
Pathophysiology of Inflammatory Bowel Disease: Innate Immune System
 , ,
, , 
{kind=link}
{kind=link}
Abstract
:1. Introduction
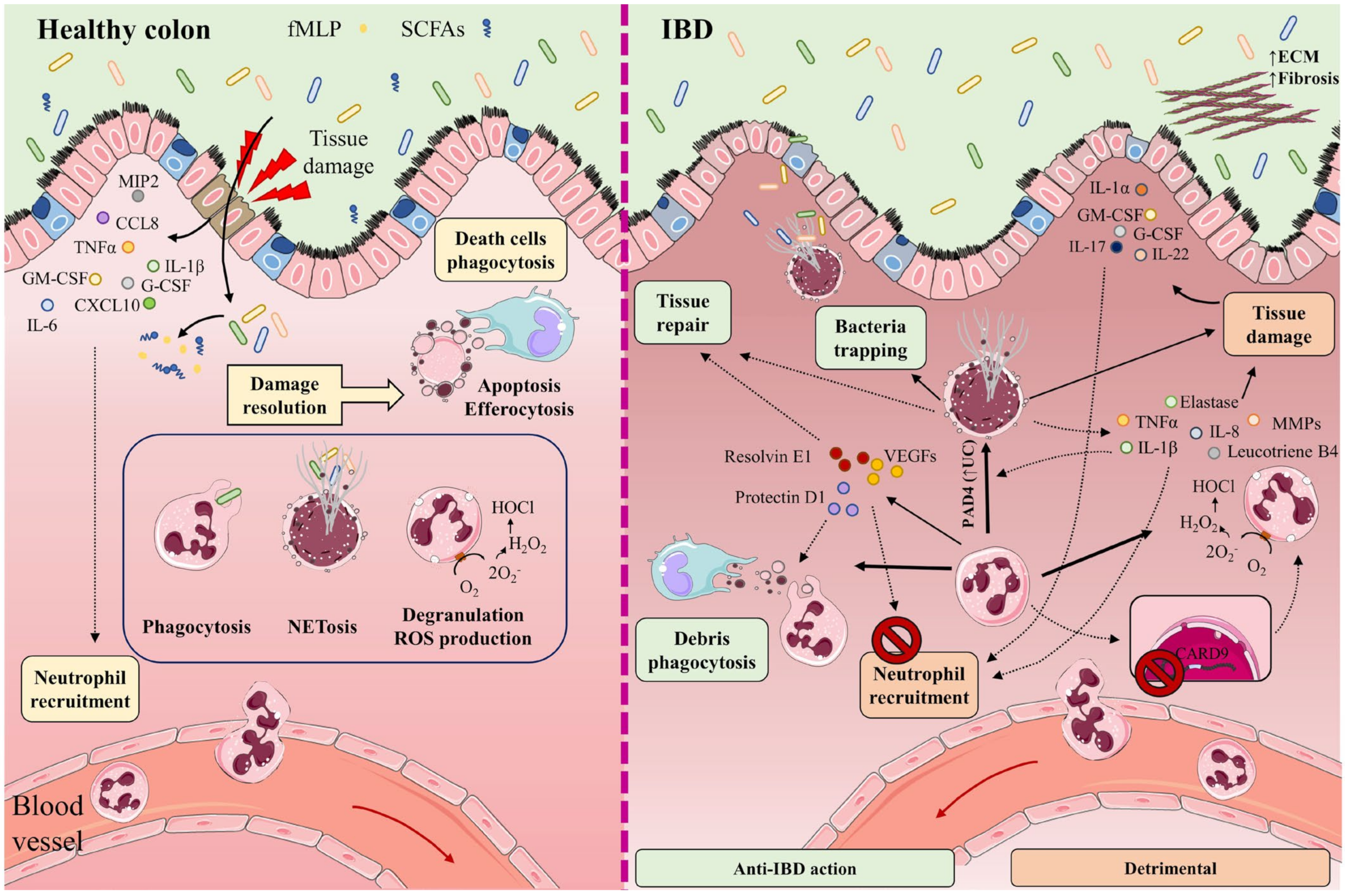
2. IBD Pathophysiology
2.1. Innate Immune Cells in the Pathogenesis of IBD
2.1.1. Neutrophils in Gut Homeostasis
2.1.2. Neutrophils in the Gut during IBD
2.1.3. Macrophages in the Gut in Steady State Conditions
2.1.4. Macrophages in the Pathogenesis of IBD
2.1.5. Innate Lymphoid Cells in IBD
2.1.6. Dendritic Cells in Homeostasis
2.1.7. Dendritic Cells in IBD
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | Antigen presenting cell |
| Card9 | Caspase recruitment domain 9 |
| CCL | Chemokine (C-C motif) ligand |
| CCR | C-C motif chemokine receptor |
| CD | Crohn’s disease |
| CitH3 | Citrullination histone H3 |
| CLA | Cutaneous lymphocyte antigen |
| CPD | Common dendritic progenitor |
| CXCL | Chemokine (C-X-C motif) ligand |
| CX3CR | Chemokine (C-X3-C motif) receptor |
| DAMPs | Damage-associated molecular patterns |
| DCs | Dendritic cells |
| DSS | Dextran sodium sulfate |
| ECM | Extracellular matrix |
| Flt3L | Fms-like tyrosine kinase 3 ligand |
| Foxp3 | Forkhead Box P3 |
| G-CSF | Granulocyte colony-stimulating factor |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| GWAS | Genome-wide association studies |
| IBD | Inflammatory bowel disease |
| IFN | Interferon |
| Ig | Immunoglobulin |
| IIC | Innate immune cells |
| iNOS | Inducible nitric oxide synthetase |
| ILCs | Innate lymphoid cells |
| IL | Interleukin |
| IRF | Interferon regulatory factor |
| MCP | Monocyte chemoattractant protein |
| MHCII | Major histocompatibility complex molecules class II |
| MIP | Macrophage inflammatory proteins |
| MMPs | Matrix metalloproteinases |
| MMT | Myofibroblast transition |
| MPO | Myeloperoxidase |
| mTOR | Mammalian target of rapamycin |
| NK | Natural killer |
| NETs | Neutrophil extracellular traps |
| NOD2 | Nucleotide binding oligomerization domain containing 2 |
| NLR | Nod-like receptors |
| PAD4 | Protein arginine deiminase 4 |
| PAMPs | Pathogen-associated molecular patterns |
| pDC | Plasmacytoid DCs |
| RA | Retinoic acid |
| PRRs | Pattern recognition receptors |
| RORγt | RAR-related orphan receptor gamma t |
| SCFAs | Short-chain fatty acids |
| scRNA-seq | Single cell RNA sequencing |
| SIPRα | Signal-regulatory protein alpha |
| STAT | Signal transducer and activator of transcription |
| Th | T-helper |
| TGF | Transforming growth factor |
| TL1A | TNF-like ligand 1 A |
| TLRs | Toll-like receptors |
| TNBS | 2,4,6-trinitrobenzene sulfonic acid |
| TNF | Tumor necrosis factor |
| Treg | T regulatory |
| Tr1 | T regulatory type 1 |
| TWAS | Transcriptome-wide association studies |
| UC | Ulcerative colitis |
| WNT | Wingless-related integration site |
| XCR1 | Chemokine (C motif) receptor 1 |
References
- Ananthakrishnan, A.N.; Bernstein, C.N.; Iliopoulos, D.; Macpherson, A.; Neurath, M.F.; Ali, R.A.R.; Vavricka, S.R.; Fiocchi, C. Environmental triggers in IBD: A review of progress and evidence. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.; Griffiths, A.; Markowitz, J.; Wilson, D.C.; Turner, D.; Russell, R.K.; Fell, J.; Ruemmele, F.M.; Walters, T.; Sherlock, M.; et al. Pediatric modification of the Montreal classification for inflammatory bowel disease: The Paris classification. Inflamm. Bowel Dis. 2011, 17, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Louis, E.; Van Kemseke, C.; Reenaers, C. Necessity of phenotypic classification of inflammatory bowel disease. Best Pract. Res. Clin. Gastroenterol. 2011, 25 (Suppl. 1), S2–S7. [Google Scholar] [CrossRef] [PubMed]
- Satsangi, J.; Silverberg, M.S.; Vermeire, S.; Colombel, J.F. The Montreal classification of inflammatory bowel disease: Controversies, consensus, and implications. Gut 2006, 55, 749–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 46–54.e42, quiz e30. [Google Scholar] [CrossRef] [Green Version]
- Ananthakrishnan, A.N. Epidemiology and risk factors for IBD. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 205–217. [Google Scholar] [CrossRef]
- Chang, J.T. Pathophysiology of Inflammatory Bowel Diseases. N. Engl. J. Med. 2020, 383, 2652–2664. [Google Scholar] [CrossRef]
- Pullan, R.D.; Thomas, G.A.; Rhodes, M.; Newcombe, R.G.; Williams, G.T.; Allen, A.; Rhodes, J. Thickness of adherent mucus gel on colonic mucosa in humans and its relevance to colitis. Gut 1994, 35, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Parikh, K.; Antanaviciute, A.; Fawkner-Corbett, D.; Jagielowicz, M.; Aulicino, A.; Lagerholm, C.; Davis, S.; Kinchen, J.; Chen, H.H.; Alham, N.K.; et al. Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature 2019, 567, 49–55. [Google Scholar] [CrossRef]
- Kinchen, J.; Chen, H.H.; Parikh, K.; Antanaviciute, A.; Jagielowicz, M.; Fawkner-Corbett, D.; Ashley, N.; Cubitt, L.; Mellado-Gomez, E.; Attar, M.; et al. Structural Remodeling of the Human Colonic Mesenchyme in Inflammatory Bowel Disease. Cell 2018, 175, 372–386.e17. [Google Scholar] [CrossRef]
- Gustafsson, J.K.; Johansson, M.E.V. The role of goblet cells and mucus in intestinal homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 785–803. [Google Scholar] [CrossRef]
- Ghilas, S.; O’Keefe, R.; Mielke, L.A.; Raghu, D.; Buchert, M.; Ernst, M. Crosstalk between epithelium, myeloid and innate lymphoid cells during gut homeostasis and disease. Front. Immunol. 2022, 13, 944982. [Google Scholar] [CrossRef]
- Bassler, K.; Schulte-Schrepping, J.; Warnat-Herresthal, S.; Aschenbrenner, A.C.; Schultze, J.L. The Myeloid Cell Compartment-Cell by Cell. Annu. Rev. Immunol. 2019, 37, 269–293. [Google Scholar] [CrossRef]
- Herrero-Fernandez, B.; Gomez-Bris, R.; Somovilla-Crespo, B.; Gonzalez-Granado, J.M. Immunobiology of Atherosclerosis: A Complex Net of Interactions. Int. J. Mol. Sci. 2019, 20, 5293. [Google Scholar] [CrossRef] [Green Version]
- Leppkes, M.; Neurath, M.F. Cytokines in inflammatory bowel diseases—Update 2020. Pharmacol. Res. 2020, 158, 104835. [Google Scholar] [CrossRef]
- Mann, E.A.; Saeed, S.A. Gastrointestinal infection as a trigger for inflammatory bowel disease. Curr. Opin. Gastroenterol. 2012, 28, 24–29. [Google Scholar] [CrossRef]
- Schroeder, B.O.; Birchenough, G.M.H.; Stahlman, M.; Arike, L.; Johansson, M.E.V.; Hansson, G.C.; Backhed, F. Bifidobacteria or Fiber Protects against Diet-Induced Microbiota-Mediated Colonic Mucus Deterioration. Cell Host Microbe 2018, 23, 27–40.e7. [Google Scholar] [CrossRef] [Green Version]
- Neurath, M.F.; Leppkes, M. Resolution of ulcerative colitis. Semin. Immunopathol. 2019, 41, 747–756. [Google Scholar] [CrossRef]
- Wen, Z.; Fiocchi, C. Inflammatory bowel disease: Autoimmune or immune-mediated pathogenesis? Clin. Dev. Immunol. 2004, 11, 195–204. [Google Scholar] [CrossRef]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef]
- Munoz, L.E.; Leppkes, M.; Fuchs, T.A.; Hoffmann, M.; Herrmann, M. Missing in action-The meaning of cell death in tissue damage and inflammation. Immunol. Rev. 2017, 280, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Yu, L.; Fang, L.; Yang, W.; Yu, T.; Miao, Y.; Chen, M.; Wu, K.; Chen, F.; Cong, Y.; et al. CD177(+) neutrophils as functionally activated neutrophils negatively regulate IBD. Gut 2018, 67, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateer, S.W.; Mathe, A.; Bruce, J.; Liu, G.; Maltby, S.; Fricker, M.; Goggins, B.J.; Tay, H.L.; Marks, E.; Burns, G.; et al. IL-6 Drives Neutrophil-Mediated Pulmonary Inflammation Associated with Bacteremia in Murine Models of Colitis. Am. J. Pathol. 2018, 188, 1625–1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, M.; El Azreq, M.A.; Pelletier, J.; Robaye, B.; Aoudjit, F.; Sevigny, J. Exacerbated intestinal inflammation in P2Y6 deficient mice is associated with Th17 activation. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2595–2605. [Google Scholar] [CrossRef]
- Kamp, M.E.; Shim, R.; Nicholls, A.J.; Oliveira, A.C.; Mason, L.J.; Binge, L.; Mackay, C.R.; Wong, C.H. G Protein-Coupled Receptor 43 Modulates Neutrophil Recruitment during Acute Inflammation. PLoS ONE 2016, 11, e0163750. [Google Scholar] [CrossRef] [Green Version]
- Correa, R.O.; Vieira, A.; Sernaglia, E.M.; Lancellotti, M.; Vieira, A.T.; Avila-Campos, M.J.; Rodrigues, H.G.; Vinolo, M.A.R. Bacterial short-chain fatty acid metabolites modulate the inflammatory response against infectious bacteria. Cell. Microbiol. 2017, 19, e12720. [Google Scholar] [CrossRef] [Green Version]
- Bedouhene, S.; Liu, M.; Senani, N.; Boussetta, T.; Pintard, C.; Dang, P.M.; El-Benna, J. Prolyl-Isomerase Pin1 Controls Key fMLP-Induced Neutrophil Functions. Biomedicines 2021, 9, 1130. [Google Scholar] [CrossRef]
- Yuen, J.; Pluthero, F.G.; Douda, D.N.; Riedl, M.; Cherry, A.; Ulanova, M.; Kahr, W.H.; Palaniyar, N.; Licht, C. NETosing Neutrophils Activate Complement Both on Their Own NETs and Bacteria via Alternative and Non-alternative Pathways. Front. Immunol. 2016, 7, 137. [Google Scholar] [CrossRef] [Green Version]
- Matoszka, N.; Dzialo, J.; Tokarz-Deptula, B.; Deptula, W. NET and NETosis—New phenomenon in immunology. Postepy. Hig. Med. Dosw. 2012, 66, 437–445. [Google Scholar] [CrossRef]
- Dinallo, V.; Marafini, I.; Di Fusco, D.; Laudisi, F.; Franze, E.; Di Grazia, A.; Figliuzzi, M.M.; Caprioli, F.; Stolfi, C.; Monteleone, I.; et al. Neutrophil Extracellular Traps Sustain Inflammatory Signals in Ulcerative Colitis. J. Crohn’s Colitis 2019, 13, 772–784. [Google Scholar] [CrossRef]
- Kaur, A.; Goggolidou, P. Ulcerative colitis: Understanding its cellular pathology could provide insights into novel therapies. J. Inflamm. 2020, 17, 15. [Google Scholar] [CrossRef] [Green Version]
- Scannell, M.; Flanagan, M.B.; de Stefani, A.; Wynne, K.J.; Cagney, G.; Godson, C.; Maderna, P. Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J. Immunol. 2007, 178, 4595–4605. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Alexander, M.; Misharin, A.V.; Budinger, G.R.S. The role of macrophages in the resolution of inflammation. J. Clin. Investig. 2019, 129, 2619–2628. [Google Scholar] [CrossRef] [Green Version]
- McCracken, J.M.; Allen, L.A. Regulation of human neutrophil apoptosis and lifespan in health and disease. J. Cell Death 2014, 7, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Liu, Y.; Shi, Y.; Zhang, J.; Liu, X.; Liu, Z.; Lv, J.; Leng, Y. The emerging role of neutrophilic extracellular traps in intestinal disease. Gut Pathog. 2022, 14, 27. [Google Scholar] [CrossRef]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef]
- Drury, B.; Hardisty, G.; Gray, R.D.; Ho, G.T. Neutrophil Extracellular Traps in Inflammatory Bowel Disease: Pathogenic Mechanisms and Clinical Translation. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 321–333. [Google Scholar] [CrossRef]
- Bennike, T.B.; Carlsen, T.G.; Ellingsen, T.; Bonderup, O.K.; Glerup, H.; Bogsted, M.; Christiansen, G.; Birkelund, S.; Stensballe, A.; Andersen, V. Neutrophil Extracellular Traps in Ulcerative Colitis: A Proteome Analysis of Intestinal Biopsies. Inflamm. Bowel Dis. 2015, 21, 2052–2067. [Google Scholar] [CrossRef]
- Maronek, M.; Gromova, B.; Liptak, R.; Konecna, B.; Pastorek, M.; Cechova, B.; Harsanyova, M.; Budis, J.; Smolak, D.; Radvanszky, J.; et al. Extracellular DNA Correlates with Intestinal Inflammation in Chemically Induced Colitis in Mice. Cells 2021, 10, 81. [Google Scholar] [CrossRef] [PubMed]
- Hansberry, D.R.; Shah, K.; Agarwal, P.; Agarwal, N. Fecal Myeloperoxidase as a Biomarker for Inflammatory Bowel Disease. Cureus 2017, 9, e1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abd El Hafez, A.; Mohamed, A.S.; Shehta, A.; Sheta, H. Neutrophil extracellular traps-associated protein peptidyl arginine deaminase 4 immunohistochemical expression in ulcerative colitis and its association with the prognostic predictors. Pathol. Res. Pract. 2020, 216, 153102. [Google Scholar] [CrossRef] [PubMed]
- Kaluzna, A.; Olczyk, P.; Komosinska-Vassev, K. The Role of Innate and Adaptive Immune Cells in the Pathogenesis and Development of the Inflammatory Response in Ulcerative Colitis. J. Clin. Med. 2022, 11, 400. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Mei, Y.; Dong, W.; Wang, J.; Huang, F.; Wu, J. Evaluation of protein arginine deiminase-4 inhibitor in TNBS- induced colitis in mice. Int. Immunopharmacol. 2020, 84, 106583. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wang, C.; Liu, Y.; Li, B.; Zhang, W.; Wang, L.; Yu, M.; Zhao, X.; Du, J.; Zhang, J.; et al. Neutrophil Extracellular Traps Induce Intestinal Damage and Thrombotic Tendency in Inflammatory Bowel Disease. J. Crohn’s Colitis 2020, 14, 240–253. [Google Scholar] [CrossRef]
- Lin, E.Y.; Lai, H.J.; Cheng, Y.K.; Leong, K.Q.; Cheng, L.C.; Chou, Y.C.; Peng, Y.C.; Hsu, Y.H.; Chiang, H.S. Neutrophil Extracellular Traps Impair Intestinal Barrier Function during Experimental Colitis. Biomedicines 2020, 8, 275. [Google Scholar] [CrossRef]
- Dong, W.; Liu, D.; Zhang, T.; You, Q.; Huang, F.; Wu, J. Oral delivery of staphylococcal nuclease ameliorates DSS induced ulcerative colitis in mice via degrading intestinal neutrophil extracellular traps. Ecotoxicol. Environ. Saf. 2021, 215, 112161. [Google Scholar] [CrossRef]
- Li, G.; Lin, J.; Zhang, C.; Gao, H.; Lu, H.; Gao, X.; Zhu, R.; Li, Z.; Li, M.; Liu, Z. Microbiota metabolite butyrate constrains neutrophil functions and ameliorates mucosal inflammation in inflammatory bowel disease. Gut Microbes 2021, 13, 1968257. [Google Scholar] [CrossRef]
- Biasi, F.; Leonarduzzi, G.; Oteiza, P.I.; Poli, G. Inflammatory bowel disease: Mechanisms, redox considerations, and therapeutic targets. Antioxid. Redox Signal. 2013, 19, 1711–1747. [Google Scholar] [CrossRef]
- Wera, O.; Lancellotti, P.; Oury, C. The Dual Role of Neutrophils in Inflammatory Bowel Diseases. J. Clin. Med. 2016, 5, 118. [Google Scholar] [CrossRef]
- Zhou, G.X.; Liu, Z.J. Potential roles of neutrophils in regulating intestinal mucosal inflammation of inflammatory bowel disease. J. Dig. Dis. 2017, 18, 495–503. [Google Scholar] [CrossRef] [Green Version]
- Pavlidis, P.; Tsakmaki, A.; Pantazi, E.; Li, K.; Cozzetto, D.; Digby-Bell, J.; Yang, F.; Lo, J.W.; Alberts, E.; Sa, A.C.C.; et al. Interleukin-22 regulates neutrophil recruitment in ulcerative colitis and is associated with resistance to ustekinumab therapy. Nat. Commun. 2022, 13, 5820. [Google Scholar] [CrossRef]
- Fenini, G.; Contassot, E.; French, L.E. Potential of IL-1, IL-18 and Inflammasome Inhibition for the Treatment of Inflammatory Skin Diseases. Front. Pharmacol. 2017, 8, 278. [Google Scholar] [CrossRef] [Green Version]
- Mao, L.; Kitani, A.; Strober, W.; Fuss, I.J. The Role of NLRP3 and IL-1beta in the Pathogenesis of Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 2566. [Google Scholar] [CrossRef] [Green Version]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [Green Version]
- Di Paolo, N.C.; Shayakhmetov, D.M. Interleukin 1alpha and the inflammatory process. Nat. Immunol. 2016, 17, 906–913. [Google Scholar] [CrossRef] [Green Version]
- Laan, M.; Prause, O.; Miyamoto, M.; Sjostrand, M.; Hytonen, A.M.; Kaneko, T.; Lotvall, J.; Linden, A. A role of GM-CSF in the accumulation of neutrophils in the airways caused by IL-17 and TNF-alpha. Eur. Respir. J. 2003, 21, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Leppkes, M.; Becker, C.; Ivanov, I.I.; Hirth, S.; Wirtz, S.; Neufert, C.; Pouly, S.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; et al. RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology 2009, 136, 257–267. [Google Scholar] [CrossRef]
- Stark, M.A.; Huo, Y.; Burcin, T.L.; Morris, M.A.; Olson, T.S.; Ley, K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity 2005, 22, 285–294. [Google Scholar] [CrossRef]
- Phillipson, M.; Kubes, P. The Healing Power of Neutrophils. Trends. Immunol. 2019, 40, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Danne, C.; Michaudel, C.; Skerniskyte, J.; Planchais, J.; Magniez, A.; Agus, A.; Michel, M.L.; Lamas, B.; Da Costa, G.; Spatz, M.; et al. CARD9 in neutrophils protects from colitis and controls mitochondrial metabolism and cell survival. Gut 2022. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Fang, X.; Song, Y.H.; He, Z.X.; Wang, Z.J.; Wang, S.L.; Li, Z.S.; Bai, Y. Neutrophil-Epithelial Crosstalk During Intestinal Inflammation. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 1257–1267. [Google Scholar] [CrossRef] [PubMed]
- McCourt, M.; Wang, J.H.; Sookhai, S.; Redmond, H.P. Proinflammatory mediators stimulate neutrophil-directed angiogenesis. Arch. Surg. 1999, 134, 1325–1331, discussion 1331–1332. [Google Scholar] [CrossRef] [PubMed]
- Schwab, J.M.; Chiang, N.; Arita, M.; Serhan, C.N. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 2007, 447, 869–874. [Google Scholar] [CrossRef] [Green Version]
- Seo, D.H.; Che, X.; Kim, S.; Kim, D.H.; Ma, H.W.; Kim, J.H.; Kim, T.I.; Kim, W.H.; Kim, S.W.; Cheon, J.H. Triggering Receptor Expressed on Myeloid Cells-1 Agonist Regulates Intestinal Inflammation via Cd177(+) Neutrophils. Front. Immunol. 2021, 12, 650864. [Google Scholar] [CrossRef]
- Clancy, D.M.; Sullivan, G.P.; Moran, H.B.T.; Henry, C.M.; Reeves, E.P.; McElvaney, N.G.; Lavelle, E.C.; Martin, S.J. Extracellular Neutrophil Proteases are Efficient Regulators of IL-1, IL-33, and IL-36 Cytokine Activity but Poor Effectors of Microbial Killing. Cell Rep. 2018, 22, 2937–2950. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Chen, Q.; Chertov, O.; Oppenheim, J.J. Human neutrophil defensins selectively chemoattract naive T and immature dendritic cells. J. Leukoc. Biol. 2000, 68, 9–14. [Google Scholar] [CrossRef]
- Cunliffe, R.N.; Kamal, M.; Rose, F.R.; James, P.D.; Mahida, Y.R. Expression of antimicrobial neutrophil defensins in epithelial cells of active inflammatory bowel disease mucosa. J. Clin. Pathol. 2002, 55, 298–304. [Google Scholar] [CrossRef]
- Yui, S.; Nakatani, Y.; Mikami, M. Calprotectin (S100A8/S100A9), an inflammatory protein complex from neutrophils with a broad apoptosis-inducing activity. Biol. Pharm. Bull. 2003, 26, 753–760. [Google Scholar] [CrossRef]
- Karmakar, M.; Minns, M.; Greenberg, E.N.; Diaz-Aponte, J.; Pestonjamasp, K.; Johnson, J.L.; Rathkey, J.K.; Abbott, D.W.; Wang, K.; Shao, F.; et al. N-GSDMD trafficking to neutrophil organelles facilitates IL-1beta release independently of plasma membrane pores and pyroptosis. Nat. Commun. 2020, 11, 2212. [Google Scholar] [CrossRef]
- Ozaki, R.; Kobayashi, T.; Okabayashi, S.; Nakano, M.; Morinaga, S.; Hara, A.; Ohbu, M.; Matsuoka, K.; Toyonaga, T.; Saito, E.; et al. Histological Risk Factors to Predict Clinical Relapse in Ulcerative Colitis with Endoscopically Normal Mucosa. J. Crohn’s Colitis 2018, 12, 1288–1294. [Google Scholar] [CrossRef]
- Surawicz, C.M.; Haggitt, R.C.; Husseman, M.; McFarland, L.V. Mucosal biopsy diagnosis of colitis: Acute self-limited colitis and idiopathic inflammatory bowel disease. Gastroenterology 1994, 107, 755–763. [Google Scholar] [CrossRef]
- Shinoda, M.; Shin-Ya, M.; Naito, Y.; Kishida, T.; Ito, R.; Suzuki, N.; Yasuda, H.; Sakagami, J.; Imanishi, J.; Kataoka, K.; et al. Early-stage blocking of Notch signaling inhibits the depletion of goblet cells in dextran sodium sulfate-induced colitis in mice. J. Gastroenterol. 2010, 45, 608–617. [Google Scholar] [CrossRef]
- Leiper, K.; Campbell, B.J.; Jenkinson, M.D.; Milton, J.; Yu, L.G.; Democratis, J.; Rhodes, J.M. Interaction between bacterial peptides, neutrophils and goblet cells: A possible mechanism for neutrophil recruitment and goblet cell depletion in colitis. Clin. Sci. 2001, 101, 395–402. [Google Scholar] [CrossRef]
- Bilyy, R.; Fedorov, V.; Vovk, V.; Leppkes, M.; Dumych, T.; Chopyak, V.; Schett, G.; Herrmann, M. Neutrophil Extracellular Traps Form a Barrier between Necrotic and Viable Areas in Acute Abdominal Inflammation. Front. Immunol. 2016, 7, 424. [Google Scholar] [CrossRef] [Green Version]
- Bain, C.C.; Schridde, A. Origin, Differentiation, and Function of Intestinal Macrophages. Front. Immunol. 2018, 9, 2733. [Google Scholar] [CrossRef] [Green Version]
- Bleriot, C.; Chakarov, S.; Ginhoux, F. Determinants of Resident Tissue Macrophage Identity and Function. Immunity 2020, 52, 957–970. [Google Scholar] [CrossRef]
- Lis-Lopez, L.; Bauset, C.; Seco-Cervera, M.; Cosin-Roger, J. Is the Macrophage Phenotype Determinant for Fibrosis Development? Biomedicines 2021, 9, 1747. [Google Scholar] [CrossRef]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef]
- Guilliams, M.; Scott, C.L. Does niche competition determine the origin of tissue-resident macrophages? Nat. Rev. Immunol. 2017, 17, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haldar, M.; Murphy, K.M. Origin, development, and homeostasis of tissue-resident macrophages. Immunol. Rev. 2014, 262, 25–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef] [Green Version]
- Perdiguero, E.G.; Geissmann, F. The development and maintenance of resident macrophages. Nat. Immunol. 2016, 17, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef]
- De Schepper, S.; Verheijden, S.; Aguilera-Lizarraga, J.; Viola, M.F.; Boesmans, W.; Stakenborg, N.; Voytyuk, I.; Schmidt, I.; Boeckx, B.; Dierckx de Casterle, I.; et al. Self-Maintaining Gut Macrophages Are Essential for Intestinal Homeostasis. Cell 2018, 175, 400–415.e13. [Google Scholar] [CrossRef] [Green Version]
- Bain, C.C.; Bravo-Blas, A.; Scott, C.L.; Perdiguero, E.G.; Geissmann, F.; Henri, S.; Malissen, B.; Osborne, L.C.; Artis, D.; Mowat, A.M. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat. Immunol. 2014, 15, 929–937. [Google Scholar] [CrossRef] [Green Version]
- Bain, C.C.; Scott, C.L.; Uronen-Hansson, H.; Gudjonsson, S.; Jansson, O.; Grip, O.; Guilliams, M.; Malissen, B.; Agace, W.W.; Mowat, A.M. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 2013, 6, 498–510. [Google Scholar] [CrossRef]
- Tamoutounour, S.; Henri, S.; Lelouard, H.; de Bovis, B.; de Haar, C.; van der Woude, C.J.; Woltman, A.M.; Reyal, Y.; Bonnet, D.; Sichien, D.; et al. CD64 distinguishes macrophages from dendritic cells in the gut and reveals the Th1-inducing role of mesenteric lymph node macrophages during colitis. Eur. J. Immunol. 2012, 42, 3150–3166. [Google Scholar] [CrossRef]
- Delfini, M.; Stakenborg, N.; Viola, M.F.; Boeckxstaens, G. Macrophages in the gut: Masters in multitasking. Immunity 2022, 55, 1530–1548. [Google Scholar] [CrossRef]
- Viola, M.F.; Boeckxstaens, G. Niche-specific functional heterogeneity of intestinal resident macrophages. Gut 2021, 70, 1383–1395. [Google Scholar] [CrossRef]
- Gabanyi, I.; Muller, P.A.; Feighery, L.; Oliveira, T.Y.; Costa-Pinto, F.A.; Mucida, D. Neuro-immune Interactions Drive Tissue Programming in Intestinal Macrophages. Cell 2016, 164, 378–391. [Google Scholar] [CrossRef] [Green Version]
- Na, Y.R.; Stakenborg, M.; Seok, S.H.; Matteoli, G. Macrophages in intestinal inflammation and resolution: A potential therapeutic target in IBD. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 531–543. [Google Scholar] [CrossRef]
- Shaw, T.N.; Houston, S.A.; Wemyss, K.; Bridgeman, H.M.; Barbera, T.A.; Zangerle-Murray, T.; Strangward, P.; Ridley, A.J.L.; Wang, P.; Tamoutounour, S.; et al. Tissue-resident macrophages in the intestine are long lived and defined by Tim-4 and CD4 expression. J. Exp. Med. 2018, 215, 1507–1518. [Google Scholar] [CrossRef]
- Muller, P.A.; Koscso, B.; Rajani, G.M.; Stevanovic, K.; Berres, M.L.; Hashimoto, D.; Mortha, A.; Leboeuf, M.; Li, X.M.; Mucida, D.; et al. Crosstalk between muscularis macrophages and enteric neurons regulates gastrointestinal motility. Cell 2014, 158, 300–313. [Google Scholar] [CrossRef] [Green Version]
- Hume, D.A.; Perry, V.H.; Gordon, S. The mononuclear phagocyte system of the mouse defined by immunohistochemical localisation of antigen F4/80: Macrophages associated with epithelia. Anat. Rec. 1984, 210, 503–512. [Google Scholar] [CrossRef]
- Hayashi, A.; Sato, T.; Kamada, N.; Mikami, Y.; Matsuoka, K.; Hisamatsu, T.; Hibi, T.; Roers, A.; Yagita, H.; Ohteki, T.; et al. A single strain of Clostridium butyricum induces intestinal IL-10-producing macrophages to suppress acute experimental colitis in mice. Cell Host Microbe 2013, 13, 711–722. [Google Scholar] [CrossRef] [Green Version]
- Danne, C.; Ryzhakov, G.; Martinez-Lopez, M.; Ilott, N.E.; Franchini, F.; Cuskin, F.; Lowe, E.C.; Bullers, S.J.; Arthur, J.S.C.; Powrie, F. A Large Polysaccharide Produced by Helicobacter hepaticus Induces an Anti-inflammatory Gene Signature in Macrophages. Cell Host Microbe 2017, 22, 733–745.e5. [Google Scholar] [CrossRef]
- Hiemstra, I.H.; Beijer, M.R.; Veninga, H.; Vrijland, K.; Borg, E.G.; Olivier, B.J.; Mebius, R.E.; Kraal, G.; den Haan, J.M. The identification and developmental requirements of colonic CD169(+) macrophages. Immunology 2014, 142, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, A.; Donaldson, D.S.; Pridans, C.; Sauter, K.A.; Hume, D.A.; Mabbott, N.A. The role of CSF1R-dependent macrophages in control of the intestinal stem-cell niche. Nat. Commun. 2018, 9, 1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadis, U.; Wahl, B.; Schulz, O.; Hardtke-Wolenski, M.; Schippers, A.; Wagner, N.; Muller, W.; Sparwasser, T.; Forster, R.; Pabst, O. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity 2011, 34, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.H.; Kamada, N.; Kim, Y.G.; Nunez, G. Microbiota-induced IL-1beta, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J. Exp. Med. 2012, 209, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Tan, S.; Shao, Z.; Wang, X. Latitudinal and longitudinal regulation of tissue macrophages in inflammatory diseases. Genes Dis. 2022, 9, 1194–1207. [Google Scholar] [CrossRef]
- Yao, H.; Tang, G. Macrophages in intestinal fibrosis and regression. Cell Immunol. 2022, 381, 104614. [Google Scholar] [CrossRef]
- Mortha, A.; Chudnovskiy, A.; Hashimoto, D.; Bogunovic, M.; Spencer, S.P.; Belkaid, Y.; Merad, M. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science 2014, 343, 1249288. [Google Scholar] [CrossRef] [Green Version]
- Spindler, M.P.; Siu, S.; Mogno, I.; Li, Z.; Yang, C.; Mehandru, S.; Britton, G.J.; Faith, J.J. Human gut microbiota stimulate defined innate immune responses that vary from phylum to strain. Cell Host Microbe 2022, 30, 1481–1498.e5. [Google Scholar] [CrossRef]
- Zegarra Ruiz, D.F.; Kim, D.V.; Norwood, K.; Saldana-Morales, F.B.; Kim, M.; Ng, C.; Callaghan, R.; Uddin, M.; Chang, L.C.; Longman, R.S.; et al. Microbiota manipulation to increase macrophage IL-10 improves colitis and limits colitis-associated colorectal cancer. Gut Microbes 2022, 14, 2119054. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Ruder, B.; Becker, C. At the Forefront of the Mucosal Barrier: The Role of Macrophages in the Intestine. Cells 2020, 9, 2162. [Google Scholar] [CrossRef]
- Miao, E.A.; Mao, D.P.; Yudkovsky, N.; Bonneau, R.; Lorang, C.G.; Warren, S.E.; Leaf, I.A.; Aderem, A. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. USA 2010, 107, 3076–3080. [Google Scholar] [CrossRef] [Green Version]
- Bortoluci, K.R.; Medzhitov, R. Control of infection by pyroptosis and autophagy: Role of TLR and NLR. Cell Mol. Life Sci. 2010, 67, 1643–1651. [Google Scholar] [CrossRef]
- Rivollier, A.; He, J.; Kole, A.; Valatas, V.; Kelsall, B.L. Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J. Exp. Med. 2012, 209, 139–155. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.D.; Smythies, L.E.; Mosteller-Barnum, M.; Sibley, D.A.; Russell, M.W.; Merger, M.; Sellers, M.T.; Orenstein, J.M.; Shimada, T.; Graham, M.F.; et al. Intestinal macrophages lack CD14 and CD89 and consequently are down-regulated for LPS- and IgA-mediated activities. J. Immunol. 2001, 167, 2651–2656. [Google Scholar] [CrossRef] [Green Version]
- Weber, B.; Saurer, L.; Schenk, M.; Dickgreber, N.; Mueller, C. CX3CR1 defines functionally distinct intestinal mononuclear phagocyte subsets which maintain their respective functions during homeostatic and inflammatory conditions. Eur. J. Immunol. 2011, 41, 773–779. [Google Scholar] [CrossRef]
- Smythies, L.E.; Sellers, M.; Clements, R.H.; Mosteller-Barnum, M.; Meng, G.; Benjamin, W.H.; Orenstein, J.M.; Smith, P.D. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J. Clin. Investig. 2005, 115, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Denning, T.L.; Wang, Y.C.; Patel, S.R.; Williams, I.R.; Pulendran, B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat. Immunol. 2007, 8, 1086–1094. [Google Scholar] [CrossRef]
- Murai, M.; Turovskaya, O.; Kim, G.; Madan, R.; Karp, C.L.; Cheroutre, H.; Kronenberg, M. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat. Immunol. 2009, 10, 1178–1184. [Google Scholar] [CrossRef] [Green Version]
- Ueda, Y.; Kayama, H.; Jeon, S.G.; Kusu, T.; Isaka, Y.; Rakugi, H.; Yamamoto, M.; Takeda, K. Commensal microbiota induce LPS hyporesponsiveness in colonic macrophages via the production of IL-10. Int. Immunol. 2010, 22, 953–962. [Google Scholar] [CrossRef]
- Zigmond, E.; Bernshtein, B.; Friedlander, G.; Walker, C.R.; Yona, S.; Kim, K.W.; Brenner, O.; Krauthgamer, R.; Varol, C.; Muller, W.; et al. Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity 2014, 40, 720–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zigmond, E.; Varol, C.; Farache, J.; Elmaliah, E.; Satpathy, A.T.; Friedlander, G.; Mack, M.; Shpigel, N.; Boneca, I.G.; Murphy, K.M.; et al. Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity 2012, 37, 1076–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Ding, M.; Huang, L.; Gilkeson, G.; Lang, R.; Jiang, W. Toll-like receptor-mediated immune responses in intestinal macrophages; implications for mucosal immunity and autoimmune diseases. Clin. Immunol. 2016, 173, 81–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.; Xu, D.; Austin, C.D.; Caplazi, P.; Senger, K.; Sun, Y.; Jeet, S.; Young, J.; Delarosa, D.; Suto, E.; et al. Function of CSF1 and IL34 in Macrophage Homeostasis, Inflammation, and Cancer. Front. Immunol. 2019, 10, 2019. [Google Scholar] [CrossRef] [PubMed]
- Zwicker, S.; Martinez, G.L.; Bosma, M.; Gerling, M.; Clark, R.; Majster, M.; Soderman, J.; Almer, S.; Bostrom, E.A. Interleukin 34: A new modulator of human and experimental inflammatory bowel disease. Clin. Sci. 2015, 129, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.M.; Ryan, G.R.; Hapel, A.J.; Dominguez, M.G.; Russell, R.G.; Kapp, S.; Sylvestre, V.; Stanley, E.R. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood 2002, 99, 111–120. [Google Scholar] [CrossRef]
- Felix, R.; Cecchini, M.G.; Hofstetter, W.; Elford, P.R.; Stutzer, A.; Fleisch, H. Impairment of macrophage colony-stimulating factor production and lack of resident bone marrow macrophages in the osteopetrotic op/op mouse. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 1990, 5, 781–789. [Google Scholar] [CrossRef]
- Wiktor-Jedrzejczak, W.; Bartocci, A.; Ferrante, A.W., Jr.; Ahmed-Ansari, A.; Sell, K.W.; Pollard, J.W.; Stanley, E.R. Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc. Natl. Acad. Sci. USA 1990, 87, 4828–4832. [Google Scholar] [CrossRef] [Green Version]
- Cecchini, M.G.; Dominguez, M.G.; Mocci, S.; Wetterwald, A.; Felix, R.; Fleisch, H.; Chisholm, O.; Hofstetter, W.; Pollard, J.W.; Stanley, E.R. Role of colony stimulating factor-1 in the establishment and regulation of tissue macrophages during postnatal development of the mouse. Development 1994, 120, 1357–1372. [Google Scholar] [CrossRef]
- MacDonald, K.P.; Palmer, J.S.; Cronau, S.; Seppanen, E.; Olver, S.; Raffelt, N.C.; Kuns, R.; Pettit, A.R.; Clouston, A.; Wainwright, B.; et al. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood 2010, 116, 3955–3963. [Google Scholar] [CrossRef]
- Hume, D.A.; Pavli, P.; Donahue, R.E.; Fidler, I.J. The effect of human recombinant macrophage colony-stimulating factor (CSF-1) on the murine mononuclear phagocyte system in vivo. J. Immunol. 1988, 141, 3405–3409. [Google Scholar] [CrossRef]
- Schridde, A.; Bain, C.C.; Mayer, J.U.; Montgomery, J.; Pollet, E.; Denecke, B.; Milling, S.W.F.; Jenkins, S.J.; Dalod, M.; Henri, S.; et al. Tissue-specific differentiation of colonic macrophages requires TGFbeta receptor-mediated signaling. Mucosal Immunol. 2017, 10, 1387–1399. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.D.; Janoff, E.N.; Mosteller-Barnum, M.; Merger, M.; Orenstein, J.M.; Kearney, J.F.; Graham, M.F. Isolation and purification of CD14-negative mucosal macrophages from normal human small intestine. J. Immunol. Methods 1997, 202, 1–11. [Google Scholar] [CrossRef]
- Cummings, R.J.; Barbet, G.; Bongers, G.; Hartmann, B.M.; Gettler, K.; Muniz, L.; Furtado, G.C.; Cho, J.; Lira, S.A.; Blander, J.M. Different tissue phagocytes sample apoptotic cells to direct distinct homeostasis programs. Nature 2016, 539, 565–569. [Google Scholar] [CrossRef]
- Vallon-Eberhard, A.; Landsman, L.; Yogev, N.; Verrier, B.; Jung, S. Transepithelial pathogen uptake into the small intestinal lamina propria. J. Immunol. 2006, 176, 2465–2469. [Google Scholar] [CrossRef] [Green Version]
- Rescigno, M.; Urbano, M.; Valzasina, B.; Francolini, M.; Rotta, G.; Bonasio, R.; Granucci, F.; Kraehenbuhl, J.P.; Ricciardi-Castagnoli, P. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2001, 2, 361–367. [Google Scholar] [CrossRef]
- Hapfelmeier, S.; Muller, A.J.; Stecher, B.; Kaiser, P.; Barthel, M.; Endt, K.; Eberhard, M.; Robbiani, R.; Jacobi, C.A.; Heikenwalder, M.; et al. Microbe sampling by mucosal dendritic cells is a discrete, MyD88-independent step in DeltainvG S. Typhimurium colitis. J. Exp. Med. 2008, 205, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Medina-Contreras, O.; Geem, D.; Laur, O.; Williams, I.R.; Lira, S.A.; Nusrat, A.; Parkos, C.A.; Denning, T.L. CX3CR1 regulates intestinal macrophage homeostasis, bacterial translocation, and colitogenic Th17 responses in mice. J. Clin. Investig. 2011, 121, 4787–4795. [Google Scholar] [CrossRef] [Green Version]
- Pabst, O.; Bernhardt, G. The puzzle of intestinal lamina propria dendritic cells and macrophages. Eur. J. Immunol. 2010, 40, 2107–2111. [Google Scholar] [CrossRef]
- Niess, J.H.; Brand, S.; Gu, X.; Landsman, L.; Jung, S.; McCormick, B.A.; Vyas, J.M.; Boes, M.; Ploegh, H.L.; Fox, J.G.; et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 2005, 307, 254–258. [Google Scholar] [CrossRef]
- Mazzini, E.; Massimiliano, L.; Penna, G.; Rescigno, M. Oral tolerance can be established via gap junction transfer of fed antigens from CX3CR1(+) macrophages to CD103(+) dendritic cells. Immunity 2014, 40, 248–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farache, J.; Koren, I.; Milo, I.; Gurevich, I.; Kim, K.W.; Zigmond, E.; Furtado, G.C.; Lira, S.A.; Shakhar, G. Luminal bacteria recruit CD103+ dendritic cells into the intestinal epithelium to sample bacterial antigens for presentation. Immunity 2013, 38, 581–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denning, T.L.; Norris, B.A.; Medina-Contreras, O.; Manicassamy, S.; Geem, D.; Madan, R.; Karp, C.L.; Pulendran, B. Functional specializations of intestinal dendritic cell and macrophage subsets that control Th17 and regulatory T cell responses are dependent on the T cell/APC ratio, source of mouse strain, and regional localization. J. Immunol. 2011, 187, 733–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panea, C.; Farkas, A.M.; Goto, Y.; Abdollahi-Roodsaz, S.; Lee, C.; Koscso, B.; Gowda, K.; Hohl, T.M.; Bogunovic, M.; Ivanov, I.I. Intestinal Monocyte-Derived Macrophages Control Commensal-Specific Th17 Responses. Cell. Rep. 2015, 12, 1314–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serbina, N.V.; Pamer, E.G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006, 7, 311–317. [Google Scholar] [CrossRef]
- El Sayed, S.; Patik, I.; Redhu, N.S.; Glickman, J.N.; Karagiannis, K.; El Naenaeey, E.S.Y.; Elmowalid, G.A.; Abd El Wahab, A.M.; Snapper, S.B.; Horwitz, B.H. CCR2 promotes monocyte recruitment and intestinal inflammation in mice lacking the interleukin-10 receptor. Sci. Rep. 2022, 12, 452. [Google Scholar] [CrossRef]
- Platt, A.M.; Bain, C.C.; Bordon, Y.; Sester, D.P.; Mowat, A.M. An independent subset of TLR expressing CCR2-dependent macrophages promotes colonic inflammation. J. Immunol. 2010, 184, 6843–6854. [Google Scholar] [CrossRef] [Green Version]
- Andres, P.G.; Beck, P.L.; Mizoguchi, E.; Mizoguchi, A.; Bhan, A.K.; Dawson, T.; Kuziel, W.A.; Maeda, N.; MacDermott, R.P.; Podolsky, D.K.; et al. Mice with a selective deletion of the CC chemokine receptors 5 or 2 are protected from dextran sodium sulfate-mediated colitis: Lack of CC chemokine receptor 5 expression results in a NK1.1+ lymphocyte-associated Th2-type immune response in the intestine. J. Immunol. 2000, 164, 6303–6312. [Google Scholar] [CrossRef] [Green Version]
- Smythies, L.E.; Maheshwari, A.; Clements, R.; Eckhoff, D.; Novak, L.; Vu, H.L.; Mosteller-Barnum, L.M.; Sellers, M.; Smith, P.D. Mucosal IL-8 and TGF-beta recruit blood monocytes: Evidence for cross-talk between the lamina propria stroma and myeloid cells. J. Leukoc. Biol. 2006, 80, 492–499. [Google Scholar] [CrossRef] [Green Version]
- Redhu, N.S.; Bakthavatchalu, V.; Conaway, E.A.; Shouval, D.S.; Tsou, A.; Goettel, J.A.; Biswas, A.; Wang, C.; Field, M.; Muller, W.; et al. Macrophage dysfunction initiates colitis during weaning of infant mice lacking the interleukin-10 receptor. elife 2017, 6, e27652. [Google Scholar] [CrossRef]
- Rugtveit, J.; Nilsen, E.M.; Bakka, A.; Carlsen, H.; Brandtzaeg, P.; Scott, H. Cytokine profiles differ in newly recruited and resident subsets of mucosal macrophages from inflammatory bowel disease. Gastroenterology 1997, 112, 1493–1505. [Google Scholar] [CrossRef]
- Thiesen, S.; Janciauskiene, S.; Uronen-Hansson, H.; Agace, W.; Hogerkorp, C.M.; Spee, P.; Hakansson, K.; Grip, O. CD14(hi)HLA-DR(dim) macrophages, with a resemblance to classical blood monocytes, dominate inflamed mucosa in Crohn’s disease. J. Leukoc. Biol. 2014, 95, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Perminow, G.; Reikvam, D.H.; Lyckander, L.G.; Brandtzaeg, P.; Vatn, M.H.; Carlsen, H.S. Increased number and activation of colonic macrophages in pediatric patients with untreated Crohn’s disease. Inflamm. Bowel Dis. 2009, 15, 1368–1378. [Google Scholar] [CrossRef]
- Grimm, M.C.; Pullman, W.E.; Bennett, G.M.; Sullivan, P.J.; Pavli, P.; Doe, W.F. Direct evidence of monocyte recruitment to inflammatory bowel disease mucosa. J. Gastroenterol. Hepatol. 1995, 10, 387–395. [Google Scholar] [CrossRef]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef]
- Kostadinova, F.I.; Baba, T.; Ishida, Y.; Kondo, T.; Popivanova, B.K.; Mukaida, N. Crucial involvement of the CX3CR1-CX3CL1 axis in dextran sulfate sodium-mediated acute colitis in mice. J. Leukoc. Biol. 2010, 88, 133–143. [Google Scholar] [CrossRef]
- Brand, S.; Hofbauer, K.; Dambacher, J.; Schnitzler, F.; Staudinger, T.; Pfennig, S.; Seiderer, J.; Tillack, C.; Konrad, A.; Goke, B.; et al. Increased expression of the chemokine fractalkine in Crohn’s disease and association of the fractalkine receptor T280M polymorphism with a fibrostenosing disease Phenotype. Am. J. Gastroenterol. 2006, 101, 99–106. [Google Scholar] [CrossRef]
- Sabate, J.M.; Ameziane, N.; Lamoril, J.; Jouet, P.; Farmachidi, J.P.; Soule, J.C.; Harnois, F.; Sobhani, I.; Jian, R.; Deybach, J.C.; et al. The V249I polymorphism of the CX3CR1 gene is associated with fibrostenotic disease behavior in patients with Crohn’s disease. Eur. J. Gastroenterol. Hepatol. 2008, 20, 748–755. [Google Scholar] [CrossRef]
- Begue, B.; Verdier, J.; Rieux-Laucat, F.; Goulet, O.; Morali, A.; Canioni, D.; Hugot, J.P.; Daussy, C.; Verkarre, V.; Pigneur, B.; et al. Defective IL10 signaling defining a subgroup of patients with inflammatory bowel disease. Am. J. Gastroenterol. 2011, 106, 1544–1555. [Google Scholar] [CrossRef]
- Franke, A.; Balschun, T.; Karlsen, T.H.; Sventoraityte, J.; Nikolaus, S.; Mayr, G.; Domingues, F.S.; Albrecht, M.; Nothnagel, M.; Ellinghaus, D.; et al. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat. Genet. 2008, 40, 1319–1323. [Google Scholar] [CrossRef]
- Glocker, E.O.; Kotlarz, D.; Boztug, K.; Gertz, E.M.; Schaffer, A.A.; Noyan, F.; Perro, M.; Diestelhorst, J.; Allroth, A.; Murugan, D.; et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med. 2009, 361, 2033–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotlarz, D.; Beier, R.; Murugan, D.; Diestelhorst, J.; Jensen, O.; Boztug, K.; Pfeifer, D.; Kreipe, H.; Pfister, E.D.; Baumann, U.; et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: Implications for diagnosis and therapy. Gastroenterology 2012, 143, 347–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, R.; Lohler, J.; Rennick, D.; Rajewsky, K.; Muller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Moran, C.J.; Walters, T.D.; Guo, C.H.; Kugathasan, S.; Klein, C.; Turner, D.; Wolters, V.M.; Bandsma, R.H.; Mouzaki, M.; Zachos, M.; et al. IL-10R polymorphisms are associated with very-early-onset ulcerative colitis. Inflamm. Bowel Dis. 2013, 19, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Spencer, S.D.; Di Marco, F.; Hooley, J.; Pitts-Meek, S.; Bauer, M.; Ryan, A.M.; Sordat, B.; Gibbs, V.C.; Aguet, M. The orphan receptor CRF2-4 is an essential subunit of the interleukin 10 receptor. J. Exp. Med. 1998, 187, 571–578. [Google Scholar] [CrossRef] [Green Version]
- Shouval, D.S.; Biswas, A.; Goettel, J.A.; McCann, K.; Conaway, E.; Redhu, N.S.; Mascanfroni, I.D.; Al Adham, Z.; Lavoie, S.; Ibourk, M.; et al. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity 2014, 40, 706–719. [Google Scholar] [CrossRef] [Green Version]
- Bernshtein, B.; Curato, C.; Ioannou, M.; Thaiss, C.A.; Gross-Vered, M.; Kolesnikov, M.; Wang, Q.; David, E.; Chappell-Maor, L.; Harmelin, A.; et al. IL-23-producing IL-10Ralpha-deficient gut macrophages elicit an IL-22-driven proinflammatory epithelial cell response. Sci. Immunol. 2019, 4, eaau6571. [Google Scholar] [CrossRef]
- Asano, K.; Takahashi, N.; Ushiki, M.; Monya, M.; Aihara, F.; Kuboki, E.; Moriyama, S.; Iida, M.; Kitamura, H.; Qiu, C.H.; et al. Intestinal CD169(+) macrophages initiate mucosal inflammation by secreting CCL8 that recruits inflammatory monocytes. Nat. Commun. 2015, 6, 7802. [Google Scholar] [CrossRef] [Green Version]
- Daley, J.M.; Brancato, S.K.; Thomay, A.A.; Reichner, J.S.; Albina, J.E. The phenotype of murine wound macrophages. J. Leukoc. Biol. 2010, 87, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Pull, S.L.; Doherty, J.M.; Mills, J.C.; Gordon, J.I.; Stappenbeck, T.S. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc. Natl. Acad. Sci. USA 2005, 102, 99–104. [Google Scholar] [CrossRef]
- Morhardt, T.L.; Hayashi, A.; Ochi, T.; Quiros, M.; Kitamoto, S.; Nagao-Kitamoto, H.; Kuffa, P.; Atarashi, K.; Honda, K.; Kao, J.Y.; et al. IL-10 produced by macrophages regulates epithelial integrity in the small intestine. Sci. Rep. 2019, 9, 1223. [Google Scholar] [CrossRef] [Green Version]
- Quiros, M.; Nishio, H.; Neumann, P.A.; Siuda, D.; Brazil, J.C.; Azcutia, V.; Hilgarth, R.; O’Leary, M.N.; Garcia-Hernandez, V.; Leoni, G.; et al. Macrophage-derived IL-10 mediates mucosal repair by epithelial WISP-1 signaling. J. Clin. Investig. 2017, 127, 3510–3520. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, F.; Bernasconi, E.; Schafer, M.; Moyat, M.; Michetti, P.; Maillard, M.H.; Velin, D. Macrophages promote epithelial repair through hepatocyte growth factor secretion. Clin. Exp. Immunol. 2013, 174, 60–72. [Google Scholar] [CrossRef]
- Ma, S.; Zhang, J.; Liu, H.; Li, S.; Wang, Q. The Role of Tissue-Resident Macrophages in the Development and Treatment of Inflammatory Bowel Disease. Front. Cell Dev. Biol. 2022, 10, 896591. [Google Scholar] [CrossRef]
- Cosin-Roger, J.; Ortiz-Masia, D.; Calatayud, S.; Hernandez, C.; Esplugues, J.V.; Barrachina, M.D. The activation of Wnt signaling by a STAT6-dependent macrophage phenotype promotes mucosal repair in murine IBD. Mucosal Immunol. 2016, 9, 986–998. [Google Scholar] [CrossRef] [Green Version]
- Seo, D.H.; Che, X.; Kwak, M.S.; Kim, S.; Kim, J.H.; Ma, H.W.; Kim, D.H.; Kim, T.I.; Kim, W.H.; Kim, S.W.; et al. Interleukin-33 regulates intestinal inflammation by modulating macrophages in inflammatory bowel disease. Sci. Rep. 2017, 7, 851. [Google Scholar] [CrossRef] [Green Version]
- Waddell, A.; Vallance, J.E.; Moore, P.D.; Hummel, A.T.; Wu, D.; Shanmukhappa, S.K.; Fei, L.; Washington, M.K.; Minar, P.; Coburn, L.A.; et al. IL-33 Signaling Protects from Murine Oxazolone Colitis by Supporting Intestinal Epithelial Function. Inflamm. Bowel Dis. 2015, 21, 2737–2746. [Google Scholar] [CrossRef] [Green Version]
- Schleier, L.; Wiendl, M.; Heidbreder, K.; Binder, M.T.; Atreya, R.; Rath, T.; Becker, E.; Schulz-Kuhnt, A.; Stahl, A.; Schulze, L.L.; et al. Non-classical monocyte homing to the gut via alpha4beta7 integrin mediates macrophage-dependent intestinal wound healing. Gut 2020, 69, 252–263. [Google Scholar] [CrossRef]
- Pakshir, P.; Hinz, B. The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol. 2018, 68–69, 81–93. [Google Scholar] [CrossRef]
- Wynn, T.A.; Barron, L. Macrophages: Master regulators of inflammation and fibrosis. Semin. Liver Dis. 2010, 30, 245–257. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenti, M.V.; Di Sabatino, A. Intestinal fibrosis. Mol. Aspects Med. 2019, 65, 100–109. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, S.; Ungaro, F.; Noviello, D.; Lovisa, S.; Peyrin-Biroulet, L.; Danese, S. Revisiting fibrosis in inflammatory bowel disease: The gut thickens. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Lagares, D. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 2020, 16, 11–31. [Google Scholar] [CrossRef]
- Franze, E.; Dinallo, V.; Laudisi, F.; Di Grazia, A.; Di Fusco, D.; Colantoni, A.; Ortenzi, A.; Giuffrida, P.; Di Carlo, S.; Sica, G.S.; et al. Interleukin-34 Stimulates Gut Fibroblasts to Produce Collagen Synthesis. J. Crohn’s Colitis 2020, 14, 1436–1445. [Google Scholar] [CrossRef]
- Cupedo, T.; Crellin, N.K.; Papazian, N.; Rombouts, E.J.; Weijer, K.; Grogan, J.L.; Fibbe, W.E.; Cornelissen, J.J.; Spits, H. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat. Immunol. 2009, 10, 66–74. [Google Scholar] [CrossRef]
- Cella, M.; Fuchs, A.; Vermi, W.; Facchetti, F.; Otero, K.; Lennerz, J.K.; Doherty, J.M.; Mills, J.C.; Colonna, M. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 2009, 457, 722–725. [Google Scholar] [CrossRef] [Green Version]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.; Mebius, R.E.; et al. Innate lymphoid cells—A proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef]
- Ignacio, A.; Breda, C.N.S.; Camara, N.O.S. Innate lymphoid cells in tissue homeostasis and diseases. World. J. Hepatol. 2017, 9, 979–989. [Google Scholar] [CrossRef]
- Eberl, G.; Colonna, M.; Di Santo, J.P.; McKenzie, A.N. Innate lymphoid cells. Innate lymphoid cells: A new paradigm in immunology. Science 2015, 348, aaa6566. [Google Scholar] [CrossRef]
- Artis, D.; Spits, H. The biology of innate lymphoid cells. Nature 2015, 517, 293–301. [Google Scholar] [CrossRef]
- Huang, Y.; Mao, K.; Germain, R.N. Thinking differently about ILCs-Not just tissue resident and not just the same as CD4(+) T-cell effectors. Immunol. Rev. 2018, 286, 160–171. [Google Scholar] [CrossRef]
- Trabanelli, S.; Gomez-Cadena, A.; Salome, B.; Michaud, K.; Mavilio, D.; Landis, B.N.; Jandus, P.; Jandus, C. Human innate lymphoid cells (ILCs): Toward a uniform immune-phenotyping. Cytom. B Clin. Cytom. 2018, 94, 392–399. [Google Scholar] [CrossRef]
- Panda, S.K.; Colonna, M. Innate Lymphoid Cells in Mucosal Immunity. Front. Immunol. 2019, 10, 861. [Google Scholar] [CrossRef] [Green Version]
- Giuffrida, P.; Corazza, G.R.; Di Sabatino, A. Old and New Lymphocyte Players in Inflammatory Bowel Disease. Dig. Dis. Sci. 2018, 63, 277–288. [Google Scholar] [CrossRef]
- Sepahi, A.; Liu, Q.; Friesen, L.; Kim, C.H. Dietary fiber metabolites regulate innate lymphoid cell responses. Mucosal Immunol. 2021, 14, 317–330. [Google Scholar] [CrossRef]
- Luo, W.; Tian, L.; Tan, B.; Shen, Z.; Xiao, M.; Wu, S.; Meng, X.; Wu, X.; Wang, X. Update: Innate Lymphoid Cells in Inflammatory Bowel Disease. Dig. Dis. Sci. 2021, 67, 56–66. [Google Scholar] [CrossRef]
- Zhou, W.; Sonnenberg, G.F. Activation and Suppression of Group 3 Innate Lymphoid Cells in the Gut. Trends Immunol. 2020, 41, 721–733. [Google Scholar] [CrossRef]
- Wu, Y.; Shen, J. Innate Lymphoid Cells in Crohn’s Disease. Front. Immunol. 2020, 11, 554880. [Google Scholar] [CrossRef]
- Diefenbach, A.; Gnafakis, S.; Shomrat, O. Innate Lymphoid Cell-Epithelial Cell Modules Sustain Intestinal Homeostasis. Immunity 2020, 52, 452–463. [Google Scholar] [CrossRef]
- Saez, A.; Gomez-Bris, R.; Herrero-Fernandez, B.; Mingorance, C.; Rius, C.; Gonzalez-Granado, J.M. Innate Lymphoid Cells in Intestinal Homeostasis and Inflammatory Bowel Disease. Int. J. Mol. Sci. 2021, 22, 7618. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Shi, S.; Ashworth, G.; Dong, C.; Liu, J.; Xing, F. ILC3 function as a double-edged sword in inflammatory bowel diseases. Cell Death Dis. 2019, 10, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz-Kuhnt, A.; Neurath, M.F.; Wirtz, S.; Atreya, I. Innate Lymphoid Cells as Regulators of Epithelial Integrity: Therapeutic Implications for Inflammatory Bowel Diseases. Front. Med. 2021, 8, 656745. [Google Scholar] [CrossRef] [PubMed]
- Peng, V.; Jaeger, N.; Colonna, M. Innate Lymphoid Cells and Inflammatory Bowel Disease. Adv. Exp. Med. Biol. 2022, 1365, 97–112. [Google Scholar] [PubMed]
- Schraml, B.U.; Reis e Sousa, C. Defining dendritic cells. Curr. Opin. Immunol. 2015, 32, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Mildner, A.; Jung, S. Development and function of dendritic cell subsets. Immunity 2014, 40, 642–656. [Google Scholar] [CrossRef] [Green Version]
- Guilliams, M.; Dutertre, C.A.; Scott, C.L.; McGovern, N.; Sichien, D.; Chakarov, S.; Van Gassen, S.; Chen, J.; Poidinger, M.; De Prijck, S.; et al. Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity 2016, 45, 669–684. [Google Scholar] [CrossRef] [Green Version]
- Dutertre, C.A.; Wang, L.F.; Ginhoux, F. Aligning bona fide dendritic cell populations across species. Cell. Immunol. 2014, 291, 3–10. [Google Scholar] [CrossRef]
- Zernecke, A. Dendritic cells in atherosclerosis: Evidence in mice and humans. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 763–770. [Google Scholar] [CrossRef] [Green Version]
- Gil-Pulido, J.; Zernecke, A. Antigen-presenting dendritic cells in atherosclerosis. Eur. J. Pharmacol. 2017, 816, 25–31. [Google Scholar] [CrossRef]
- Greter, M.; Helft, J.; Chow, A.; Hashimoto, D.; Mortha, A.; Agudo-Cantero, J.; Bogunovic, M.; Gautier, E.L.; Miller, J.; Leboeuf, M.; et al. GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity 2012, 36, 1031–1046. [Google Scholar] [CrossRef] [Green Version]
- Ushach, I.; Zlotnik, A. Biological role of granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) on cells of the myeloid lineage. J. Leukoc. Biol. 2016, 100, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef]
- Mellman, I.; Steinman, R.M. Dendritic cells: Specialized and regulated antigen processing machines. Cell 2001, 106, 255–258. [Google Scholar] [CrossRef] [Green Version]
- Palucka, K.; Banchereau, J. Dendritic cells: A link between innate and adaptive immunity. J. Clin. Immunol. 1999, 19, 12–25. [Google Scholar] [CrossRef]
- Palucka, K.; Banchereau, J. Linking innate and adaptive immunity. Nat. Med. 1999, 5, 868–870. [Google Scholar] [CrossRef]
- Bousso, P. T-cell activation by dendritic cells in the lymph node: Lessons from the movies. Nat. Rev. Immunol. 2008, 8, 675–684. [Google Scholar] [CrossRef]
- Steinman, R.M. Decisions about dendritic cells: Past, present, and future. Annu. Rev. Immunol. 2012, 30, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Lelouard, H.; Fallet, M.; de Bovis, B.; Meresse, S.; Gorvel, J.P. Peyer’s patch dendritic cells sample antigens by extending dendrites through M cell-specific transcellular pores. Gastroenterology 2012, 142, 592–601.e3. [Google Scholar] [CrossRef]
- Ohta, T.; Sugiyama, M.; Hemmi, H.; Yamazaki, C.; Okura, S.; Sasaki, I.; Fukuda, Y.; Orimo, T.; Ishii, K.J.; Hoshino, K.; et al. Crucial roles of XCR1-expressing dendritic cells and the XCR1-XCL1 chemokine axis in intestinal immune homeostasis. Sci. Rep. 2016, 6, 23505. [Google Scholar] [CrossRef]
- Johansson-Lindbom, B.; Svensson, M.; Pabst, O.; Palmqvist, C.; Marquez, G.; Forster, R.; Agace, W.W. Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. J. Exp. Med. 2005, 202, 1063–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agace, W.W.; Persson, E.K. How vitamin A metabolizing dendritic cells are generated in the gut mucosa. Trends Immunol. 2012, 33, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, M.; Hoshii, T.; Fujii, H.; Koyasu, S.; Hirao, A.; Matsuda, S. Cutting edge: mTORC1 in intestinal CD11c+ CD11b+ dendritic cells regulates intestinal homeostasis by promoting IL-10 production. J. Immunol. 2012, 188, 4736–4740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Hassi, H.O.; Mann, E.R.; Sanchez, B.; English, N.R.; Peake, S.T.; Landy, J.; Man, R.; Urdaci, M.; Hart, A.L.; Fernandez-Salazar, L.; et al. Altered human gut dendritic cell properties in ulcerative colitis are reversed by Lactobacillus plantarum extracellular encrypted peptide STp. Mol. Nutr. Food Res. 2014, 58, 1132–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, A.L.; Al-Hassi, H.O.; Rigby, R.J.; Bell, S.J.; Emmanuel, A.V.; Knight, S.C.; Kamm, M.A.; Stagg, A.J. Characteristics of intestinal dendritic cells in inflammatory bowel diseases. Gastroenterology 2005, 129, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, M.K.; Brynjolfsson, S.F.; Dige, A.; Uronen-Hansson, H.; Borjesson, L.G.; Bengtsson, J.L.; Gudjonsson, S.; Ohman, L.; Agnholt, J.; Sjovall, H.; et al. Macrophage and dendritic cell subsets in IBD: ALDH+ cells are reduced in colon tissue of patients with ulcerative colitis regardless of inflammation. Mucosal Immunol. 2016, 9, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Faleiro, R.; Liu, J.; Karunarathne, D.; Edmundson, A.; Winterford, C.; Nguyen, T.H.; Simms, L.A.; Radford-Smith, G.; Wykes, M. Crohn’s disease is facilitated by a disturbance of programmed death-1 ligand 2 on blood dendritic cells. Clin. Transl. Immunology 2019, 8, e01071. [Google Scholar] [CrossRef]
- Sawai, C.M.; Serpas, L.; Neto, A.G.; Jang, G.; Rashidfarrokhi, A.; Kolbeck, R.; Sanjuan, M.A.; Reizis, B.; Sisirak, V. Plasmacytoid Dendritic Cells Are Largely Dispensable for the Pathogenesis of Experimental Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 2475. [Google Scholar] [CrossRef]
- Mishima, Y.; Sartor, R.B. Manipulating resident microbiota to enhance regulatory immune function to treat inflammatory bowel diseases. J. Gastroenterol. 2020, 55, 4–14. [Google Scholar] [CrossRef] [Green Version]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef]
- Matsuno, H.; Kayama, H.; Nishimura, J.; Sekido, Y.; Osawa, H.; Barman, S.; Ogino, T.; Takahashi, H.; Haraguchi, N.; Hata, T.; et al. CD103+ Dendritic Cell Function Is Altered in the Colons of Patients with Ulcerative Colitis. Inflamm. Bowel Dis. 2017, 23, 1524–1534. [Google Scholar] [CrossRef] [Green Version]
- Pool, L.; Rivollier, A.; Agace, W.W. Deletion of IRF4 in Dendritic Cells Leads to Delayed Onset of T Cell-Dependent Colitis. J. Immunol. 2020, 204, 1047–1055. [Google Scholar] [CrossRef]
- Muzaki, A.R.; Tetlak, P.; Sheng, J.; Loh, S.C.; Setiagani, Y.A.; Poidinger, M.; Zolezzi, F.; Karjalainen, K.; Ruedl, C. Intestinal CD103(+)CD11b(-) dendritic cells restrain colitis via IFN-gamma-induced anti-inflammatory response in epithelial cells. Mucosal Immunol. 2016, 9, 336–351. [Google Scholar] [CrossRef] [Green Version]
- Welty, N.E.; Staley, C.; Ghilardi, N.; Sadowsky, M.J.; Igyarto, B.Z.; Kaplan, D.H. Intestinal lamina propria dendritic cells maintain T cell homeostasis but do not affect commensalism. J. Exp. Med. 2013, 210, 2011–2024. [Google Scholar] [CrossRef] [Green Version]
- Furey, T.S.; Sethupathy, P.; Sheikh, S.Z. Redefining the IBDs using genome-scale molecular phenotyping. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 296–311. [Google Scholar] [CrossRef]
- Graham, D.B.; Xavier, R.J. Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature 2020, 578, 527–539. [Google Scholar] [CrossRef]
- De Lange, K.M.; Moutsianas, L.; Lee, J.C.; Lamb, C.A.; Luo, Y.; Kennedy, N.A.; Jostins, L.; Rice, D.L.; Gutierrez-Achury, J.; Ji, S.G.; et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 2017, 49, 256–261. [Google Scholar] [CrossRef] [Green Version]
- Loddo, I.; Romano, C. Inflammatory Bowel Disease: Genetics, Epigenetics, and Pathogenesis. Front. Immunol. 2015, 6, 551. [Google Scholar] [CrossRef] [Green Version]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Mitsialis, V.; Wall, S.; Liu, P.; Ordovas-Montanes, J.; Parmet, T.; Vukovic, M.; Spencer, D.; Field, M.; McCourt, C.; Toothaker, J.; et al. Single-Cell Analyses of Colon and Blood Reveal Distinct Immune Cell Signatures of Ulcerative Colitis and Crohn’s Disease. Gastroenterology 2020, 159, 591–608.e10. [Google Scholar] [CrossRef]
- Martin, J.C.; Chang, C.; Boschetti, G.; Ungaro, R.; Giri, M.; Grout, J.A.; Gettler, K.; Chuang, L.S.; Nayar, S.; Greenstein, A.J.; et al. Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell 2019, 178, 1493–1508.e20. [Google Scholar] [CrossRef] [PubMed]
- Corridoni, D.; Antanaviciute, A.; Gupta, T.; Fawkner-Corbett, D.; Aulicino, A.; Jagielowicz, M.; Parikh, K.; Repapi, E.; Taylor, S.; Ishikawa, D.; et al. Single-cell atlas of colonic CD8(+) T cells in ulcerative colitis. Nat. Med. 2020, 26, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.S.; He, Z.; Tsai, M.S.; Olvera, J.G.; Omilusik, K.D.; Duong, H.G.; Kim, E.S.; Limary, A.E.; Jin, W.; Milner, J.J.; et al. Heterogeneity and clonal relationships of adaptive immune cells in ulcerative colitis revealed by single-cell analyses. Sci. Immunol. 2020, 5, eabb4432. [Google Scholar] [CrossRef] [PubMed]
- Smillie, C.S.; Biton, M.; Ordovas-Montanes, J.; Sullivan, K.M.; Burgin, G.; Graham, D.B.; Herbst, R.H.; Rogel, N.; Slyper, M.; Waldman, J.; et al. Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 2019, 178, 714–730.e22. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.J.; Hou, K.; Dey, K.K.; Sakaue, S.; Jagadeesh, K.A.; Weinand, K.; Taychameekiatchai, A.; Rao, P.; Pisco, A.O.; Zou, J.; et al. Polygenic enrichment distinguishes disease associations of individual cells in single-cell RNA-seq data. Nat. Genet. 2022, 54, 1572–1580. [Google Scholar] [CrossRef]
- Saul, D.; Leite Barros, L.; Wixom, A.Q.; Gellhaus, B.; Gibbons, H.R.; Faubion, W.A.; Kosinsky, R.L. Cell Type-Specific Induction of Inflammation-Associated Genes in Crohn’s Disease and Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 3082. [Google Scholar] [CrossRef]
- Li, G.; Zhang, B.; Hao, J.; Chu, X.; Wiestler, M.; Cornberg, M.; Xu, C.J.; Liu, X.; Li, Y. Identification of Novel Population-Specific Cell Subsets in Chinese Ulcerative Colitis Patients Using Single-Cell RNA Sequencing. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 99–117. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhang, T.; Yang, H. ScRNA-seq identified the metabolic reprogramming of human colonic immune cells in different locations and disease states. Biochem. Biophys. Res. Commun. 2022, 604, 96–103. [Google Scholar] [CrossRef]
- Diez-Obrero, V.; Moratalla-Navarro, F.; Ibanez-Sanz, G.; Guardiola, J.; Rodriguez-Moranta, F.; Obon-Santacana, M.; Diez-Villanueva, A.; Dampier, C.H.; Devall, M.; Carreras-Torres, R.; et al. Transcriptome-Wide Association Study for Inflammatory Bowel Disease Reveals Novel Candidate Susceptibility Genes in Specific Colon Subsites and Tissue Categories. J. Crohn’s Colitis 2022, 16, 275–285. [Google Scholar] [CrossRef]
- Valenzuela, P.L.; Amo, C.; Sanchez-Martinez, G.; Torrontegi, E.; Vazquez-Carrion, J.; Montalvo, Z.; Lucia, A.; de la Villa, P. Enhancement of Mood but not Performance in Elite Athletes With Transcranial Direct-Current Stimulation. Int. J. Sports Physiol. Perform. 2019, 14, 310–316. [Google Scholar] [CrossRef]
- Fukunaga, S.; Kuwaki, K.; Mitsuyama, K.; Takedatsu, H.; Yoshioka, S.; Yamasaki, H.; Yamauchi, R.; Mori, A.; Kakuma, T.; Tsuruta, O.; et al. Detection of calprotectin in inflammatory bowel disease: Fecal and serum levels and immunohistochemical localization. Int. J. Mol. Med. 2018, 41, 107–118. [Google Scholar] [CrossRef] [Green Version]
- Muthas, D.; Reznichenko, A.; Balendran, C.A.; Bottcher, G.; Clausen, I.G.; Karrman Mardh, C.; Ottosson, T.; Uddin, M.; MacDonald, T.T.; Danese, S.; et al. Neutrophils in ulcerative colitis: A review of selected biomarkers and their potential therapeutic implications. Scand. J. Gastroenterol. 2017, 52, 125–135. [Google Scholar] [CrossRef]
- Barry, R.; Ruano-Gallego, D.; Radhakrishnan, S.T.; Lovell, S.; Yu, L.; Kotik, O.; Glegola-Madejska, I.; Tate, E.W.; Choudhary, J.S.; Williams, H.R.T.; et al. Faecal neutrophil elastase-antiprotease balance reflects colitis severity. Mucosal Immunol. 2020, 13, 322–333. [Google Scholar] [CrossRef] [Green Version]
- Jablaoui, A.; Kriaa, A.; Mkaouar, H.; Akermi, N.; Soussou, S.; Wysocka, M.; Woloszyn, D.; Amouri, A.; Gargouri, A.; Maguin, E.; et al. Fecal Serine Protease Profiling in Inflammatory Bowel Diseases. Front. Cell. Infect. Microbiol. 2020, 10, 21. [Google Scholar] [CrossRef]
- Kumar, K.G.; Trevaskis, J.L.; Lam, D.D.; Sutton, G.M.; Koza, R.A.; Chouljenko, V.N.; Kousoulas, K.G.; Rogers, P.M.; Kesterson, R.A.; Thearle, M.; et al. Identification of adropin as a secreted factor linking dietary macronutrient intake with energy homeostasis and lipid metabolism. Cell Metab. 2008, 8, 468–481. [Google Scholar] [CrossRef] [Green Version]
- Simac, P.; Perkovic, D.; Bozic, I.; Bilopavlovic, N.; Martinovic, D.; Bozic, J. Serum Adropin Levels in Patients with Rheumatoid Arthritis. Life 2022, 12, 169. [Google Scholar] [CrossRef]
- Brnic, D.; Martinovic, D.; Zivkovic, P.M.; Tokic, D.; Tadin Hadjina, I.; Rusic, D.; Vilovic, M.; Supe-Domic, D.; Tonkic, A.; Bozic, J. Serum adropin levels are reduced in patients with inflammatory bowel diseases. Sci. Rep. 2020, 10, 9264. [Google Scholar] [CrossRef]
- Zivkovic, P.M.; Matetic, A.; Tadin Hadjina, I.; Rusic, D.; Vilovic, M.; Supe-Domic, D.; Borovac, J.A.; Mudnic, I.; Tonkic, A.; Bozic, J. Serum Catestatin Levels and Arterial Stiffness Parameters Are Increased in Patients with Inflammatory Bowel Disease. J. Clin. Med. 2020, 9, 628. [Google Scholar] [CrossRef] [Green Version]
- Muntjewerff, E.M.; Tang, K.; Lutter, L.; Christoffersson, G.; Nicolasen, M.J.T.; Gao, H.; Katkar, G.D.; Das, S.; Ter Beest, M.; Ying, W.; et al. Chromogranin A regulates gut permeability via the antagonistic actions of its proteolytic peptides. Acta Physiol. 2021, 232, e13655. [Google Scholar] [CrossRef]
- Rabbi, M.F.; Labis, B.; Metz-Boutigue, M.H.; Bernstein, C.N.; Ghia, J.E. Catestatin decreases macrophage function in two mouse models of experimental colitis. Biochem. Pharmacol. 2014, 89, 386–398. [Google Scholar] [CrossRef]
- Mahata, S.K.; Mahata, M.; Fung, M.M.; O’Connor, D.T. Catestatin: A multifunctional peptide from chromogranin A. Regul. Pept. 2010, 162, 33–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, W.I.; Ghia, J.E. Gut hormones: Emerging role in immune activation and inflammation. Clin. Exp. Immunol. 2010, 161, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Eissa, N.; Hussein, H.; Mesgna, R.; Bonin, S.; Hendy, G.N.; Metz-Boutigue, M.H.; Bernstein, C.N.; Ghia, J.E. Catestatin Regulates Epithelial Cell Dynamics to Improve Intestinal Inflammation. Vaccines 2018, 6, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabbi, M.F.; Munyaka, P.M.; Eissa, N.; Metz-Boutigue, M.H.; Khafipour, E.; Ghia, J.E. Human Catestatin Alters Gut Microbiota Composition in Mice. Front. Microbiol. 2016, 7, 2151. [Google Scholar] [CrossRef] [Green Version]
- Zigmond, E.; Jung, S. Intestinal macrophages: Well educated exceptions from the rule. Trends Immunol. 2013, 34, 162–168. [Google Scholar] [CrossRef]
- Yona, S.; Kim, K.W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Gisbert, J.P.; Chaparro, M. Primary Failure to an Anti-TNF Agent in Inflammatory Bowel Disease: Switch (to a Second Anti-TNF Agent) or Swap (for Another Mechanism of Action)? J. Clin. Med. 2021, 10, 5318. [Google Scholar] [CrossRef]
- Yanai, H.; Hanauer, S.B. Assessing response and loss of response to biological therapies in IBD. Am. J. Gastroenterol. 2011, 106, 685–698. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saez, A.; Herrero-Fernandez, B.; Gomez-Bris, R.; Sánchez-Martinez, H.; Gonzalez-Granado, J.M. Pathophysiology of Inflammatory Bowel Disease: Innate Immune System. Int. J. Mol. Sci. 2023, 24, 1526. https://doi.org/10.3390/ijms24021526
Saez A, Herrero-Fernandez B, Gomez-Bris R, Sánchez-Martinez H, Gonzalez-Granado JM. Pathophysiology of Inflammatory Bowel Disease: Innate Immune System. International Journal of Molecular Sciences. 2023; 24(2):1526. https://doi.org/10.3390/ijms24021526
Chicago/Turabian StyleSaez, Angela, Beatriz Herrero-Fernandez, Raquel Gomez-Bris, Hector Sánchez-Martinez, and Jose M. Gonzalez-Granado. 2023. "Pathophysiology of Inflammatory Bowel Disease: Innate Immune System" International Journal of Molecular Sciences 24, no. 2: 1526. https://doi.org/10.3390/ijms24021526
APA StyleSaez, A., Herrero-Fernandez, B., Gomez-Bris, R., Sánchez-Martinez, H., & Gonzalez-Granado, J. M. (2023). Pathophysiology of Inflammatory Bowel Disease: Innate Immune System. International Journal of Molecular Sciences, 24(2), 1526. https://doi.org/10.3390/ijms24021526