
Transcriptome Analysis Reveals the Defense Mechanism of Cotton against Verticillium dahliae Induced by Hypovirulent Fungus Gibellulopsis nigrescens CEF08111

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results
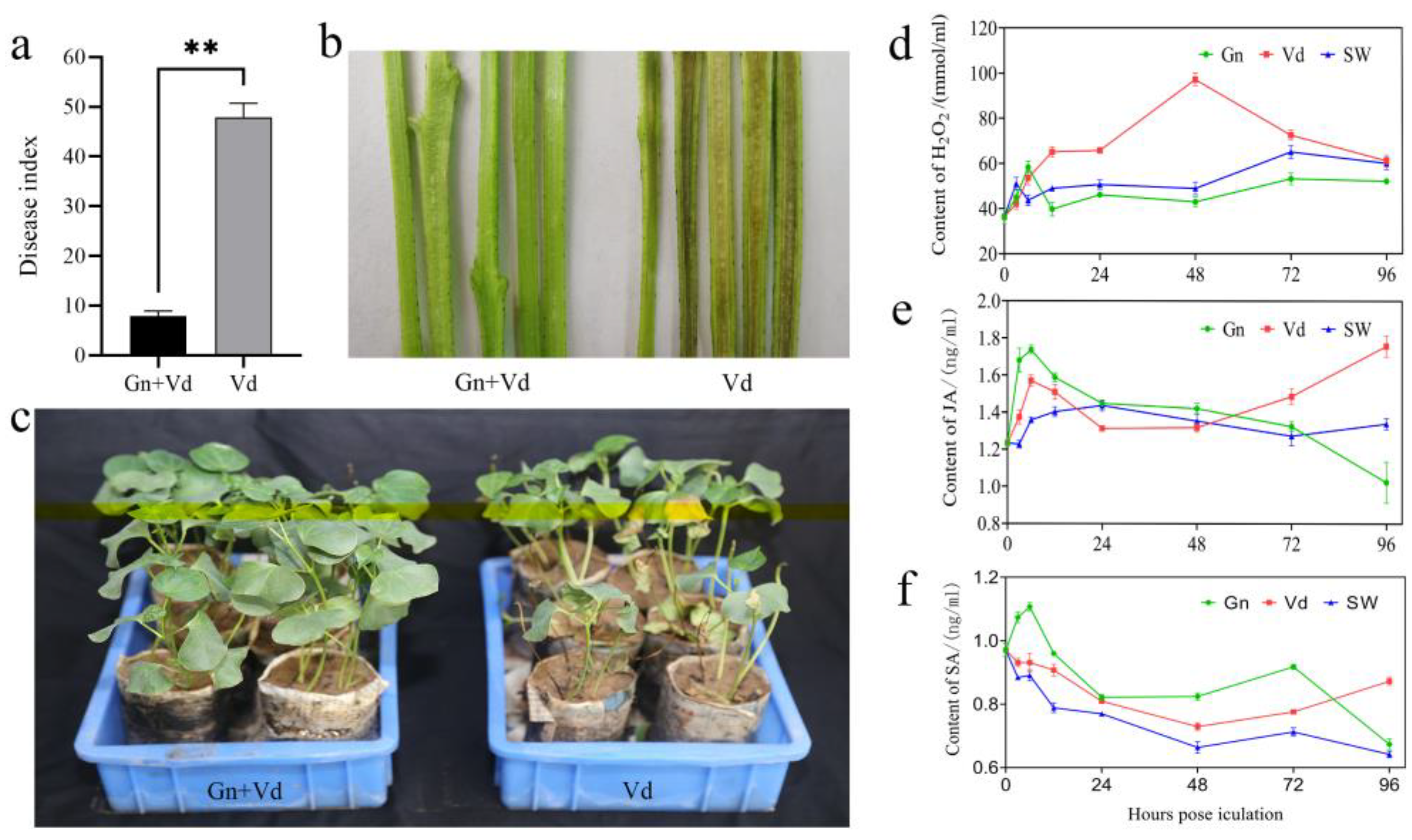
2.1. Control Effect of CEF08111 on Verticillium Wilt of Cotton
2.2. Content of H2O2, JA and SA
2.3. RNA Sequencing and Transcript Identification
2.4. DEGs of Cotton Resistance to Verticillium Wilt Induced by CEF08111
2.5. GO Enrichment Analyses of DEGs
2.6. KEGG Enrichment Analyses of DEGs
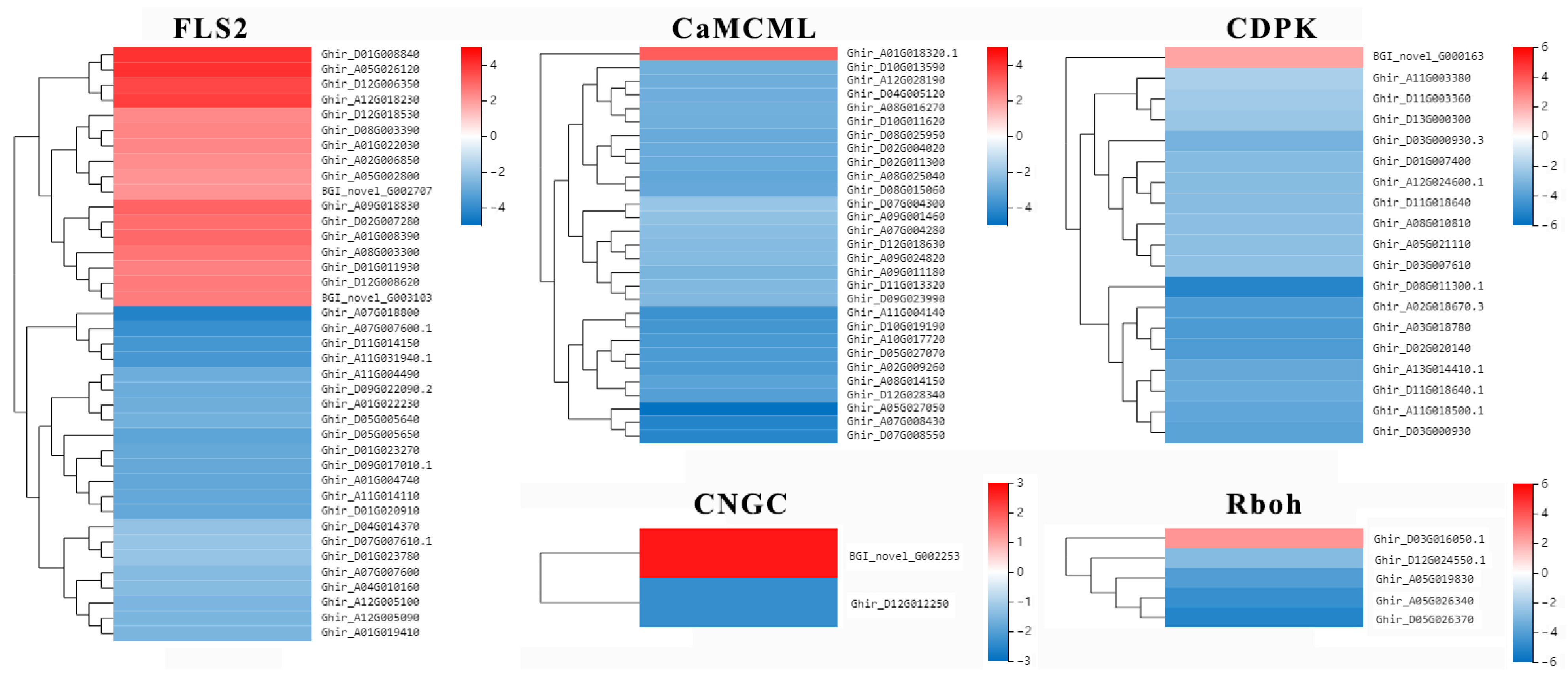
2.7. Putative R Genes Involved in Resistance to Verticillium Wilt
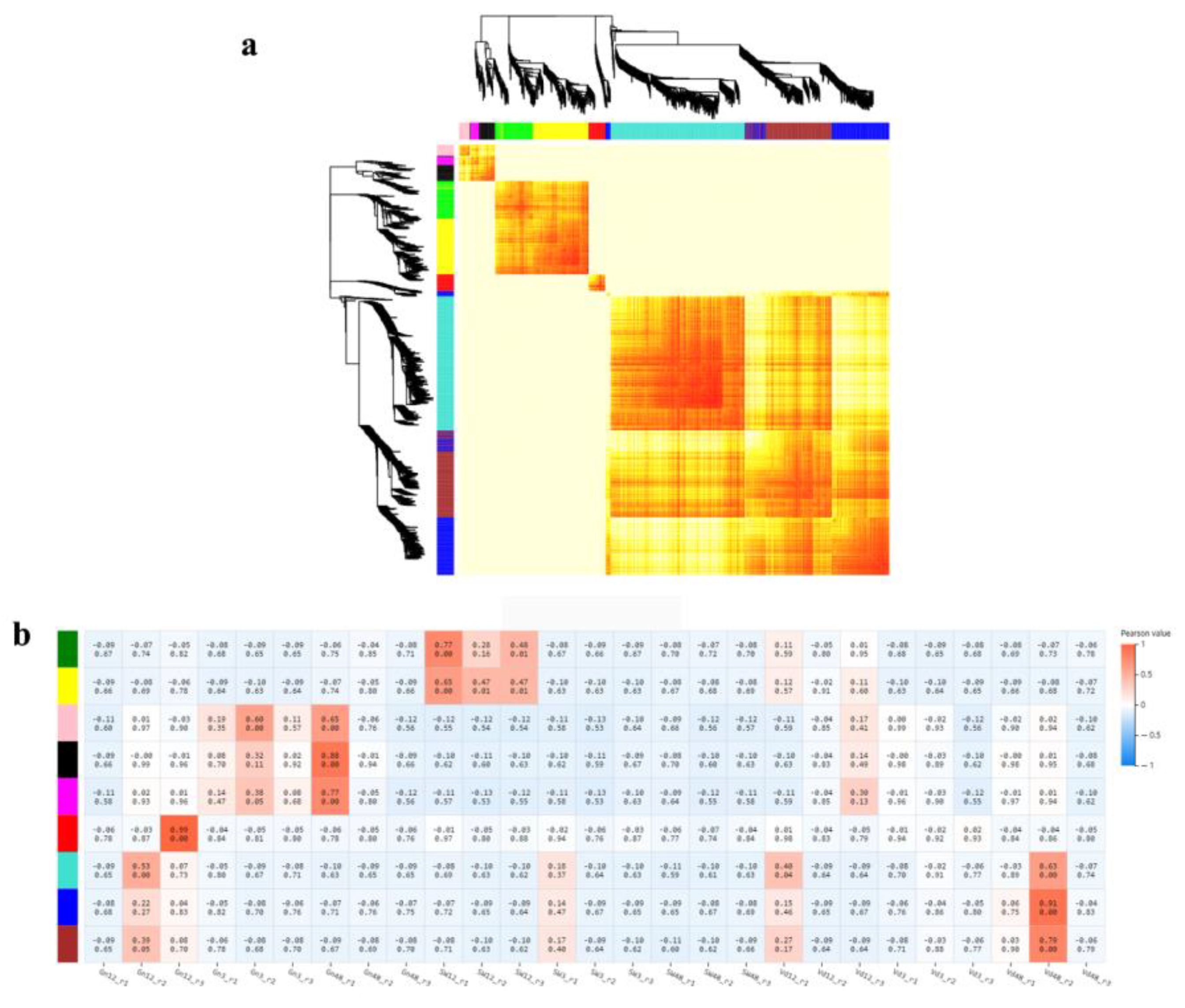
2.8. Gene Co-Expression Network Analysis
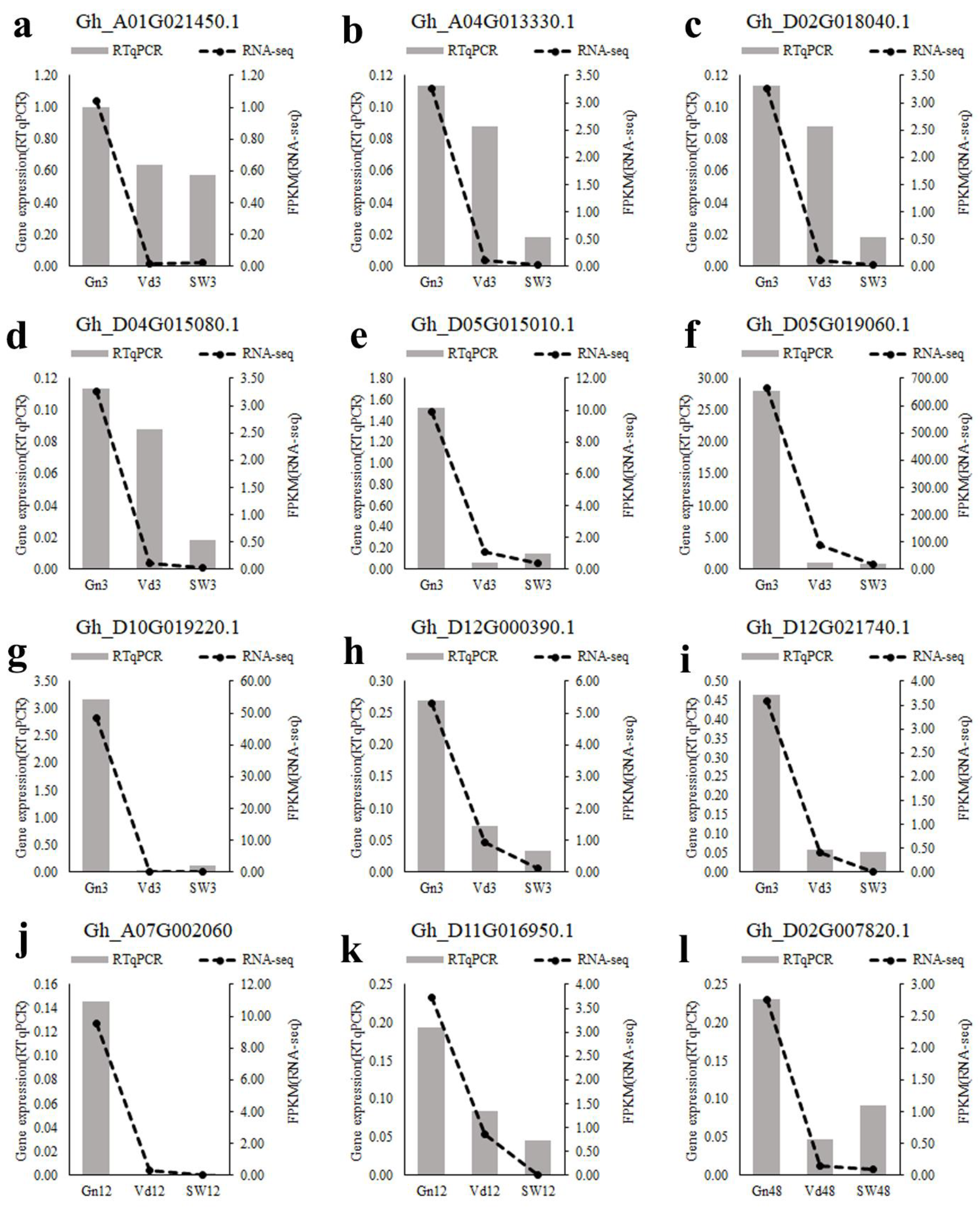
2.9. Verification of RNA-Seq Analysis by qRT-PCR
3. Discussion
4. Materials and Methods
4.1. Fungal Strain Culture
4.2. Cotton Inoculation Treatment
4.3. Control Effect of the CEF08111 on Verticillium wilt of Cotton
4.4. H2O2 Measurement
4.5. JA and SA Measurement
4.6. RNA Sequencing (RNA-Seq)
4.7. Screening and Analysis of Differentially Expressed Genes (DEGs)
4.8. Gene Co-Expression Network Analysis
4.9. Quantitative Reverse-Transcription-PCR (qRT-PCR) Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bruton, B.D.; Fish, W.W.; Subbarao, K.V.; Isakeit, T. First report of Verticillium wilt of watermelon in the texas high plains. Plant Dis. 2007, 91, 1053. [Google Scholar] [CrossRef] [PubMed]
- Klosterman, S.J.; Atallah, Z.K.; Vallad, G.E.; Subbarao, K.V. Diversity, pathogenicity, and management of Verticillium species. Annu. Rev. Phytopathol. 2009, 47, 39–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pegg, B.; Brady, G. Verticillium Wilts; CABI Publishing: New York, NY, USA, 2002. [Google Scholar]
- Zhu, H.; Song, J.; Dhar, N.; Shan, Y.; Ma, X.; Wang, X.; Chen, J.; Dai, X.; Li, R.; Wang, Z. Transcriptome analysis of a cotton cultivar provides insights into the differentially expressed genes underlying heightened resistance to the devastating Verticillium wilt. Cells 2021, 10, 2961. [Google Scholar] [CrossRef] [PubMed]
- Aini, N.; Jibril, A.N.; Liu, S.; Han, P.; Pan, Z.; Zhu, L.; Nie, X. Advances and prospects of genetic mapping of Verticillium wilt resistance in cotton. J. Cotton Res. 2022, 5, 5. [Google Scholar] [CrossRef]
- Sun, Q.; Jiang, H.; Zhu, X.; Wang, W.; He, X.; Shi, Y.; Yuan, Y.; Du, X.; Cai, Y. Analysis of sea-island cotton and upland cotton in response to Verticillium dahliae infection by RNA sequencing. BMC Genom. 2013, 14, 852. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liang, C.; Wu, S.; Zhang, X.; Tang, J.; Jian, G.; Jiao, G.; Li, F.; Chu, C. Significant Improvement of Cotton Verticillium Wilt Resistance by Manipulating the Expression of Gastrodia Antifungal Proteins. Mol. Plant. 2016, 9, 1436–1439. [Google Scholar] [CrossRef] [Green Version]
- Shaban, M.; Miao, Y.; Ullah, A.; Khan, A.Q.; Menghwar, H.; Khan, A.H.; Ahmed, M.M.; Tabassum, M.A.; Zhu, L. Physiological and molecular mechanism of defense in cotton against Verticillium dahliae. Plant Physiol. Biochem. 2018, 125, 193–204. [Google Scholar] [CrossRef]
- Yuan, Y.; Feng, H.; Wang, L.; Li, Z.; Shi, Y.; Zhao, L.; Feng, Z.; Zhu, H. Potential of endophytic fungi isolated from cotton roots for biological control against Verticillium wilt disease. PLoS ONE 2017, 12, e170557. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wang, L.; Feng, Z.; Zhao, L.; Shi, Y.; Zhu, H. Diversity of endophytic fungi from different Verticillium-wilt-resistant Gossypium hirsutum and evaluation of antifungal activity against Verticillium dahliae in vitro. J. Microbiol. Biotechnol. 2014, 24, 1149–1161. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Feng, Z.; Feng, H.; Li, Z.; Shi, Y.; Zhao, L.; Zhu, H.; Yang, J.; Zhao, L. Control effect of endophytic fungus Chaetomium globosum CEF-082 against Verticillium wilt in Gossypium hirsutum. Acta Phytopathol. Sin. 2016, 46, 697–706. [Google Scholar] [CrossRef]
- Varo, A.; Raya-Ortega, M.C.; Trapero, A. Selection and evaluation of micro-organisms for biocontrol of Verticillium dahliae in olive. J. Appl. Microbiol. 2016, 121, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lee, S.Y.; Cho, Y.J.; Ghim, S.Y.; Jung, H.Y. Mediation of induced systemic resistance by the plant growth-promoting rhizobacteria Bacillus pumilus S2-3-2. Mol. Biol. Rep. 2020, 47, 8429–8438. [Google Scholar] [CrossRef] [PubMed]
- Berg, G.; Fritze, A.; Roskot, N.; Smalla, K. Evaluation of potential biocontrol rhizobacteria from different host plants of Verticillium dahliae Kleb. J. Appl. Microbiol. 2001, 91, 963–971. [Google Scholar] [CrossRef]
- Han, Q.; Wu, F.; Wang, X.; Qi, H.; Shi, L.; Ren, A.; Liu, Q.; Zhao, M.; Tang, C. The bacterial lipopeptide iturins induce Verticillium dahliae cell death by affecting fungal signalling pathways and mediate plant defence responses involved in pathogen-associated molecular pattern-triggered immunity. Environ. Microbiol. 2015, 17, 1166–1188. [Google Scholar] [CrossRef] [PubMed]
- Veloso, J.; Diaz, J. Fusarium oxysporum Fo47 confers protection to pepper plants against Verticillium dahliae and Phytophthora eapsici, and induces the expression of defence genes. Plant Pathol. 2012, 61, 281–288. [Google Scholar] [CrossRef]
- Ding, T.; Su, B.; Chen, X.; Xie, S.; Gu, S.; Wang, Q.; Huang, D.; Jiang, H. An endophytic bacterial strain isolated from Eucommia ulm oides inhibits southern corn leaf blight. Front. Microbiol. 2017, 8, 903. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Feng, Z.; Li, Z.; Shi, Y.; Zhao, L.; Yang, J. Characterization of two fungal isolates from cotton and evaluation of their potential for biocontrol of Verticillium wilt of cotton. J. Phytopathol. 2013, 161, 70–77. [Google Scholar] [CrossRef]
- Vagelas, I.; Leontopoulos, S. Cross-protection of cotton against Verticillium wilt by Verticillium nigrescens. Emir. J. Food Agr. 2015, 27, 687–691. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wang, D.; Zhao, X.; Zhao, J.; Jing, L.; Zhou, H. Screening of attenuated Verticillium dahliae and optimization of conditions for inducing interaction protection of sunflower. Chin. J. Biol. Control. 2019, 35, 112–119. [Google Scholar] [CrossRef]
- Melouk, H.A. Verticillium nigrescens from peppermint. Phytopathology 1974, 64, 1267. [Google Scholar] [CrossRef]
- He, P.; Shan, L.; Sheen, J. Elicitation and suppression of microbe-associated molecular pattern-triggered immunity in plant-microbe interactions. Cell Microbiol. 2007, 9, 1385–1396. [Google Scholar] [CrossRef]
- Mackey, D.; Mcfall, A.J. MAMPs and MIMPs: Proposed classifications for inducers of innate immunity. Mol. Microbiol. 2006, 61, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T.; Zeh, H.J.; Rubartelli, A.; Sparvero, L.J.; Amoscato, A.A.; Washburn, N.R.; Devera, M.E.; Liang, X.; Tor, M.; Billiar, T. The grateful dead: Damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol. Rev. 2007, 220, 60–81. [Google Scholar] [CrossRef]
- Alper, S.; Mcbride, S.J.; Lackford, B.; Freedman, J.H.; Schwartz, D.A. Specificity and complexity of the Caenorhabditis elegans innate immune response. Mol. Cell. Biol. 2007, 27, 5544–5553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemaitre, B.; Hoffmann, J. The host defense of Drosophila melanogaster. Annu. Rev. Immunol. 2007, 25, 697–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Wu, Y.; Gao, M.; Zhang, J.; Kong, Q.; Liu, Y.; Ba, H.; Zhou, J.; Zhang, Y. Disruption of PAMP-induced MAP kinase cascade by a Pseudomonas syringae effector activates plant immunity mediated by the NB-LRR protein SUMM2. Cell Host Microbe 2012, 11, 253–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trda, L.; Fernandez, O.; Boutrot, F.; Heloir, M.C.; Kelloniemi, J.; Daire, X.; Adrian, M.; Clement, C.; Zipfel, C.; Dorey, S.; et al. The grapevine flagellin receptor VvFLS2 differentially recognizes flagellin-derived epitopes from the endophytic growth-promoting bacterium Burkholderia phytofirmans and plant pathogenic bacteria. New Phytol. 2014, 201, 1371–1384. [Google Scholar] [CrossRef]
- Ma, Z.; Song, T.; Zhu, L.; Ye, W.; Wang, Y.; Shao, Y.; Dong, S.; Zhang, Z.; Dou, D.; Zheng, X.; et al. A phytophthora sojae glycoside hydrolase 12 protein is a major virulence factor during soybean infection and is recognized as a PAMP. Plant Cell 2015, 27, 2057–2072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, B.; Shan, L. ROS open roads to roundworm infection. Sci. Signal. 2014, 7, e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantu, D.; Vicente, A.R.; Labavitch, J.M.; Bennett, A.B.; Powell, A.L. Strangers in the matrix: Plant cell walls and pathogen susceptibility. Trends Plant Sci. 2008, 13, 610–617. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, e17. [Google Scholar] [CrossRef]
- Guo, S.; Zuo, Y.; Zhang, Y.; Wu, C.; Su, W.; Jin, W.; Yu, H.; An, Y.; Li, Q. Large-scale transcriptome comparison of sunflower genes responsive to Verticillium dahliae. BMC Genom. 2017, 18, 42. [Google Scholar] [CrossRef] [Green Version]
- Tan, G.; Liu, K.; Kang, J.; Xu, K.; Zhang, Y.; Hu, L.; Zhang, J.; Li, C. Transcriptome analysis of the compatible interaction of tomato with Verticillium dahliae using RNA-Sequencing. Front. Plant Sci. 2015, 6, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiu, S.H.; Bleecker, A.B. Receptor-like kinases from Arabidopsis form a monophyletic gene family related to animal receptor kinases. Proc. Natl. Acad. Sci. USA 2001, 98, 10763–10768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, M.; Heard, W.; Mbengue, M.; Robatzek, S. The INs and OUTs of pattern recognition receptors at the cell surface. Curr. Opin. Plant Biol. 2012, 15, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wang, E.; Liu, J. CERK1, more than a co-receptor in plant-microbe interactions. New Phytol. 2022, 234, 1606–1613. [Google Scholar] [CrossRef]
- Zhang, D.; Bao, Y.; Sun, Y.; Yang, H.; Zhao, T.; Li, H.; Du, C.; Jiang, J.; Li, J.; Xie, L.; et al. Comparative transcriptome analysis reveals the response mechanism of Cf-16-mediated resistance to Cladosporium fulvum infection in tomato. BMC Plant Biol. 2020, 20, 33. [Google Scholar] [CrossRef] [Green Version]
- Bredow, M.; Monaghan, J. Regulation of plant immune signaling by calcium-dependent protein kinases. Mol. Plant-Microbe Interact. 2019, 32, 6–19. [Google Scholar] [CrossRef] [Green Version]
- Asano, T.; Hayashi, N.; Kobayashi, M.; Aoki, N.; Miyao, A.; Mitsuhara, I.; Ichikawa, H.; Komatsu, S.; Hirochika, H.; Kikuchi, S.; et al. A rice calcium-dependent protein kinase OsCPK12 oppositely modulates salt-stress tolerance and blast disease resistance. Plant J. 2012, 69, 26–36. [Google Scholar] [CrossRef]
- Dubiella, U.; Seybold, H.; Durian, G.; Komander, E.; Lassig, R.; Witte, C.P.; Schulze, W.X.; Romeis, T. Calcium-dependent protein kinase/NADPH oxidase activation circuit is required for rapid defense signal propagation. Proc. Natl. Acad. Sci. USA 2013, 110, 8744–8749. [Google Scholar] [CrossRef]
- Asai, T.; Tena, G.; Plotnikova, J.; Willmann, M.R.; Chiu, W.L.; Gomez-Gomez, L.; Boller, T.; Ausubel, F.M.; Sheen, J. MAP kinase signalling cascade in Arabidopsis innate immunity. Nature 2002, 415, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Wang, S.; Zhou, Y.; Bai, J.; Huang, G.; Liu, X.; Zhang, Y.; Tang, D.; Lu, D. Transcriptional regulation of the immune receptor FLS2 controls the ontogeny of plant innate immunity. Plant Cell 2018, 30, 2779–2794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Mcadams, S.A.; Bryan, G.T.; Hershey, H.P.; Valent, B. Direct interaction of resistance gene and avirulence gene products confers rice blast resistance. EMBO J. 2000, 19, 4004–4014. [Google Scholar] [CrossRef] [PubMed]
- Mackey, D.; Holt, B.R.; Wiig, A.; Dangl, J.L. RIN4 interacts with Pseudomonas syringae type III effector molecules and is required for RPM1-mediated resistance in Arabidopsis. Cell 2002, 108, 743–754. [Google Scholar] [CrossRef] [Green Version]
- Berens, M.L.; Berry, H.M.; Mine, A.; Argueso, C.T.; Tsuda, K. Evolution of hormone signaling networks in plant defense. Annu. Rev. Phytopathol. 2017, 55, 401–425. [Google Scholar] [CrossRef]
- Mao, Y.B.; Liu, Y.Q.; Chen, D.Y.; Chen, F.Y.; Fang, X.; Hong, G.J.; Wang, L.J.; Wang, J.W.; Chen, X.Y. Jasmonate response decay and defense metabolite accumulation contributes to age-regulated dynamics of plant insect resistance. Nat. Commun. 2017, 8, 13925. [Google Scholar] [CrossRef] [Green Version]
- Klessig, D.F.; Choi, H.W.; Dempsey, D.A. Systemic acquired resistance and salicylic acid: Past, present, and future. Mol. Plant-Microbe Interact. 2018, 31, 871–888. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Zhou, X.; Zhao, Y.; Zhu, S.; Wu, L.; He, Y.; Ping, X.; Lu, X.; Huang, W.; Qian, J.; et al. Colonization of endophyte Acremonium sp. D212 in Panax notoginseng and rice mediated by auxin and jasmonic acid. J. Integr. Plant Biol. 2020, 62, 1433–1451. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Z.; Feng, Z.; Feng, H.; Zhao, L.; Shi, Y.; Hu, X.; Zhu, H. Isolation and functional analysis of the pathogenicity-related gene VdPR3 from Verticillium dahliae on cotton. Curr. Genet. 2015, 61, 555–566. [Google Scholar] [CrossRef]
- Tiryaki, D.; Aydin, I.; Atici, O. Psychrotolerant bacteria isolated from the leaf apoplast of cold-adapted wild plants improve the cold resistance of bean (Phaseolus vulgaris L.) under low temperature. Cryobiology 2019, 86, 111–119. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanseverino, W.; Hermoso, A.; D’Alessandro, R.; Vlasova, A.; Andolfo, G.; Frusciante, L.; Lowy, E.; Roma, G.; Ercolano, M.R. PRGdb 2.0: Towards a community-based database model for the analysis of R-genes in plants. Nucleic Acids Res. 2013, 41, D1167–D1171. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Zhang, H.; Kong, L.; Gao, G.; Luo, J. PlantTFDB 3.0: A portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 2014, 42, D1182–D1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway ID | Pathway | Number of DEGs | p-Value | Q-Value |
|---|---|---|---|---|
| ko00010 | Glycolysis/gluconeogenesis | 14 | 9.84 × 10−8 | 6.99 × 10−6 |
| ko00940 | Phenylpropanoid biosynthesis | 16 | 1.35 × 10−5 | 0.000320 |
| ko00073 | Cutin, suberine and wax biosynthesis | 8 | 2.54 × 10−5 | 0.000450 |
| ko00520 | Amino sugar and nucleotide sugar metabolism | 13 | 0.000111 | 0.001309 |
| ko00511 | Other glycan degradation | 7 | 0.000465 | 0.004716 |
| ko00460 | Cyanoamino acid metabolism | 9 | 0.000668 | 0.005926 |
| ko04626 | Plant–pathogen interaction | 209 | 1.73 × 10−24 | 2.20 × 10−22 |
| ko04016 | MAPK signaling pathway—plant | 166 | 1.68 × 10−15 | 1.06 × 10−13 |
| ko04075 | Plant hormone signal transduction | 154 | 6.81 × 10−8 | 2.88 × 10−6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, Z.; Wei, F.; Feng, H.; Zhang, Y.; Zhao, L.; Zhou, J.; Xie, J.; Jiang, D.; Zhu, H. Transcriptome Analysis Reveals the Defense Mechanism of Cotton against Verticillium dahliae Induced by Hypovirulent Fungus Gibellulopsis nigrescens CEF08111. Int. J. Mol. Sci. 2023, 24, 1480. https://doi.org/10.3390/ijms24021480
Feng Z, Wei F, Feng H, Zhang Y, Zhao L, Zhou J, Xie J, Jiang D, Zhu H. Transcriptome Analysis Reveals the Defense Mechanism of Cotton against Verticillium dahliae Induced by Hypovirulent Fungus Gibellulopsis nigrescens CEF08111. International Journal of Molecular Sciences. 2023; 24(2):1480. https://doi.org/10.3390/ijms24021480
Chicago/Turabian StyleFeng, Zili, Feng Wei, Hongjie Feng, Yalin Zhang, Lihong Zhao, Jinglong Zhou, Jiatao Xie, Daohong Jiang, and Heqin Zhu. 2023. "Transcriptome Analysis Reveals the Defense Mechanism of Cotton against Verticillium dahliae Induced by Hypovirulent Fungus Gibellulopsis nigrescens CEF08111" International Journal of Molecular Sciences 24, no. 2: 1480. https://doi.org/10.3390/ijms24021480
APA StyleFeng, Z., Wei, F., Feng, H., Zhang, Y., Zhao, L., Zhou, J., Xie, J., Jiang, D., & Zhu, H. (2023). Transcriptome Analysis Reveals the Defense Mechanism of Cotton against Verticillium dahliae Induced by Hypovirulent Fungus Gibellulopsis nigrescens CEF08111. International Journal of Molecular Sciences, 24(2), 1480. https://doi.org/10.3390/ijms24021480