
Effect of Pore Defects on Uniaxial Mechanical Properties of Bulk Hexagonal Hydroxyapatite: A Molecular Dynamics Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
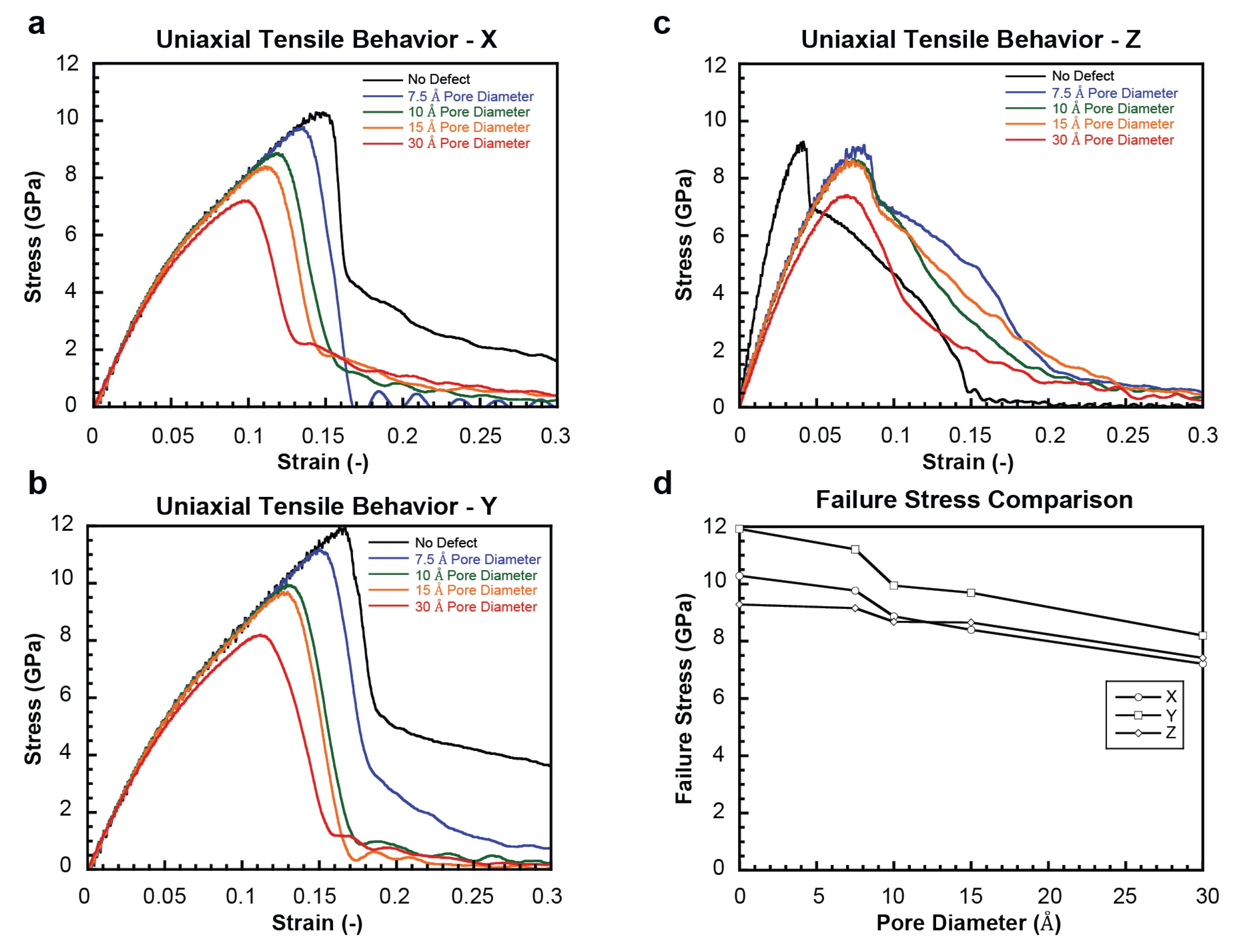
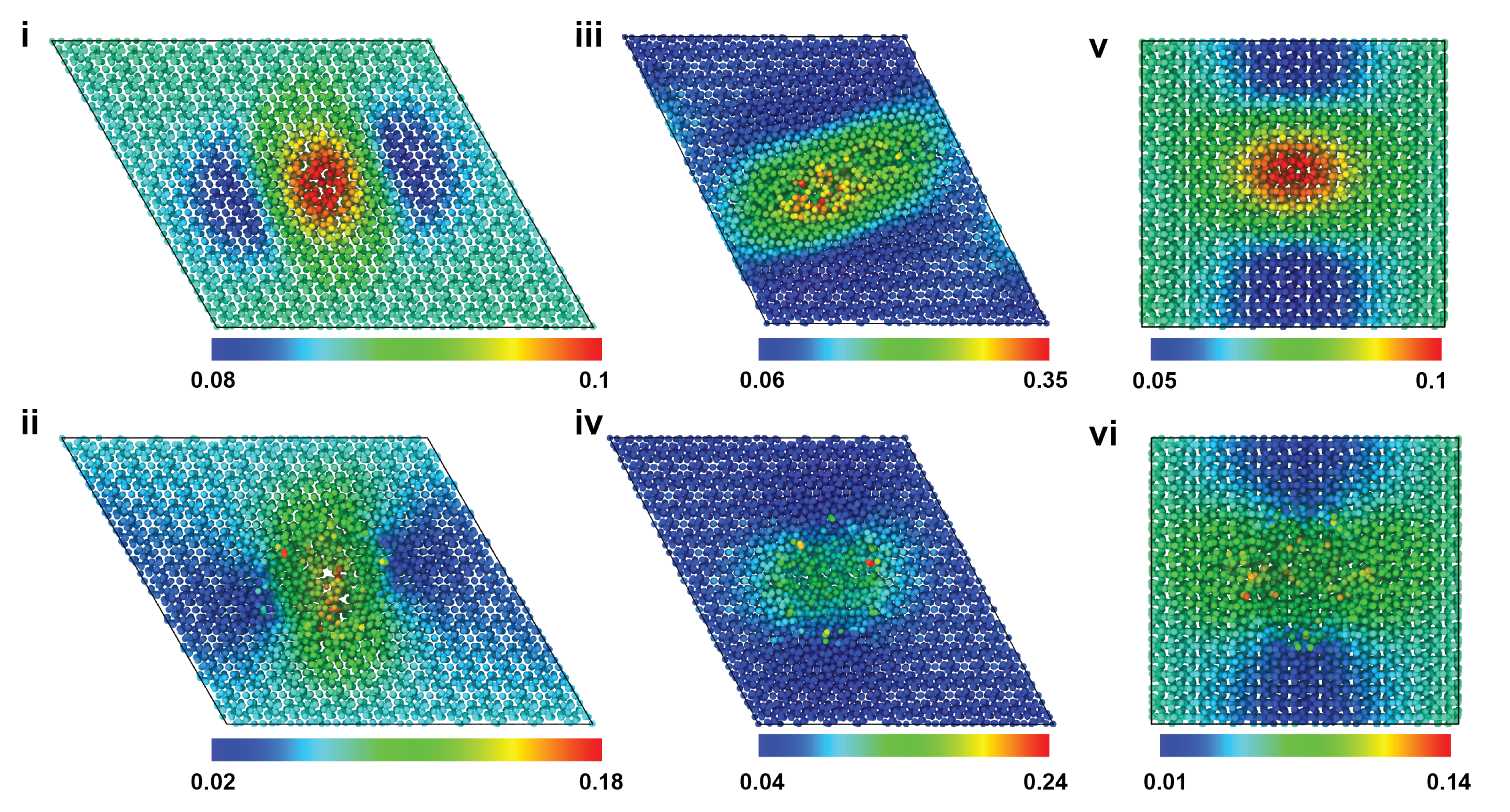
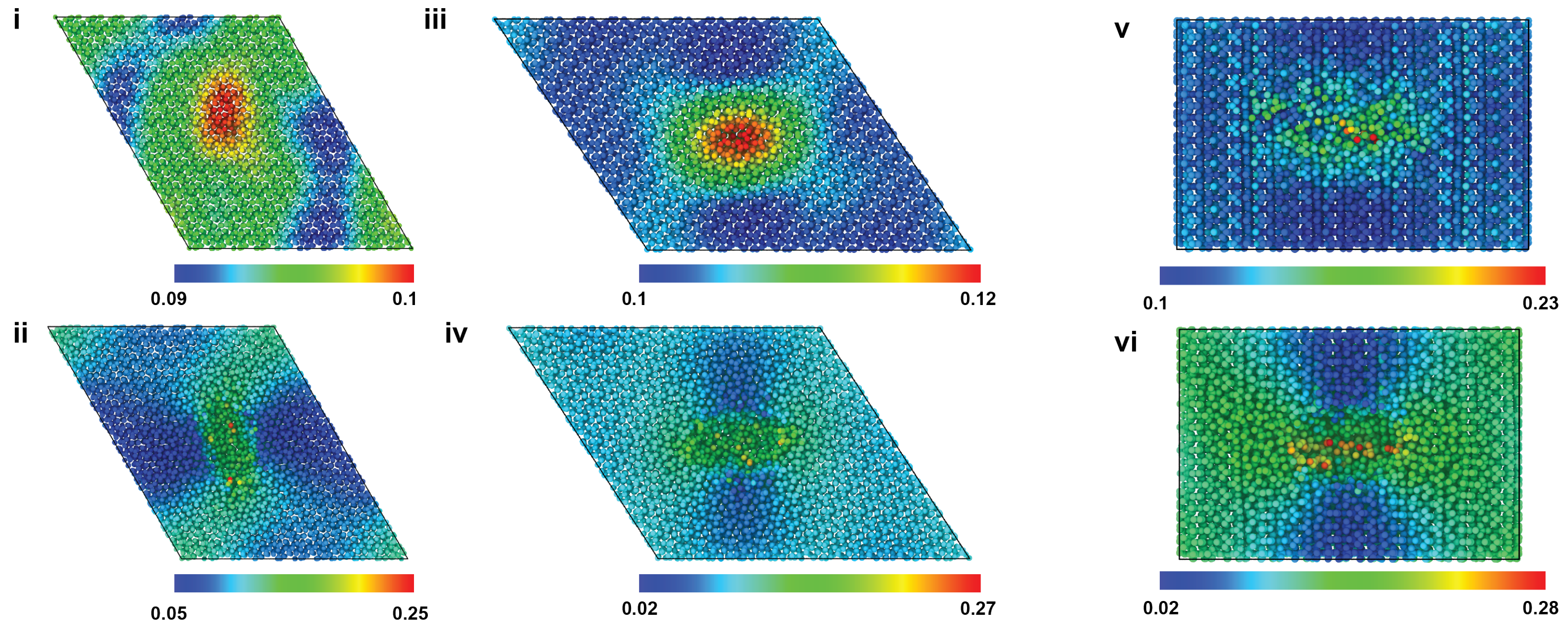
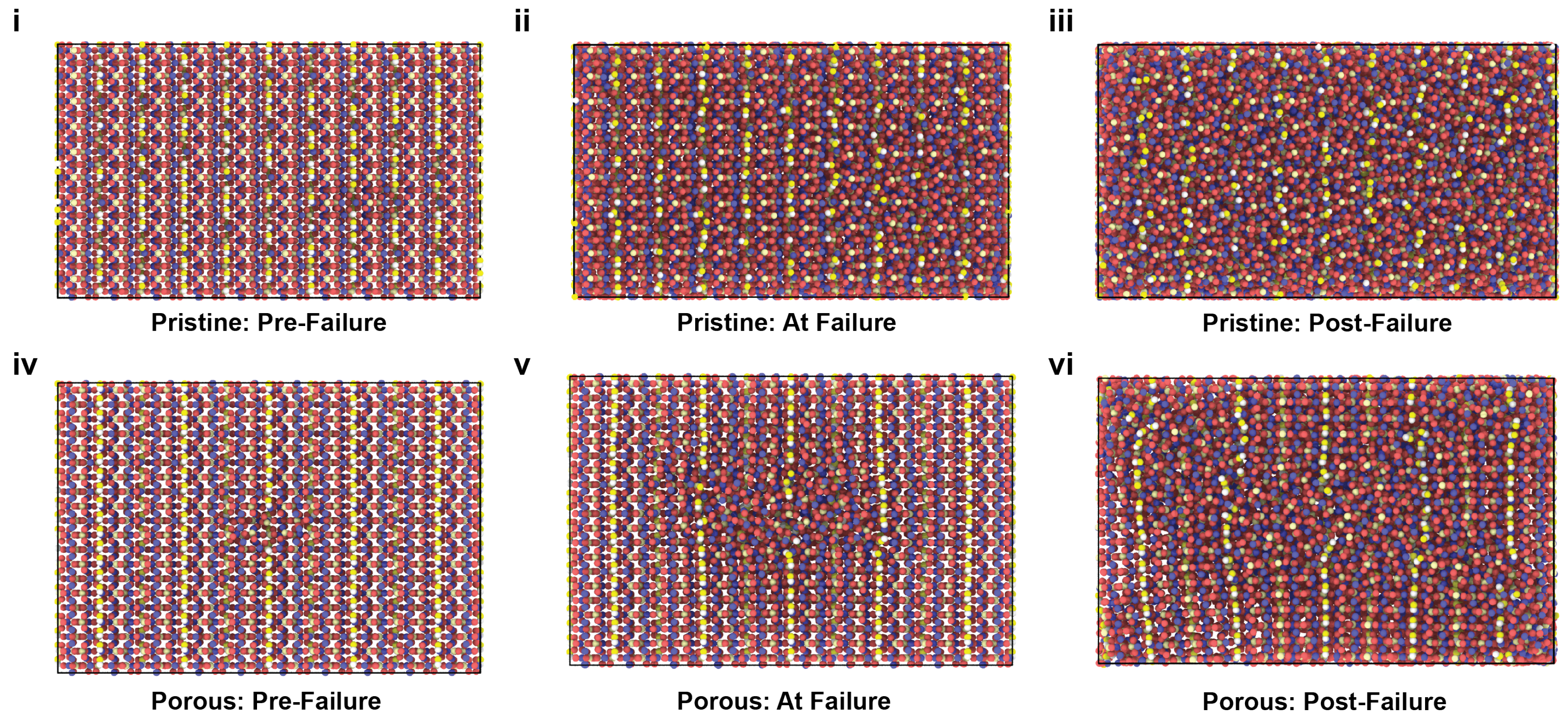
2.1. Uniaxial Tensile Behavior
2.2. Uniaxial Compressive Behavior
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HAP | Hydroxyapatite |
| MD | Molecular Dynamics |
| CVFF | Consistent Valence Force Field |
References
- Rho, J.Y.; Kuhn-Spearing, L.; Zioupos, P. Mechanical properties and the hierarchical structure of bone. Med. Eng. Phys. 1998, 20, 92–102. [Google Scholar] [CrossRef]
- Huang, W.; Restrepo, D.; Jung, J.Y.; Su, F.Y.; Liu, Z.; Ritchie, R.O.; McKittrick, J.; Zavattieri, P.; Kisailus, D. Multiscale Toughening Mechanisms in Biological Materials and Bioinspired Designs. Adv. Mater. 2019, 31, 1901561. [Google Scholar] [CrossRef] [PubMed]
- Stock, S. The Mineral–Collagen Interface in Bone. Calcif. Tissue Int. 2015, 97, 262–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Tommaso, D.; Prakash, M.; Lemaire, T.; Lewerenz, M.; De Leeuw, N.H.; Naili, S. Molecular Dynamics Simulations of Hydroxyapatite Nanopores in Contact with Electrolyte Solutions: The Effect of Nanoconfinement and Solvated Ions on the Surface Reactivity and the Structural, Dynamical, and Vibrational Properties of Water. Crystals 2017, 7, 57. [Google Scholar] [CrossRef] [Green Version]
- An, S.H.; Matsumoto, T.; Miyajima, H.; Nakahira, A.; Kim, K.H.; Imazato, S. Porous zirconia/hydroxyapatite scaffolds for bone reconstruction. Dent. Mater. 2012, 28, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, P.; Agrawal, D. Sol-gel derived hydroxyapatite coatings on titanium substrates. Surf. Coat. Technol. 2011, 206, 360–365. [Google Scholar] [CrossRef]
- Costa, D.O.; Prowse, P.D.; Chrones, T.; Sims, S.M.; Hamilton, D.W.; Rizkalla, A.S.; Dixon, S.J. The differential regulation of osteoblast and osteoclast activity by surface topography of hydroxyapatite coatings. Biomaterials 2013, 34, 7215–7226. [Google Scholar] [CrossRef]
- Deville, S.; Saiz, E.; Tomsia, A.P. Freeze casting of hydroxyapatite scaffolds for bone tissue engineering. Biomaterials 2006, 27, 5480–5489. [Google Scholar] [CrossRef] [Green Version]
- Mygind, T.; Stiehler, M.; Baatrup, A.; Li, H.; Zou, X.; Flyvbjerg, A.; Kassem, M.; Bunger, C. Mesenchymal stem cell ingrowth and differentiation on coralline hydroxyapatite scaffolds. Biomaterials 2007, 28, 1036–1047. [Google Scholar] [CrossRef]
- Harun, W.; Asri, R.; Alias, J.; Zulkifli, F.; Kadirgama, K.; Ghani, S.; Shariffuddin, J. A comprehensive review of hydroxyapatite-based coatings adhesion on metallic biomaterials. Ceram. Int. 2018, 44, 1250–1268. [Google Scholar] [CrossRef]
- Lacefield, W.R. Hydroxyapatite Coatings. Ann. N. Y. Acad. Sci. 1988, 523, 72–80. [Google Scholar] [CrossRef]
- Ong, J.L.; Chan, D.C.N. Hydroxyapatite and Their Use As Coatings in Dental Implants: A Review. Crit. Rev. Trade Biomed. Eng. 2000, 28, 667–707. [Google Scholar] [CrossRef]
- Kwon, Y.W.; Clumpner, B.R. Multiscale modeling of human bone. Multiscale Multidiscip. Model. Exp. Des. 2018, 1, 133–143. [Google Scholar] [CrossRef]
- Viswanath, B.; Raghavan, R.; Ramamurty, U.; Ravishankar, N. Mechanical properties and anisotropy in hydroxyapatite single crystals. Scr. Mater. 2007, 57, 361–364. [Google Scholar] [CrossRef]
- Saber-Samandari, S.; Gross, K.A. Micromechanical properties of single crystal hydroxyapatite by nanoindentation. Acta Biomater. 2009, 5, 2206–2212. [Google Scholar] [CrossRef] [PubMed]
- Zamiri, A.; De, S. Mechanical properties of hydroxyapatite single crystals from nanoindentation data. J. Mech. Behav. Biomed. Mater. 2011, 4, 146–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libonati, F.; Nair, A.K.; Vergani, L.; Buehler, M.J. Fracture mechanics of hydroxyapatite single crystals under geometric confinement. J. Mech. Behav. Biomed. Mater. 2013, 20, 184–191. [Google Scholar] [CrossRef]
- Ou, X.; Han, Q. Molecular dynamics simulations of the mechanical properties of monoclinic hydroxyapatite. J. Mol. Model. 2014, 20, 2505. [Google Scholar] [CrossRef]
- Nair, A.K.; Gautieri, A.; Chang, S.W.; Buehler, M.J. Molecular mechanics of mineralized collagen fibrils in bone. Nat. Commun. 2013, 4, 1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Z.; Gautieri, A.; Nair, A.K.; Inbar, H.; Buehler, M.J. Thickness of Hydroxyapatite Nanocrystal Controls Mechanical Properties of the Collagen–Hydroxyapatite Interface. Langmuir 2012, 28, 1982–1992. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.D.; Salehinia, I. Study of nanoscale deformation mechanisms in bulk hexagonal hydroxyapatite under uniaxial loading using molecular dynamics. J. Mech. Behav. Biomed. Mater. 2020, 110, 103894. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO-the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2009, 18, 015012. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Heinz, H.; Lin, T.J.; Kishore Mishra, R.; Emami, F.S. Thermodynamically Consistent Force Fields for the Assembly of Inorganic, Organic, and Biological Nanostructures: The INTERFACE Force Field. Langmuir 2013, 29, 1754–1765. [Google Scholar] [CrossRef] [PubMed]
- Heinz, H. Adsorption of biomolecules and polymers on silicates, glasses, and oxides: Mechanisms, predictions, and opportunities by molecular simulation. Curr. Opin. Chem. Eng. 2016, 11, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Emami, F.S.; Puddu, V.; Berry, R.J.; Varshney, V.; Patwardhan, S.V.; Perry, C.C.; Heinz, H. Force Field and a Surface Model Database for Silica to Simulate Interfacial Properties in Atomic Resolution. Chem. Mater. 2014, 26, 2647–2658. [Google Scholar] [CrossRef]
- Lin, T.J. Force Field Parameters and Atomistic Surface Models for Hydroxyapatite and Analysis of Biomolecular Adsorption at Aqueous Interfaces; The University of Akron: Akron, OH, USA, 2013. [Google Scholar]
- Lin, T.J.; Heinz, H. Accurate Force Field Parameters and pH Resolved Surface Models for Hydroxyapatite to Understand Structure, Mechanics, Hydration, and Biological Interfaces. J. Phys. Chem. C 2016, 120, 4975–4992. [Google Scholar] [CrossRef] [Green Version]
- Ewald, P.P. Die Berechnung optischer und elektrostatischer Gitterpotentiale. Ann. Phys. 1921, 369, 253–287. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Snyder, A.D.; Salehinia, I. Effect of Pore Defects on Uniaxial Mechanical Properties of Bulk Hexagonal Hydroxyapatite: A Molecular Dynamics Study. Int. J. Mol. Sci. 2023, 24, 1535. https://doi.org/10.3390/ijms24021535
Snyder AD, Salehinia I. Effect of Pore Defects on Uniaxial Mechanical Properties of Bulk Hexagonal Hydroxyapatite: A Molecular Dynamics Study. International Journal of Molecular Sciences. 2023; 24(2):1535. https://doi.org/10.3390/ijms24021535
Chicago/Turabian StyleSnyder, Alexander D., and Iman Salehinia. 2023. "Effect of Pore Defects on Uniaxial Mechanical Properties of Bulk Hexagonal Hydroxyapatite: A Molecular Dynamics Study" International Journal of Molecular Sciences 24, no. 2: 1535. https://doi.org/10.3390/ijms24021535
APA StyleSnyder, A. D., & Salehinia, I. (2023). Effect of Pore Defects on Uniaxial Mechanical Properties of Bulk Hexagonal Hydroxyapatite: A Molecular Dynamics Study. International Journal of Molecular Sciences, 24(2), 1535. https://doi.org/10.3390/ijms24021535