
Spotlight on P2X7 Receptor PET Imaging: A Bright Target or a Failing Star?



Abstract
:1. Introduction
P2X7R Species Differences and Polymorphism
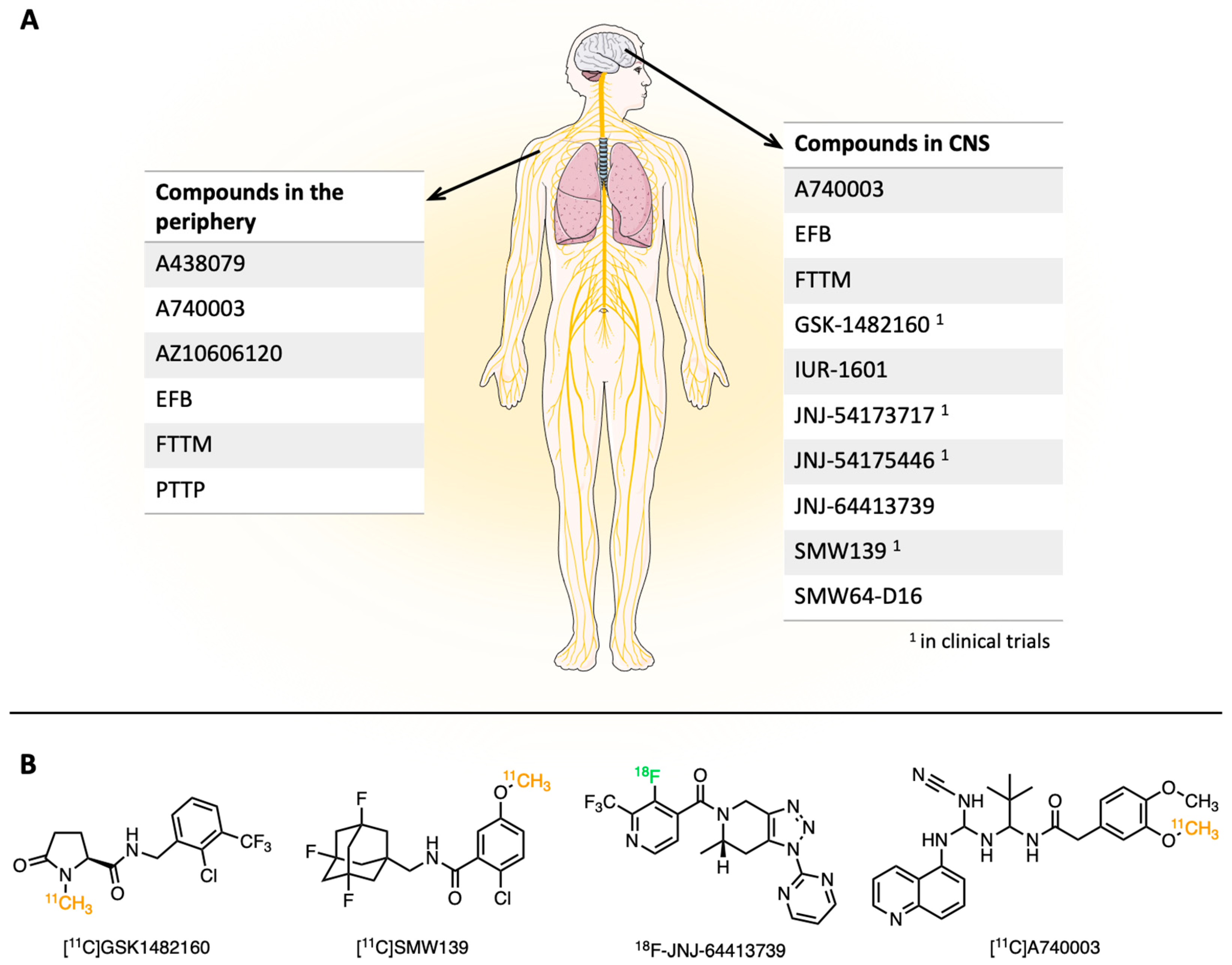
2. P2X7 Receptor Imaging Tracers
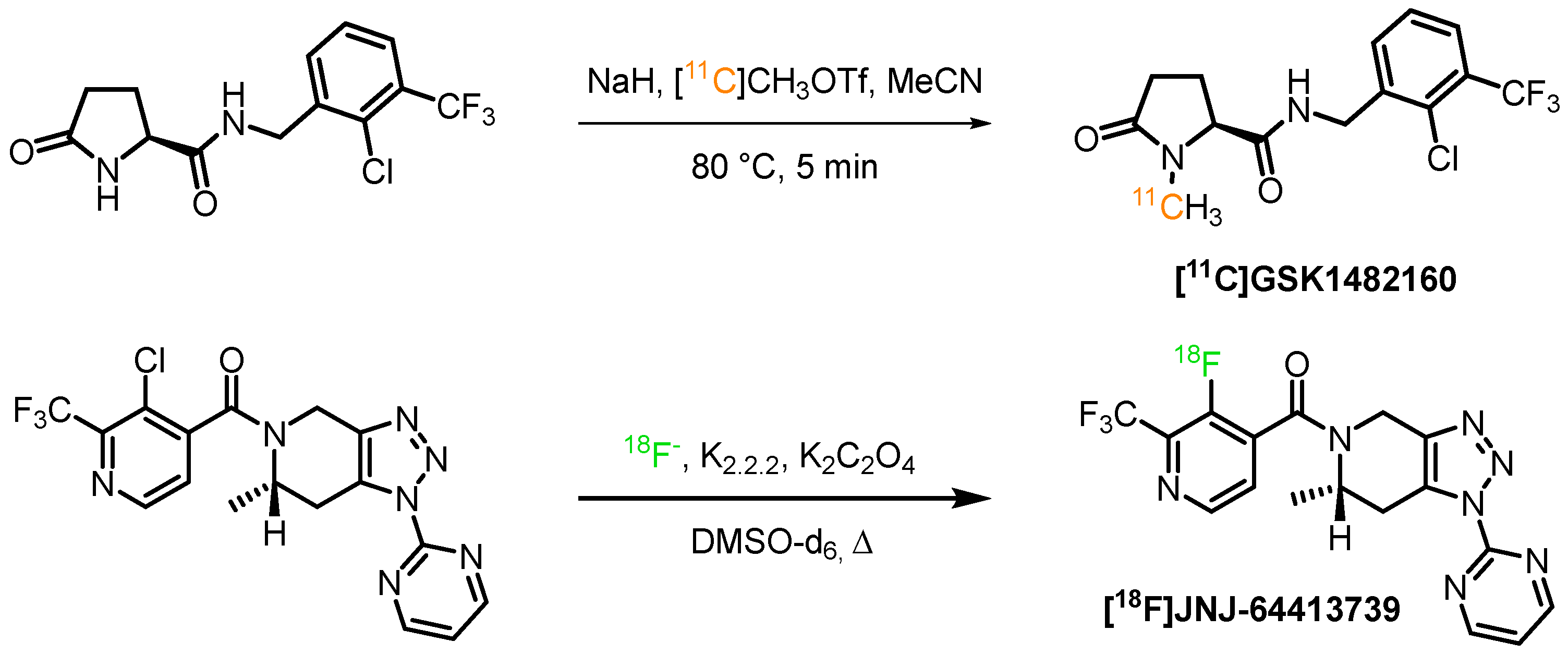
2.1. Radiolabelling Strategies

2.2. P2X7 Receptor Tracers for the PET Imaging in CNS
2.3. P2X7 Receptor Tracers for the Imaging Applications Outside of CNS
3. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Praetorius, H.A.; Leipziger, J. Intrarenal Purinergic Signaling in the Control of Renal Tubular Transport. Annu. Rev. Physiol. 2010, 72, 377–393. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic nerves. Pharmacol. Rev. 1972, 24, 509–581. [Google Scholar] [PubMed]
- Burnstock, G. Purinergic signalling: From discovery to current developments. Exp. Physiol. 2014, 99, 16–34. [Google Scholar] [CrossRef] [Green Version]
- Di Virgilio, F. Purines, Purinergic Receptors, and Cancer. Cancer Res. 2012, 72, 5441–5447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbracchio, M.P.; Burnstock, G.; Verkhratsky, A.; Zimmermann, H. Purinergic signalling in the nervous system: An overview. Trends Neurosci. 2009, 32, 19–29. [Google Scholar] [CrossRef]
- North, R.A. Molecular Physiology of P2X Receptors. Physiol. Rev. 2002, 82, 1013–1067. [Google Scholar] [CrossRef] [Green Version]
- Habermacher, C.; Dunning, K.; Chataigneau, T.; Grutter, T. Molecular structure and function of P2X receptors. Neuropharmacology 2016, 104, 18–30. [Google Scholar] [CrossRef]
- Karasawa, A.; Kawate, T. Structural basis for subtype-specific inhibition of the P2X7 receptor. eLife 2016, 5, e22153. [Google Scholar] [CrossRef]
- McCarthy, A.E.; Yoshioka, C.; Mansoor, S.E. Full-length P2X7 structures reveal how palmitoylation prevents channel desensitization. Cell 2019, 179, 659–670. [Google Scholar] [CrossRef]
- Surprenant, A.; Rassendren, F.; Kawashima, E.; North, R.A.; Buell, G. The Cytolytic P2Z Receptor for Extracellular ATP Identified as a P2X Receptor (P2X7). Science 1996, 272, 735–738. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Schmalzing, G.; Markwardt, F. The Elusive P2X7 Macropore. Trends Cell Biol. 2018, 28, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Colomar, A.; Amédée, T. ATP stimulation of P2X7 receptors activates three different ionic conductances on cultured mouse Schwann cells. Eur. J. Neurosci. 2001, 14, 927–936. [Google Scholar] [CrossRef]
- Colomar, A.; Marty, V.; Médina, C.; Combe, C.; Parnet, P.; Amédée, T. Maturation and release of interleukin-1β by lipopolysaccharide-primed mouse Schwann cells require the stimulation of P2X7 receptors. J. Biol. Chem. 2003, 278, 30732–30740. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.; Jacobson, K.A. Purinergic Signaling in Liver Pathophysiology. Front. Endocrinol. 2021, 12, 718429. [Google Scholar] [CrossRef]
- Wang, N.; Agrawal, A.; Jørgensen, N.R.; Gartland, A. P2X7 receptor regulates osteoclast function and bone loss in a mouse model of osteoporosis. Sci. Rep. 2018, 8, 3507. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.-F.; Tang, Y.; Illes, P. Astrocytic and Oligodendrocytic P2X7 Receptors Determine Neuronal Functions in the CNS. Front. Mol. Neurosci. 2021, 14, 9. [Google Scholar] [CrossRef]
- Illes, P.; Verkhratsky, A.; Burnstock, G.; Franke, H. P2X receptors and their roles in astroglia in the central and peripheral nervous system. Neuroscientist 2012, 18, 422–438. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Biber, K. The microglial ATP-gated ion channel P2X7 as a CNS drug target. Glia 2016, 64, 1772–1787. [Google Scholar] [CrossRef]
- Mehta, V.B.; Hart, J.; Wewers, M.D. ATP-stimulated Release of Interleukin (IL)-1β and IL-18 Requires Priming by Lipopolysaccharide and Is Independent of Caspase-1 Cleavage. J. Biol. Chem. 2001, 276, 3820–3826. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, D.; Chiozzi, P.; Falzoni, S.; Dal Susino, M.; Melchiorri, L.; Baricordi, O.R.; Di Virgilio, F. Extracellular ATP triggers IL-1 beta release by activating the purinergic P2Z receptor of human macrophages. J. Immunol. 1997, 159, 1451–1458. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Dal Ben, D.; Sarti, A.C.; Giuliani, A.L.; Falzoni, S. The P2X7 Receptor in Infection and Inflammation. Immunity 2017, 47, 15–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnstock, G.; Nistri, A.; Khakh, B.S.; Giniatullin, R. ATP-gated P2X receptors in health and disease. Front. Cell. Neurosci. 2014, 8, 2013–2014. [Google Scholar] [CrossRef] [Green Version]
- Savio, L.E.B.; Mello, P.D.A.; da Silva, C.G.; Coutinho-Silva, R. The P2X7 receptor in inflammatory diseases: Angel or demon? Front. Pharmacol. 2018, 9, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adinolfi, E.; Capece, M.; Amoroso, F.; De Marchi, E.; Franceschini, A. Emerging Roles of P2X Receptors in Cancer. Curr. Med. Chem. 2015, 22, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, W.C.; Jenko, K.J.; Hines, C.S.; Lyoo, C.H.; Corona, W.; Morse, C.L.; Zoghbi, S.S.; Hyde, T.; Kleinman, J.E.; Pike, V.W.; et al. A genetic polymorphism for translocator protein 18 kDa affects both in vitro and in vivo radioligand binding in human brain to this putative biomarker of neuroinflammation. J. Cereb. Blood Flow Metab. 2013, 33, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Notter, T.; Coughlin, J.M.; Sawa, A.; Meyer, U. Reconceptualization of translocator protein as a biomarker of neuroinflammation in psychiatry. Mol. Psychiatry 2018, 23, 36–47. [Google Scholar] [CrossRef]
- Roger, S.; Jelassi, B.; Couillin, I.; Pelegrin, P.; Besson, P.; Jiang, L.-H. Understanding the roles of the P2X7 receptor in solid tumour progression and therapeutic perspectives. Biochim. Biophys. Acta (BBA) Biomembr. 2015, 1848, 2584–2602. [Google Scholar] [CrossRef] [Green Version]
- Giannuzzo, A.; Saccomano, M.; Napp, J.; Ellegaard, M.; Alves, F.; Novak, I. Targeting of the P2X7 receptor in pancreatic cancer and stellate cells. Int. J. Cancer 2016, 139, 2540–2552. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.K.; Jantaratnotai, N.; Serrano-Perez, M.C.; McGeer, P.L.; McLarnon, J.G. Block of purinergic P2X7R inhibits tumor growth in a C6 glioma brain tumor animal model. J. Neuropathol. Exp. Neurol. 2011, 70, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Avanzato, D.; Genova, T.; Fiorio Pla, A.; Bernardini, M.; Bianco, S.; Bussolati, B.; Mancardi, D.; Giraudo, E.; Maione, F.; Cassoni, P.; et al. Activation of P2X7 and P2Y11 purinergic receptors inhibits migration and normalizes tumor-derived endothelial cells via cAMP signaling. Sci. Rep. 2016, 6, 32602. [Google Scholar] [CrossRef]
- Xia, J.; Yu, X.; Tang, L.; Li, G.; He, T. P2X7 receptor stimulates breast cancer cell invasion and migration via the AKT pathway. Oncol. Rep. 2015, 34, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Moesta, A.K.; Li, X.-Y.; Smyth, M.J. Targeting CD39 in cancer. Nat. Rev. Immunol. 2020, 20, 739–755. [Google Scholar] [CrossRef]
- Gao, Z.; Dong, K.; Zhang, H. The roles of CD73 in cancer. BioMed. Res. Int. 2014, 2014, 460654. [Google Scholar] [CrossRef] [Green Version]
- Vigano, S.; Alatzoglou, D.; Irving, M.; Ménétrier-Caux, C.; Caux, C.; Romero, P.; Coukos, G. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front. Immunol. 2019, 10, 925. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Qiu, R.; Wang, W.; Liu, J.; Jin, X.; Li, Y.; Li, L.; Lei, B. P2X7 Receptor Antagonist Attenuates Retinal Inflammation and Neovascularization Induced by Oxidized Low-Density Lipoprotein. Oxid. Med. Cell. Longev. 2021, 2021, 5520644. [Google Scholar] [CrossRef]
- Martínez-Cuesta, M.Á.; Blanch-Ruiz, M.A.; Ortega-Luna, R.; Sánchez-López, A.; Álvarez, Á. Structural and Functional Basis for Understanding the Biological Significance of P2X7 Receptor. Int. J. Mol. Sci. 2020, 21, 8454. [Google Scholar] [CrossRef]
- Available online: http://www.uniprot.org/ (accessed on 1 January 2023).
- Rassendren, F.; Buell, G.N.; Virginio, C.; Collo, G.; North, R.A.; Surprenant, A. The permeabilizing ATP receptor, P2X7. Cloning and expression of a human cDNA. J. Biol. Chem. 1997, 272, 5482–5486. [Google Scholar] [CrossRef] [Green Version]
- Chessell, I.P.; Simon, J.; Hibell, A.D.; Michel, A.D.; Barnard, E.A.; Humphrey, P.P.A. Cloning and functional characterisation of the mouse P2X7 receptor. FEBS Lett. 1998, 439, 26–30. [Google Scholar] [CrossRef] [Green Version]
- Caseley, E.A.; Muench, S.P.; Baldwin, S.A.; Simmons, K.; Fishwick, C.W.; Jiang, L.-H. Docking of competitive inhibitors to the P2X7 receptor family reveals key differences responsible for changes in response between rat and human. Bioorg. Med. Chem. Lett. 2015, 25, 3164–3167. [Google Scholar] [CrossRef] [Green Version]
- Burnstock, G.; Verkhratsky, A. Purinergic Signalling and the Nervous System; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012; ISBN 978-3-642-28862-3. [Google Scholar]
- Reemtsma, K. Xenotransplantation: A Historical Perspective. ILAR J. 1995, 37, 9–12. [Google Scholar] [CrossRef]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richmond, A.; Su, Y. Mouse xenograft models vs GEM models for human cancer therapeutics. Dis. Model. Mech. 2008, 1, 78–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Li, F.; Wang, L.; Lou, Y. A438079 affects colorectal cancer cell proliferation, migration, apoptosis, and pyroptosis by inhibiting the P2X7 receptor. Biochem. Biophys. Res. Commun. 2021, 558, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Yang, X.; Wang, L.; Wang, R.; Yang, F.; Wang, H.; Liu, X.; Ren, Q.; Zhang, Y.; Zhu, X.; et al. P2X7 promotes the progression of MLL-AF9 induced acute myeloid leukemia by upregulation of Pbx3. Haematologica 2021, 106, 1278–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.; Seol, H.S.; Chang, S. The Generation and Application of Patient-Derived Xenograft Model for Cancer Research. Cancer Res. Treat. 2018, 50, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, R.; Stokes, L.; Sluyter, R. The p2x7 receptor channel: Recent developments and the use of p2x7 antagonists in models of disease. Pharmacol. Rev. 2014, 66, 638–675. [Google Scholar] [CrossRef] [Green Version]
- Sluyter, R. The P2X7 Receptor BT—Protein Reviews; Atassi, M.Z., Ed.; Springer: Singapore, 2017; Volume 19, pp. 17–53. ISBN 978-981-10-7611-4. [Google Scholar]
- Adriouch, S.; Dox, C.; Welge, V.; Seman, M.; Koch-Nolte, F.; Haag, F. Cutting edge: A natural P451L mutation in the cytoplasmic domain impairs the function of the mouse P2X7 receptor. J. Immunol. 2002, 169, 4108–4112. [Google Scholar] [CrossRef] [Green Version]
- Pegoraro, A.; De Marchi, E.; Adinolfi, E. P2X7 Variants in Oncogenesis. Cells 2021, 10, 189. [Google Scholar] [CrossRef]
- Baas, T. Paradoxical P2X7. Sci. Exch. 2012, 5, 512. [Google Scholar] [CrossRef]
- Keystone, E.C.; Wang, M.M.; Layton, M.; Hollis, S.; McInnes, I.B. Clinical evaluation of the efficacy of the P2X7 purinergic receptor antagonist AZD9056 on the signs and symptoms of rheumatoid arthritis in patients with active disease despite treatment with methotrexate or sulphasalazine. Ann. Rheum. Dis. 2012, 71, 1630–1635. [Google Scholar] [CrossRef]
- Stock, T.C.; Bloom, B.J.; Wei, N.; Ishaq, S.; Park, W.; Wang, X.; Gupta, P.; Mebus, C.A. Efficacy and safety of CE-224,535, an antagonist of P2X7 receptor, in treatment of patients with rheumatoid arthritis inadequately controlled by methotrexate. J. Rheumatol. 2012, 39, 720–727. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, Y.C. P2X7 receptor antagonists: A patent review (2010–2015). Expert Opin. Ther. Pat. 2017, 27, 257–267. [Google Scholar] [CrossRef]
- Burnstock, G.; Knight, G.E. The potential of P2X7 receptors as a therapeutic target, including inflammation and tumour progression. Purinergic Signal. 2018, 14, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Ali, Z.; Laurijssens, B.; Ostenfeld, T.; McHugh, S.; Stylianou, A.; Scott-Stevens, P.; Hosking, L.; Dewit, O.; Richardson, J.C.; Chen, C. Pharmacokinetic and pharmacodynamic profiling of a P2X7 receptor allosteric modulator GSK1482160 in healthy human subjects. Br. J. Clin. Pharmacol. 2013, 75, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef] [Green Version]
- Bleeker, F.E.; Molenaar, R.J.; Leenstra, S. Recent advances in the molecular understanding of glioblastoma. J. Neurooncol. 2012, 108, 11–27. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Li, Q.; Song, W.; Peng, X.; Zhao, R. P2X7 receptor: A critical regulator and potential target for breast cancer. J. Mol. Med. 2021, 99, 349–358. [Google Scholar] [CrossRef]
- Li, Q.; Zhu, X.; Song, W.; Peng, X.; Zhao, R. The P2X7 purinergic receptor: A potential therapeutic target for lung cancer. J. Cancer Res. Clin. Oncol. 2020, 146, 2731–2741. [Google Scholar] [CrossRef]
- Rech, J.C.; Bhattacharya, A.; Letavic, M.A.; Savall, B.M. The evolution of P2X7 antagonists with a focus on CNS indications. Bioorg. Med. Chem. Lett. 2016, 26, 3838–3845. [Google Scholar] [CrossRef]
- Pevarello, P.; Bovolenta, S.; Tarroni, P.; Za, L.; Severi, E.; Torino, D.; Vitalone, R. P2X7 antagonists for CNS indications: Recent patent disclosures. Pharm. Pat. Anal. 2017, 6, 61–76. [Google Scholar] [CrossRef]
- Wolf, A.P.; Redvanly, C.S. Carbon-11 and radiopharmaceuticals. Int. J. Appl. Radiat. Isot. 1977, 28, 29–48. [Google Scholar] [CrossRef] [PubMed]
- Schubiger, P.A.; Lehmann, L.; Friebe, M.; Yang, D.J. PET Chemistry: The Driving Force in Molecular Imaging. J. Nucl. Med. 2007, 48, 1750. [Google Scholar] [CrossRef] [Green Version]
- Någren, K.; Müller, L.; Halldin, C.; Swahn, C.G.; Lehikoinen, P. Improved synthesis of some commonly used PET radioligands by the use of [11C]methyl triflate. Nucl. Med. Biol. 1995, 22, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Lundkvist, C.; Sandell, J.; Någren, K.; Pike, V.W.; Halldin, C. Improved syntheses of the PET radioligands, [11C]FLB 457, [11C]MDL 100907 and [11C]β-CIT-FE, by the use of [11C]methyl triflate. J. Label. Compd. Radiopharm. 1998, 41, 545–556. [Google Scholar] [CrossRef]
- Han, J.; Liu, H.; Liu, C.; Jin, H.; Perlmutter, J.S.; Egan, T.M.; Tu, Z. Pharmacologic characterizations of a P2X7 receptor-specific radioligand, [11 C]GSK1482160 for neuroinflammatory response. Nucl. Med. Commun. 2017, 38, 372–382. [Google Scholar] [CrossRef] [Green Version]
- Guillaume, M.; Luxen, A.; Nebeling, B.; Argentini, M.; Clark, J.C.; Pike, V.W. Recommendations for fluorine-18 production. Int. J. Radiat. Appl. Instrum. Part A Appl. Radiat. Isot. 1991, 42, 749–762. [Google Scholar] [CrossRef]
- Koole, M.; Schmidt, M.E.; Hijzen, A.; Ravenstijn, P.; Vandermeulen, C.; Van Weehaeghe, D.; Serdons, K.; Celen, S.; Bormans, G.; Ceusters, M.; et al. 18F-JNJ-64413739, a Novel PET Ligand for the P2X7 Ion Channel: Radiation Dosimetry, Kinetic Modeling, Test-Retest Variability, and Occupancy of the P2X7 Antagonist JNJ-54175446. J. Nucl. Med. 2019, 60, 683–690. [Google Scholar] [CrossRef] [Green Version]
- Green, M.; Hutchins, G.; Fletcher, J.; Territo, W.; Polson, H.; Trussell, H.; Wissmann, C.; Zheng, Q.-H.; Gao, M.; WANG, M.I.N.; et al. Distribution of the P2X7-receptor-targeted [11C]GSK1482160 radiopharmaceutical in normal human subjects. J. Nucl. Med. 2018, 59, 1009. [Google Scholar]
- Qi, G.; Weinzimmer, D.; Labaree, D.; Carson, R.; Yu-Shin, D.; Ridler, K.; Gunn, R.N.; Rabiner, E.A.; Passchier, J.; Bennacef, I. Imaging P2X7 receptor using PET. J. Label. Compd. Rad. 2011, 54, S298. [Google Scholar]
- Gao, M.; Wang, M.; Green, M.A.; Hutchins, G.D.; Zheng, Q.-H. Synthesis of [(11)C]GSK1482160 as a new PET agent for targeting P2X(7) receptor. Bioorg. Med. Chem. Lett. 2015, 25, 1965–1970. [Google Scholar] [CrossRef] [Green Version]
- Territo, P.R.; Meyer, J.A.; Peters, J.S.; Riley, A.A.; McCarthy, B.P.; Gao, M.; Min, W.; Green, M.A.; Zheng, Q.H.; Hutchins, G.D. Characterization of 11C-GSK1482160 for Targeting the P2X7 receptor as a biomarker for neuroinflammation. J. Nucl. Med. 2017, 58, 458–465. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Wang, M.; Glick-Wilson, B.E.; Meyer, J.A.; Peters, J.S.; Territo, P.R.; Green, M.A.; Hutchins, G.D.; Zarrinmayeh, H.; Zheng, Q.-H. Synthesis and preliminary biological evaluation of a novel P2X7R radioligand [18F]IUR-1601. Bioorg. Med. Chem. Lett. 2018, 28, 1603–1609. [Google Scholar] [CrossRef]
- Gao, M.; Wang, M.; Meyer, J.A.; Territo, P.R.; Hutchins, G.D.; Zarrinmayeh, H.; Zheng, Q.-H. Synthesis and in vitro biological evaluation of new P2X7R radioligands [11C]halo-GSK1482160 analogs. Bioorg. Med. Chem. Lett. 2019, 29, 1476–1480. [Google Scholar] [CrossRef]
- Wilkinson, S.M.; Barron, M.L.; O’Brien-Brown, J.; Janssen, B.; Stokes, L.; Werry, E.L.; Chishty, M.; Skarratt, K.K.; Ong, J.A.; Hibbs, D.E. Pharmacological evaluation of novel bioisosteres of an adamantanyl benzamide P2X7 receptor antagonist. ACS Chem. Neurosci. 2017, 8, 2374–2380. [Google Scholar] [CrossRef] [Green Version]
- Janssen, B.; Vugts, D.J.; Wilkinson, S.M.; Ory, D.; Chalon, S.; Hoozemans, J.J.M.; Schuit, R.C.; Beaino, W.; Kooijman, E.J.M.; Van Den Hoek, J. Identification of the allosteric P2X7 receptor antagonist [11C] SMW139 as a PET tracer of microglial activation. Sci. Rep. 2018, 8, 6580. [Google Scholar] [CrossRef]
- Fantoni, E.R.; Dal Ben, D.; Falzoni, S.; Di Virgilio, F.; Lovestone, S.; Gee, A. Design, synthesis and evaluation in an LPS rodent model of neuroinflammation of a novel 18F-labelled PET tracer targeting P2X7. EJNMMI Res. 2017, 7, 31. [Google Scholar] [CrossRef] [Green Version]
- Van Weehaeghe, D.; Koole, M.; Schmidt, M.E.; Deman, S.; Jacobs, A.H.; Souche, E.; Serdons, K.; Sunaert, S.; Bormans, G.; Vandenberghe, W.; et al. [11C]JNJ54173717, a novel P2X7 receptor radioligand as marker for neuroinflammation: Human biodistribution, dosimetry, brain kinetic modelling and quantification of brain P2X7 receptors in patients with Parkinson’s disease and healthy volunteers. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2051–2064. [Google Scholar] [CrossRef]
- Crabbé, M.; Van Der Perren, A.; Bollaerts, I.; Kounelis, S.; Baekelandt, V.; Bormans, G.; Casteels, C.; Moons, L.; Laere, K. Van Increased P2X7 receptor binding is associated with neuroinflammation in acute but not chronic rodent models for Parkinson’s disease. Front. Neurosci. 2019, 13, 799. [Google Scholar] [CrossRef] [Green Version]
- Savall, B.M.; Wu, D.; De Angelis, M.; Carruthers, N.I.; Ao, H.; Wang, Q.; Lord, B.; Bhattacharya, A.; Letavic, M.A. Synthesis, SAR, and Pharmacological Characterization of Brain Penetrant P2X7 Receptor Antagonists. ACS Med. Chem. Lett. 2015, 6, 671–676. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Miao, X.; Ravenstijn, P.; Hijzen, A.; Schmidt, M.E.; Nandy, P.; Zhou, H. Translational Model-Informed Dose Selection for a Human Positron Emission Tomography Imaging Study of JNJ-54175446, a P2X7 Receptor Antagonist. Clin. Transl. Sci. 2020, 13, 309–317. [Google Scholar] [CrossRef]
- Ory, D.; Celen, S.; Gijsbers, R.; Van Den Haute, C.; Postnov, A.; Koole, M.; Vandeputte, C.; Andrés, J.I.; Alcazar, J.; De Angelis, M.; et al. Preclinical evaluation of a P2X7 receptor-selective radiotracer: PET studies in a rat model with local overexpression of the human p2x7 receptor and in nonhuman primates. J. Nucl. Med. 2016, 57, 1436–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, H.; Zhang, W.; Chen, G.; Xia, C.; Szardenings, K.; Bhattacharya, A.; Lord, B.; Letavic, M.; Andres, J.I. 394. Development and Preclinical Evaluation of [18F]JNJ-64413739 as a PET Radioligand for P2X7 Receptors. Biol. Psychiatry 2017, 81, S161. [Google Scholar] [CrossRef]
- Kolb, H.C.; Barret, O.; Bhattacharya, A.; Chen, G.; Constantinescu, C.; Huang, C.; Letavic, M.; Tamagnan, G.; Xia, C.A.; Zhang, W.; et al. Preclinical Evaluation and Nonhuman Primate Receptor Occupancy Study of 18F-JNJ-64413739, a PET Radioligand for P2X7 Receptors. J. Nucl. Med. 2019, 60, 1154–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.; Lin, Q.; Xu, Z.; Zhao, Y.; Cheng, Y.; Shi, D.; Fu, W.; Yang, T.; Shi, H.; Cheng, D. P2X7 receptor-specific radioligand 18F-FTTM for atherosclerotic plaque PET imaging. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 2595–2604. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Lin, Q.; Xu, Z.; Fu, W.; Shi, D.; Cheng, Y.; Yang, T.; Liu, G.; Shi, H.; Cheng, D. Longitudinal Positron Emission Tomography Imaging with P2X7 Receptor-Specific Radioligand 18F-FTTM in a Kainic Acid Rat Model of Temporal Lobe Epilepsy. ACS Chem. Neurosci. 2022, 13, 3512–3522. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Lin, Q.; Hu, B.; Zhang, Y.; Chen, W.; Zhu, J.; Zhao, Y.; Choi, H.S.; Shi, H.; Cheng, D. P2X7 PET Radioligand 18F-PTTP for Differentiation of Lung Tumor from Inflammation. J. Nucl. Med. 2019, 60, 930–936. [Google Scholar] [CrossRef] [Green Version]
- Swanson, D.M.; Savall, B.M.; Coe, K.J.; Schoetens, F.; Koudriakova, T.; Skaptason, J.; Wall, J.; Rech, J.; Deng, X.; De Angelis, M. Identification of (R)-(2-Chloro-3-(trifluoromethyl)phenyl)(1-(5-fluoropyridin-2-yl)-4-methyl-6,7-dihydro-1H-imidazo[4,5-c]pyridin-5(4H)-yl)methanone (JNJ 54166060), a Small Molecule Antagonist of the P2X7 receptor. J. Med. Chem. 2016, 59, 8535–8548. [Google Scholar] [CrossRef]
- Janssen, B.; Vugts, D.J.; Funke, U.; Spaans, A.; Schuit, R.C.; Kooijman, E.; Rongen, M.; Perk, L.R.; Lammertsma, A.A.; Windhorst, A.D. Synthesis and initial preclinical evaluation of the P2X7 receptor antagonist [11C]A-740003 as a novel tracer of neuroinflammation. J. Label. Compd. Radiopharm. 2014, 57, 509–516. [Google Scholar] [CrossRef]
- Abdi, M.H.; Beswick, P.J.; Billinton, A.; Chambers, L.J.; Charlton, A.; Collins, S.D.; Collis, K.L.; Dean, D.K.; Fonfria, E.; Gleave, R.J.; et al. Discovery and structure–activity relationships of a series of pyroglutamic acid amide antagonists of the P2X7 receptor. Bioorg. Med. Chem. Lett. 2010, 20, 5080–5084. [Google Scholar] [CrossRef]
- Janssen, B.; Ory, D.; Wilkinson, S.M.; Vugts, D.J.; Kooijman, E.J.; Verbeek, J.; Funke, U.; Molenaar, G.T.; Kruijer, P.S.; Lammertsma, A.A. Initial evaluation of P2X(7)R antagonists [C-11]A-740003 and [C-11]SMW64-D16 as PET tracers of microglial activation in neuroinflammation. J. Label. Comp. Radiopharm. 2015, 58, S277. [Google Scholar]
- Beaino, W.; Janssen, B.; Kooijman, E.; Vos, R.; Schuit, R.; Kassiou, M.; Vugts, D.; de Vries, H.; Windhorst, A. Neuroinflammation imaging with the P2X(7)R PET tracer [C-11]SMW139 in the experimental autoimmune encephalomyelitis (EAE) model. J. Label. Compd. Radiopharm. 2019, 62, S73–S74. [Google Scholar]
- Hagens, M.H.J.; Golla, S.S.V.; Janssen, B.; Vugts, D.J.; Beaino, W.; Windhorst, A.D.; O’Brien-Brown, J.; Kassiou, M.; Schuit, R.C.; Schwarte, L.A. The P2X7 receptor tracer [11C] SMW139 as an in vivo marker of neuroinflammation in multiple sclerosis: A first-in man study. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 379–389. [Google Scholar] [CrossRef] [Green Version]
- Van Weehaeghe, D.; Van Schoor, E.; Koole, M.; De Vocht, J.; Ceelen, S.; Declercq, L.; Thal, D.R.; Van Damme, P.; Bormans, G.; Van Laere, K. [C-11]JNJ717 P2X7 receptor PET as a novel neuroinflammation target: Ex vivo and in vivo comparison with [F-18]DPA714 in human ALS. J. Cereb. Blood Flow Metab. 2019, 39, 120–121. [Google Scholar]
- Berdyyeva, T.; Xia, C.; Taylor, N.; He, Y.; Chen, G.; Huang, C.; Zhang, W.; Kolb, H.; Letavic, M.; Bhattacharya, A.; et al. PET Imaging of the P2X7 Ion Channel with a Novel Tracer [18F]JNJ-64413739 in a Rat Model of Neuroinflammation. Mol. Imaging Biol. 2019, 21, 871–878. [Google Scholar] [CrossRef] [Green Version]
- Donnelly-Roberts, D.L.; Namovic, M.T.; Surber, B.; Vaidyanathan, S.X.; Perez-Medrano, A.; Wang, Y.; Carroll, W.A.; Jarvis, M.F. [3H]A-804598 ([3H]2-cyano-1-[(1S)-1-phenylethyl]-3-quinolin-5-ylguanidine) is a novel, potent, and selective antagonist radioligand for P2X7 receptors. Neuropharmacology 2009, 56, 223–229. [Google Scholar] [CrossRef]
- Adinolfi, E.; Capece, M.; Franceschini, A.; Falzoni, S.; Giuliani, A.L.; Rotondo, A.; Sarti, A.C.; Bonora, M.; Syberg, S.; Corigliano, D. Accelerated tumor progression in mice lacking the ATP receptor P2X7. Cancer Res. 2015, 75, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Adinolfi, E.; Raffaghello, L.; Giuliani, A.L.; Cavazzini, L.; Capece, M.; Chiozzi, P.; Bianchi, G.; Kroemer, G.; Pistoia, V.; Di Virgilio, F. Expression of P2X7 receptor increases in vivo tumor growth. Cancer Res. 2012, 72, 2957–2969. [Google Scholar] [CrossRef] [Green Version]
- Jelassi, B.; Chantome, A.; Alcaraz-Perez, F.; Baroja-Mazo, A.; Cayuela, M.L.; Pelegrin, P.; Surprenant, A.; Roger, S. P2X7 receptor activation enhances SK3 channels-and cystein cathepsin-dependent cancer cells invasiveness. Oncogene 2011, 30, 2108–2122. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Compound | Structure | Ki/IC50-Values | Model | Refs. |
|---|---|---|---|---|
| [11C]GSK1482160 |  | Ki = 5.14 ± 0.85 nM (hP2X7R, HEK-293) | Rodent model of MS; First-in-human P.E.T. studies | [68,71,72,73,74,75] |
| [11C](F/Br/I)-GSK1482160 |  | Ki = 54.2 nM (F), 2.5 nM (Br), 1.9 nM (I) (hP2X7R, HEK-293) | In vitro binding assay on HEK293-hP2X7R cells | [76] |
| [18F]IUR-1601 |  | Ki = 4.31 ± 0.92 nM (hP2X7R, HEK-293) | 5xFAD mouse model of AD, but no results reported | [75] |
| [11C]SMW139 |  | IC50 = 24.5 ± 5.5 nM (hP2X7R, 1321N1) IC50 = 158 ± 44 nM (rP2X7R, 1321N1) | EAE in vivo rat model; first in-human study (5 MS patients, 5 healthy subjects) | [77,78] |
| [11C]SMW64-D16 |  | pA2 = 7.2 (hP2X7R, 1321N1) | In vivo LPS-injected rat model of neuroinflammation | [79] |
| [11C]JNJ-54173717 |  | IC50 = 4.2 nM (hP2X7R) IC50 = 7.6nM (rP2X7R) | First in-human PET studies In vitro [11C]JNJ-717 and [18F]DPA-714 Autoradiography (PD, rodent model) | [80,81,82,83,84] |
| [18F]-JNJ-64413739 |  | IC50 = 1.0 nM (hP2X7R, 1321N1) IC50 = 2.0 nM (rP2X7R, 1321N1) | data from early human clinical studies and a monkey PET study | [85,86] |
| [18F]FTTM |  | Kd = 25.35 ± 3.47 nM | atherosclerotic plaque imaging in a ApoE mouse model Kainic acid rat model of temporal lobe epilepsy | [87,88] |
| [18F]-PTTP |  | IC50 = 4.0 nM (hP2X7R, 1321N1) IC50 = 7.0 nM (rP2X7R, 1321N1) | In vivo mouse model with implanted human lung cancer cells | [89,90] |
| [11C]A740003 |  | IC50 = 18 nM (hP2X7R, HEK-293) IC50 = 40 nM (rP2X7R, HEK-293) Ki = 0.1 nM (hP2X7R, HEK-293) Ki = 8.67 nM (rP2X7R, HEK-293) Ki = 97.2 nM | In vivo healthy male Wistar rats and ox-LDL mouse model | [35,91] |
| [18F]EFB |  | Ki = 2.9 nM (hP2X7R, HEK-293) Ki = 36.1 nM (rP2X7R, HEK-293) Ki = 547 nM (mP2X7R, HEK-293) | In vivo LPS-injected rat model of neuroinflammation | [79] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, S.; Isaak, A.; Junker, A. Spotlight on P2X7 Receptor PET Imaging: A Bright Target or a Failing Star? Int. J. Mol. Sci. 2023, 24, 1374. https://doi.org/10.3390/ijms24021374
Schmidt S, Isaak A, Junker A. Spotlight on P2X7 Receptor PET Imaging: A Bright Target or a Failing Star? International Journal of Molecular Sciences. 2023; 24(2):1374. https://doi.org/10.3390/ijms24021374
Chicago/Turabian StyleSchmidt, Stephan, Andreas Isaak, and Anna Junker. 2023. "Spotlight on P2X7 Receptor PET Imaging: A Bright Target or a Failing Star?" International Journal of Molecular Sciences 24, no. 2: 1374. https://doi.org/10.3390/ijms24021374
APA StyleSchmidt, S., Isaak, A., & Junker, A. (2023). Spotlight on P2X7 Receptor PET Imaging: A Bright Target or a Failing Star? International Journal of Molecular Sciences, 24(2), 1374. https://doi.org/10.3390/ijms24021374