong, Y.M. Evaluation of the Temporal Association between Kawasaki Disease and Viral Infections in South Korea. Korean Circ. J. 2014, 44, 250–254. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, D.; Cerda, J.; Perret, C.; Borzutzky, A.; Hoyos-Bachiloglu, R. Asociación temporal entre la circulación de virus respiratorios y hospitalizaciones por enfermedad de Kawasaki [Temporal association between the circulation of respiratory viruses and hospitalizations due to Kawasaki disease]. Rev. Chil. Infectol. 2021, 38, 152–160. (In Spanish) [Google Scholar] [CrossRef]
- Kang, J.M.; Jung, J.; Kim, Y.E.; Huh, K.; Hong, J.; Kim, D.W.; Kim, M.Y.; Jung, S.Y.; Kim, J.H.; Ahn, J.G. Temporal Correlation Between Kawasaki Disease and Infectious Diseases in South Korea. JAMA Netw. Open 2022, 5, e2147363. [Google Scholar] [CrossRef]
- Mir, T.H. Thrombotic microangiopathy (aHUS/iTTP) reported so far in Covid-9 patients: The virus alone or an omnium gatherum of mechanisms and etiologies? Crit. Rev. Oncol. Hematol. 2021, 162, 103347. [Google Scholar] [CrossRef] [PubMed]
- Nussinovitch, U.; Shoenfeld, Y. The clinical and diagnostic significance of anti-myosin autoantibodies in cardiac disease. Clin. Rev. Allergy Immunol. 2013, 44, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Passariello, M.; Vetrei, C.; Amato, F.; De Lorenzo, C. Interactions of spike-RBD of SARS-CoV-2 and Platelet Factor 4: New insights in the etiopathogenesis of thrombosis. Int. J. Mol. Sci. 2021, 22, 8562. [Google Scholar] [CrossRef] [PubMed]
- Vojdani, A.; Kharrazian, D. Potential antigenic cross-reactivity between SARS-CoV-2 and human tissue with a possible link to an increase in autoimmune diseases. Clin. Immun. 2020, 217, 108480. [Google Scholar] [CrossRef]
- Vojdani, A.; Vojdani, E.; Kharrazian, D. Reaction of human monoclonal antibodies to SARS-CoV-2 proteins with tissue antigens: Implications for autoimmune diseases. Front. Immunol. 2021, 11, 617089. [Google Scholar] [CrossRef]
- Wallukat, G.; Hohberger, B.; Wenzel, K.; Fürst, J.; Schulze-Rothe, S.; Wallukat, A.; Hönicke, A.S.; Müller, J. Functional autoantibodies against G-protein coupled receptors in patients with persistent Long-COVID-19 symptoms. J. Transl. Autoimmun. 2021, 4, 100100. [Google Scholar] [CrossRef]
- Root-Bernstein, R.; Fairweather, D. Unresolved issues in theories of autoimmune disease using myocarditis as a framework. J. Theor. Biol. 2015, 375, 101–123. [Google Scholar] [CrossRef] [Green Version]
- Root-Bernstein, R.; Fairweather, D. Complexities in the relationship between infection and autoimmunity. Curr. Allergy Asthma Rep. 2014, 14, 407. [Google Scholar] [CrossRef] [Green Version]
- Damian, R.T. A theory of immunoselection for eclipsed antigens of parasites and its implications for the problem of antigenic polymorphism in man. J. Parasitol. 1962, 48, 16. [Google Scholar]
- Fujinami, R.S.; Oldstone, M.B.; Wroblewska, Z.; Frankel, M.E.; Koprowski, H. Molecular mimicry in virus infection: Crossreaction of measles virus phosphoprotein or of herpes simplex virus protein with human intermediate filaments. Proc. Natl. Acad. Sci. USA 1983, 80, 2346–2350. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, M.H.; Meyeserian, M. An immunological cross-reaction between group A streptococcal cells and human heart tissue. Lancet 1962, 1, 706. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.W.; McCormack, J.M.; Talaber, L.R.; Harley, J.B.; Ayoub, E.M.; Muneer, R.S.; Chun, L.T.; Reddy, D.V. Human monoclonal antibodies reactive with antigens of the group A Streptococcus and human heart. J. Immunol. 1988, 141, 2760–2766. [Google Scholar] [CrossRef] [PubMed]
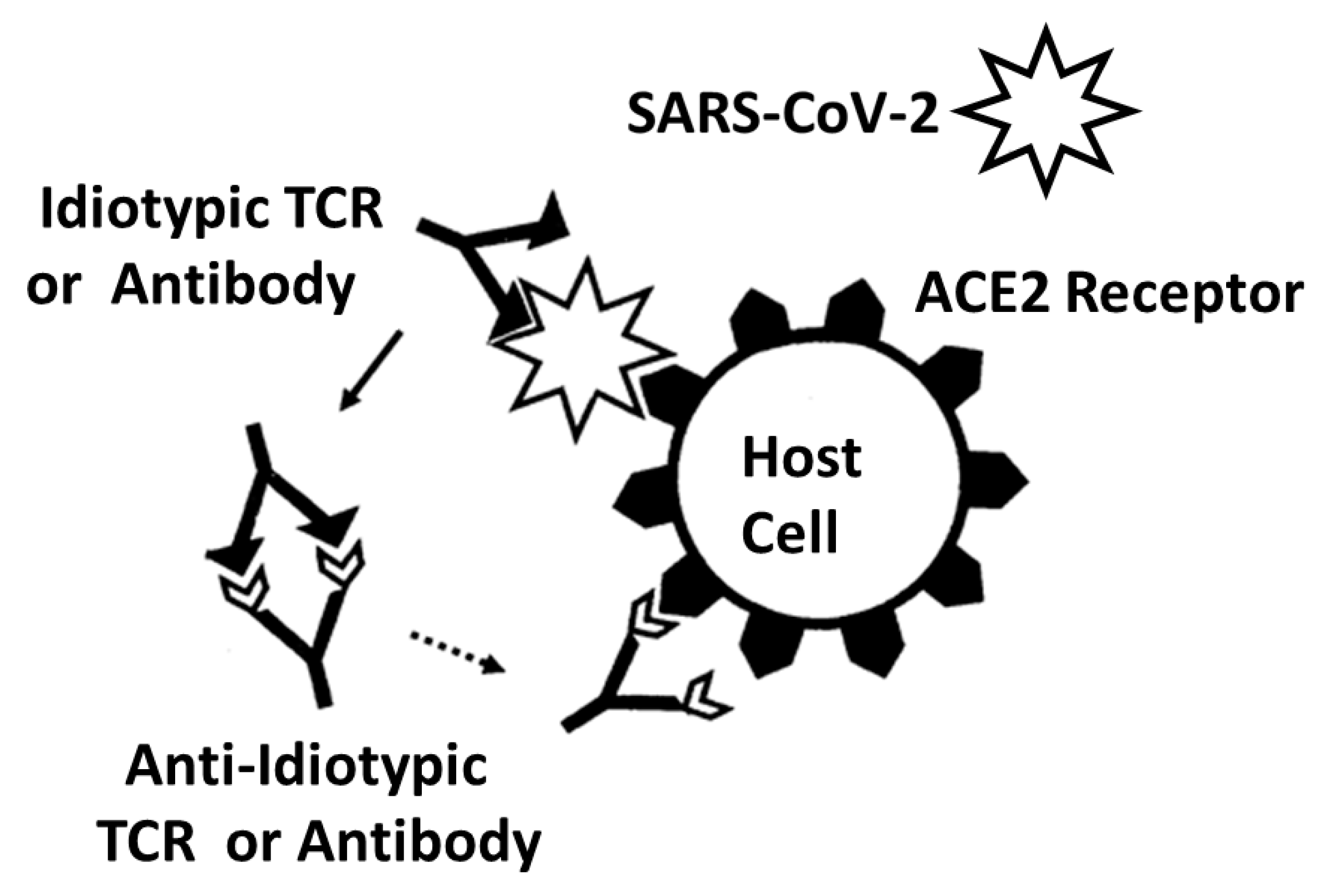
- Plotz, P.H. Autoantibodies are anti-idiotype antibodies to antiviral antibodies. Lancet 1983, 2, 824–826. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, M. Anti-idiotypic antibodies and autoantibodies. Ann. N. Y. Acad. Sci. 1983, 418, 363–378. [Google Scholar] [CrossRef]
- Tzioufas, A.G.; Routsias, J.G. Idiotype, anti-idiotype network of autoantibodies: Pathogenetic considerations and clinical application. Autoimmun. Rev. 2010, 9, 631–633. [Google Scholar] [CrossRef]
- Junior, A.G.; Tolouei, S.E.L.; Dos Reis Lívero, F.A.; Gasparotto, F.; Boeing, T.; de Souza, P. Natural Agents Modulating ACE-2: A Review of Compounds with Potential against SARS-CoV-2 Infections. Curr. Pharm. Des. 2021, 27, 1588–1596. [Google Scholar] [CrossRef]
- Fujinami, R.S.; von Herrath, M.G.; Christen, U.; Whitton, J.L. Molecular mimicry, bystander activation, or viral persistence: Infections and autoimmune disease. Clin. Microbiol. Rev. 2006, 19, 80. [Google Scholar] [CrossRef] [Green Version]
- Tandon, R.; Sharma, M.; Chandrasekhar, Y.; Kotb, M.; Yacoub, M.H.; Narula, J. Revisiting the pathogenesis of rheumatic fever and carditis. Nat. Rev. Cardiol. 2013, 10, 171–177. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Synergistic Activation of Toll-Like and NOD Receptors by Complementary Antigens as Facilitators of Autoimmune Disease: Review, Model and Novel Predictions. Int. J. Mol. Sci. 2020, 21, 4645. [Google Scholar] [CrossRef]
- Root-Bernstein, R. How to Make a Non-Antigenic Protein (Auto) Antigenic: Molecular Complementarity Alters Antigen Processing and Activates Adaptive-Innate Immunity Synergy. Anticancer Agents Med. Chem. 2015, 15, 1242–1259. [Google Scholar] [CrossRef]
- Root-Bernstein, R.; Couturier, J. Antigenic complementarity in the origins of autoimmunity: A general theory illustrated with a case study of idiopathic thrombocytopenia purpura. Clin. Dev. Immunol. 2006, 13, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.; Vonck, J.; Podufaly, A. Antigenic complementarity between coxsackie virus and streptococcus in the induction of rheumatic heart disease and autoimmune myocarditis. Autoimmunity 2009, 42, 1–16. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Antigenic complementarity in the induction of autoimmunity: A general theory and review. Autoimmun. Rev. 2007, 6, 272–277. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Innate Receptor Activation Patterns Involving TLR and NLR Synergisms in COVID-19, ALI/ARDS and Sepsis Cytokine Storms: A Review and Model Making Novel Predictions and Therapeutic Suggestions. Int. J. Mol. Sci. 2021, 22, 2108. [Google Scholar] [CrossRef]
- Pendergraft, W.F., III.; Badhwar, A.K.; Preston, G.A. Autoantigen complementarity and its contributions to hallmarks of autoimmune disease. J. Theor. Biol. 2015, 375, 88–94. [Google Scholar] [CrossRef]
- Yang, J.; Bautz, D.J.; Lionaki, S.; Hogan, S.L.; Chin, H.; Tisch, R.M.; Schmitz, J.L.; Pressler, B.M.; Jennette, J.C.; Falk, R.J.; et al. ANCA patients have T cells responsive to complementary PR-3 antigen. Kidney Int. 2008, 74, 1159–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bautz, D.J.; Preston, G.A.; Lionaki, S.; Hewins, P.; Wolberg, A.S.; Yang, J.J.; Hogan, S.L.; Chin, H.; Moll, S.; Jennette, J.C.; et al. Antibodies with dual reactivity to plasminogen and complementary PR3 in PR3-ANCA vasculitis. J. Am. Soc. Nephrol. 2008, 19, 2421–2429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preston, G.; Falk, R. Autoimmunity: Does autoantigen complementarity underlie PR3-ANCA AAV? Nat. Rev. Rheumatol. 2011, 7, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Hewins, P.; Belmonte, F.; Jennette, J.C.; Falk, R.J.; Preston, G.A. Longitudinal studies of patients with ANCA vasculitis demonstrate concurrent reactivity to complementary PR3 protein segments cPR3m and cPR3C and with no reactivity to cPR3N. Autoimmunity 2011, 44, 98–106. [Google Scholar] [CrossRef]
- Reynolds, J.; Preston, G.A.; Pressler, B.M.; Hewins, P.; Brown, M.; Roth, A.; Alderman, E.; Bunch, D.; Jennette, J.C.; Cook, H.T.; et al. Autoimmunity to the alpha 3 chain of type IV collagen in glomerulonephritis is triggered by ‘autoantigen complementarity’. J. Autoimmun. 2015, 59, 8–18. [Google Scholar] [CrossRef] [Green Version]
- Konstantinov, K.N.; Ulff-Møller, C.J.; Tzamaloukas, A.H. Infections and antineutrophil cytoplasmic antibodies: Triggering mechanisms. Autoimmun. Rev. 2015, 14, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Fujita, J. SARS-CoV-2 as a causative agent of idiopathic interstitial pneumonia and interstitial pneumonia associated with collagen vascular disorders. Respir. Investig. 2020, 58, 427–429. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.C.; May, J.W.; Cunningham, M.W.; Jones, O.Y. Multisystem Inflammatory Syndrome in Children (MIS-C), a Post-viral Myocarditis and Systemic Vasculitis-A Critical Review of Its Pathogenesis and Treatment. Front. Pediatr. 2020, 8, 626182. [Google Scholar] [CrossRef] [PubMed]
- Jerne, N.K. Towards a network theory of the immune system. Ann. Immunol. 1974, 125C, 373–389. [Google Scholar]
- Rowley, A.H.; Shulman, S.T.; Garcia, F.L.; Guzman-Cottrill, J.A.; Miura, M.; Lee, H.L.; Baker, S.C. Cloning the arterial IgA antibody response during acute Kawasaki disease. J. Immunol. 2005, 175, 8386–8391. [Google Scholar] [CrossRef] [Green Version]
- Rowley, A.H.; Shulman, S.T.; Spike, B.T.; Mask, C.A.; Baker, S.C. Oligoclonal IgA response in the vascular wall in acute Kawasaki disease. J. Immunol. 2001, 166, 1334–1343. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.H.; Park, I.H.; Shin, J.S.; Kim, D.S. Immunoglobulin V(H) chain gene analysis of peripheral blood IgM-producing B cells in patients with Kawasaki disease. Yonsei Med. J. 2009, 50, 493–504. [Google Scholar] [CrossRef] [Green Version]
- Thindwa, D.; Quesada, M.G.; Liu, Y.; Bennett, J.; Cohen, C.; Knoll, M.D.; von Gottberg, A.; Hayford, K.; Flasche, S. Use of seasonal influenza and pneumococcal polysaccharide vaccines in older adults to reduce COVID-19 mortality. Vaccine 2020, 38, 5398–5401. [Google Scholar] [CrossRef]
- Nunes, M.C.; Cutland, C.L.; Klugman, K.P.; Madhi, S.A. Pneumococcal Conjugate Vaccine Protection against Corona-virus-Associated Pneumonia Hospitalization in Children Living with and without HIV. mBio 2021, 12, e02347-20. [Google Scholar] [CrossRef]
- Jehi, L.; Ji, X.; Milinovich, A.; Erzurum, S.; Rubin, B.P.; Gordon, S.; Young, J.B.; Kattan, M.W. Individualizing Risk Prediction for Positive Coronavirus Disease 2019 Testing. Chest 2020, 158, 1364–1375. [Google Scholar] [CrossRef]
- Pawlowski, C.; Puranik, A.; Bandi, H.; Venkatakrishnan, A.J.; Agarwal, V.; Kennedy, R.; O’Horo, J.C.; Gores, G.J.; Williams, A.W.; Halamka, J.; et al. Exploratory analysis of immunization records highlights decreased SARS-CoV-2 rates in individuals with recent non-COVID-19 vaccinations. Sci. Rep. 2021, 11, 4741. [Google Scholar] [CrossRef] [PubMed]
- Noale, M.; Trevisan, C.; Maggi, S.; Incalzi, R.A.; Pedone, C.; Di Bari, M.; Adorni, F.; Jesuthasan, N.; Sojic, A.; Galli, M.; et al. The Association between Influenza and Pneumococcal Vaccinations and SARS-Cov-2 Infection: Data from the EPICOVID19 Web-Based Survey. Vaccines 2020, 8, 471. [Google Scholar] [CrossRef] [PubMed]
- Lewnard, A.J.; Bruxvoort, K.J.; Fischer, H.; Hong, V.X.; Grant, L.R.; Jódar, L.; Gessner, B.D.; Tartof, S.Y. Prevention of COVID-19 among older adults receiving pneumococcal conjugate vaccine suggests interactions between Streptococcus pneumoniae and SARS-CoV-2 in the respiratory tract. J. Infect. Dis. 2021, 128, 1710–1720. [Google Scholar] [CrossRef]
- Sumbul, B.; Sumbul, H.E.; Okyay, R.A.; Gülümsek, E.; ¸Sahin, A.R.; Boral, B.; Koçyiğit, B.F.; Alfishawy, M.; Gold, J.; Tasdogan, A.M. Is there a link between pre-existing antibodies acquired due to childhood vaccinations or past infections and COVID-19? A case control study. PeerJ 2021, 9, e10910. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R. Age and Location in Severity of COVID-19 Pathology: Do Lactoferrin and Pneumococcal Vaccination Explain Low Infant Mortality and Regional Differences? BioEssays 2020, 42, 2000076. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Possible Cross-Reactivity between SARS-CoV-2 Proteins, CRM197 and Proteins in Pneumococcal Vaccines May Protect Against Symptomatic SARS-CoV-2 Disease and Death. Vaccines 2020, 8, 559. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Pneumococcal and Influenza Vaccination Rates and Pneumococcal Invasive Disease Rates Set Geograph-ical and Ethnic Population Susceptibility to Serious COVID-19 Cases and Deaths. Vaccines 2021, 9, 474. [Google Scholar] [CrossRef]
- Cambier, S.; Metzemaekers, M.; de Carvalho, A.C.; Nooyens, A.; Jacobs, C.; Vanderbeke, L.; Malengier-Devlies, B.; Gouwy, M.; Heylen, E.; Meersseman, P.; et al. Atypical response to bacterial coinfection and persistent neutrophilic bronchoalveolar inflammation distinguish critical COVID-19 from influenza. JCI Insight 2022, 7, e155055. [Google Scholar] [CrossRef]
- Malik, A.; Tóth, E.N.; Teng, M.S.; Hurst, J.; Watt, E.; Wise, L.; Kent, N.; Bartram, J.; Grandjean, L.; Dominguez-Villar, M.; et al. Distorted TCR repertoires define multisystem inflammatory syndrome in children. PLoS ONE 2022, 17, e0274289. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Anosmia-hyposmia and dysgeusia in COVID-19 may be due to SARS-CoV-2 protein mimicry of olfactory receptors. Rhinol. Online 2020, 3, 148–151. [Google Scholar] [CrossRef]
- Britanova, O.V.; Putintseva, E.V.; Shugay, M.; Merzlyak, E.M.; Turchaninova, M.A.; Staroverov, D.B.; Bolotin, D.A.; Lukyanov, S.; Bogdanova, E.A.; Mamedov, I.Z.; et al. Age-related decrease in TCR repertoire diversity measured with deep and normalized sequence profiling. J. Immunol. 2014, 192, 2689–2698. [Google Scholar] [CrossRef] [Green Version]
- Nolan, S.; Vignali, M.; Klinger, M.; Dines, J.; Kaplan, I.; Svejnoha, E.; Craft, T.; Boland, K.; Pesesky, M.; Gittelman, R.M.; et al. A Large-Scale Database of T-Cell Receptor Beta (TCRb) Sequences and Binding Associations from Natural and Synthetic Exposure to SARS-CoV-2. 2020. Available online: https://clients.adaptivebiotech.com/pub/covid-2020 (accessed on 20 November 2022).
- Scott, C.S.; Richards, S.J.; Roberts, B.E. Patterns of membrane TcR alpha beta and TcR gamma delta chain expression by normal blood CD4+CD8-, CD4-CD8+, CD4-CD8dim+ and CD4-CD8- lymphocytes. Immunology 1990, 70, 351–356. [Google Scholar]
- Shomuradova, A.S.; Vagida, M.S.; Sheetikov, S.A.; Zornikova, K.V.; Kiryukhin, D.; Titov, A.; Peshkova, I.O.; Khmelevskaya, A.; Dianov, D.V.; Malasheva, M.; et al. SARS-CoV-2 Epitopes Are Recognized by a Public and Diverse Repertoire of Human T Cell Receptors. Immunity 2020, 53, 1245–1257.e5. [Google Scholar] [CrossRef]
- Sidhom, J.W.; Baras, A.S. Analysis of SARS-CoV-2 specific T-cell receptors in ImmuneCode reveals cross-reactivity to immunodominant Influenza M1 epitope. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Wang, X.M.; Xing, X.; Xu, Z.; Zhang, C.; Song, J.W.; Fan, X.; Xia, P.; Fu, J.L.; Wang, S.Y.; et al. Single-cell landscape of immunological responses in patients with COVID-19. Nat. Immunol. 2020, 21, 1107–1118. [Google Scholar] [CrossRef]
- Porritt, R.A.; Paschold, L.; Rivas, M.N.; Cheng, M.H.; Yonker, L.M.; Chandnani, H.; Lopez, M.; Simnica, D.; Schultheiß, C.; Santiskulvong, C.; et al. HLA class I-associated expansion of TRBV11-2 T cells in multisystem inflammatory syndrome in children. J. Clin. Investig. 2021, 131, e146614. [Google Scholar] [CrossRef]
- Leung, D.Y.; Giorno, R.C.; Kazemi, L.V.; Flynn, P.A.; Busse, J.B. Evidence for superantigen involvement in cardiovascular injury due to Kawasaki syndrome. J. Immunol. 1995, 155, 5018–5021. [Google Scholar] [CrossRef]
















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Root-Bernstein, R.; Churchill, E.; Oliverio, S. T Cell Receptor Sequences Amplified during Severe COVID-19 and Multisystem Inflammatory Syndrome in Children Mimic SARS-CoV-2, Its Bacterial Co-Infections and Host Autoantigens. Int. J. Mol. Sci. 2023, 24, 1335. https://doi.org/10.3390/ijms24021335
Root-Bernstein R, Churchill E, Oliverio S. T Cell Receptor Sequences Amplified during Severe COVID-19 and Multisystem Inflammatory Syndrome in Children Mimic SARS-CoV-2, Its Bacterial Co-Infections and Host Autoantigens. International Journal of Molecular Sciences. 2023; 24(2):1335. https://doi.org/10.3390/ijms24021335
Chicago/Turabian StyleRoot-Bernstein, Robert, Elizabeth Churchill, and Shelby Oliverio. 2023. "T Cell Receptor Sequences Amplified during Severe COVID-19 and Multisystem Inflammatory Syndrome in Children Mimic SARS-CoV-2, Its Bacterial Co-Infections and Host Autoantigens" International Journal of Molecular Sciences 24, no. 2: 1335. https://doi.org/10.3390/ijms24021335
APA StyleRoot-Bernstein, R., Churchill, E., & Oliverio, S. (2023). T Cell Receptor Sequences Amplified during Severe COVID-19 and Multisystem Inflammatory Syndrome in Children Mimic SARS-CoV-2, Its Bacterial Co-Infections and Host Autoantigens. International Journal of Molecular Sciences, 24(2), 1335. https://doi.org/10.3390/ijms24021335