1. Introduction
The acquisition, distribution and elimination of metal micronutrients, such as iron (Fe) and copper (Cu), are essential for life [
1]. Metabolic diseases such as diabetes, obesity and non-alcoholic fatty liver usually involve the interaction of multiple underlying molecular mechanisms [
2,
3,
4]. Multiple lines of evidence have shown that metabolic diseases including obesity and diabetes are usually accompanied by the dysfunctional regulation of various metal ions [
5,
6]. Among them, copper is central to many important biological processes, including mitochondrial respiration, antioxidant defense and biocompound synthesis [
1,
7], and is particularly closely associated with the severity and progression of diabetes [
8]. Serum copper was significantly increased in diabetic patients and an STZ-induced diabetic rat model [
8,
9,
10]. Experiments in vitro and in vitro showed that coronary perfusion with a low concentration of copper ions could significantly reduce cardiac function [
11], and the bivalent copper chelator triethylenetetramine could significantly improve cardiac function in rats with diabetic cardiomyopathy [
12,
13]. This experimental evidence indicated that copper homeostasis might play an important role in maintaining cardiac function. However, how copper ions influence cardiac function in diabetic cardiomyopathy remains elusive [
10,
14,
15].
Recently, a unique insight into programmed cell death, referred to as cuproptosis, was proposed in tumor cells and ATP7B
−/− mice [
16]. This copper-induced cell death is characterized by, on the one hand, the decrease in lipoylation of DLAT and DLST and the oligomerization of lipoylated proteins of the TCA cycle induced by the direct copper binding and, on the other hand, destabilization and overall reduction in iron–sulfur (Fe–S) cluster proteins, which together lead to proteotoxic stress and mitochondrial dysfunction [
16]. The Fe–S cluster protein FDX1 and lipoylation are key regulators of cuproptosis [
16]. However, cuproptosis in cardiomyocytes has not been reported, and whether cuproptosis is involved in diabetic cardiomyopathy is still undetermined.
Cu homeostasis is tightly maintained in all organisms through mechanisms of uptake, transport, storage and excretion at a precise scale [
1]. The upper limit of loosely bound copper is restricted to an almost vanishingly low level of less than a single atom per cell [
17]. Redox Cu(II) bound to plasma protein carriers (ceruloplasmin) is predominant in the blood. Cu(II) is reduced to Cu(I) by reductases and transported into the cytoplasm by the high-affinity importer SLC31A1 (CTR1). The imported Cu(I) binds metallothioneins (GSH, etc.) and metallochaperones (superoxide dismutase, etc.), which distribute Cu(I) to different subcellular locations [
1,
18]. Copper ionophores such as elesclomol are copper-binding small molecules that shuttle copper into the cell, ignoring the ion concentration gradient. Therefore, they are useful tools to study copper toxicity [
7,
19]. A series of studies have indicated that the mechanism of copper ionophore-induced cell death involves intracellular copper accumulation and not the effect of the small molecule chaperones themselves [
16].
Advanced glycosylation end products (AGEs) are a heterogeneous group of compounds that include more than 20 different products which are produced by glycation, called the Maillard reaction [
20]. Glucose, fructose or more reactive dicarbonyls react nonenzymatically with nucleophilic groups on proteins, preferentially with ε-amino groups of lysines and N-terminal-amino groups, following a succession of rearrangements of intermediate compounds and subsequently converting to stable AGEs [
20,
21]. AGEs react with new proteins, perpetuating and propagating oxidative modifications and producing new AGEs crosslinks and then accumulating gradually [
21]. Although this reaction is very slow under physiological conditions, this protein modification by glucose is significantly accelerated under the hyperglycemic conditions of diabetes and is considered one of the major factors in the development of diabetic complications [
21,
22,
23]. More importantly, oxidative reactions catalyzed by Cu
2+ and Fe
3+ redox metal ions are involved in AGEs formation under in vivo conditions, but these metal ions are sequestered in specific metal transporters and other metalloproteins in blood plasma or the cellular cytoplasm [
21]. Copper metalloproteins after glycation reactions undergo significant conformational changes and even fragmentation, causing the release of bound metal, completing a positive-feedback loop [
24,
25]. AGEs could promote cardiomyocyte death through calcium overload, oxidative stress or excessive autophagy [
26,
27,
28]. However, it is unclear whether cuproptosis is involved in AGEs-induced cardiomyocyte death. Thus, we investigated the potential effect of cuproptosis on AGEs-induced cardiomyocyte dysfunction in diabetic cardiomyopathy.
3. Discussion
Metal dyshomeostasis at various levels of an organism is a common denominator and cause or consequence of many illnesses [
5]. Defective copper regulation mediates cardiovascular damage through two general processes that occur simultaneously in the same individual: elevation of Cu(II)-mediated pro-oxidant stress and impairment of copper-catalyzed antioxidant defense mechanisms [
4,
10]. Low concentrations of copper ions through coronary perfusion could significantly reduce cardiac function [
11]. Excess copper was also proven to be mobilized following myocardial ischemia and facilitate tissue injury by catalyzing the production of hydroxyl radicals [
32,
33]. Long-term exposure to copper induces apoptosis in mouse hearts [
34], but it is still unclear whether or how excess copper depresses cardiac function. Meanwhile, multiple lines of evidence have shown that the Cu(II)-selective chelators triethylenetetramine (TETA) and trientine could ameliorate left-ventricular hypertrophy and cardiac dysfunction in diabetic cardiomyopathy [
12,
13]. In this study, we found that copper in the serum and myocardium of STZ-induced and db/db spontaneous diabetic mice were both significantly increased. These results were consistent with those of previous studies [
8,
9,
10,
14]. This indicated that diabetes might facilitate copper overload, which could influence the cardiomyocyte function directly in diabetes. Diabetic cardiomyopathy is characterized by cardiomyocyte death, followed by cardiomyocyte hypertrophy, fibroblast proliferation and extracellular matrix increase, resulting in myocardial remodeling and cardiac dysfunction. However, the present limited study showed that copper aggravated diabetic cardiomyopathy mainly by increasing the extracellular matrix by activating TGF-beta/Smad signaling or oxidative stress [
3]. It remaines elusive how copper directly influences the function of cardiomyocytes in diabetic cardiomyopathy [
10,
14,
15], and there is no relevant study on the underpinning mechanism of copper overload-induced cuproptosis in diabetic cardiomyopathy.
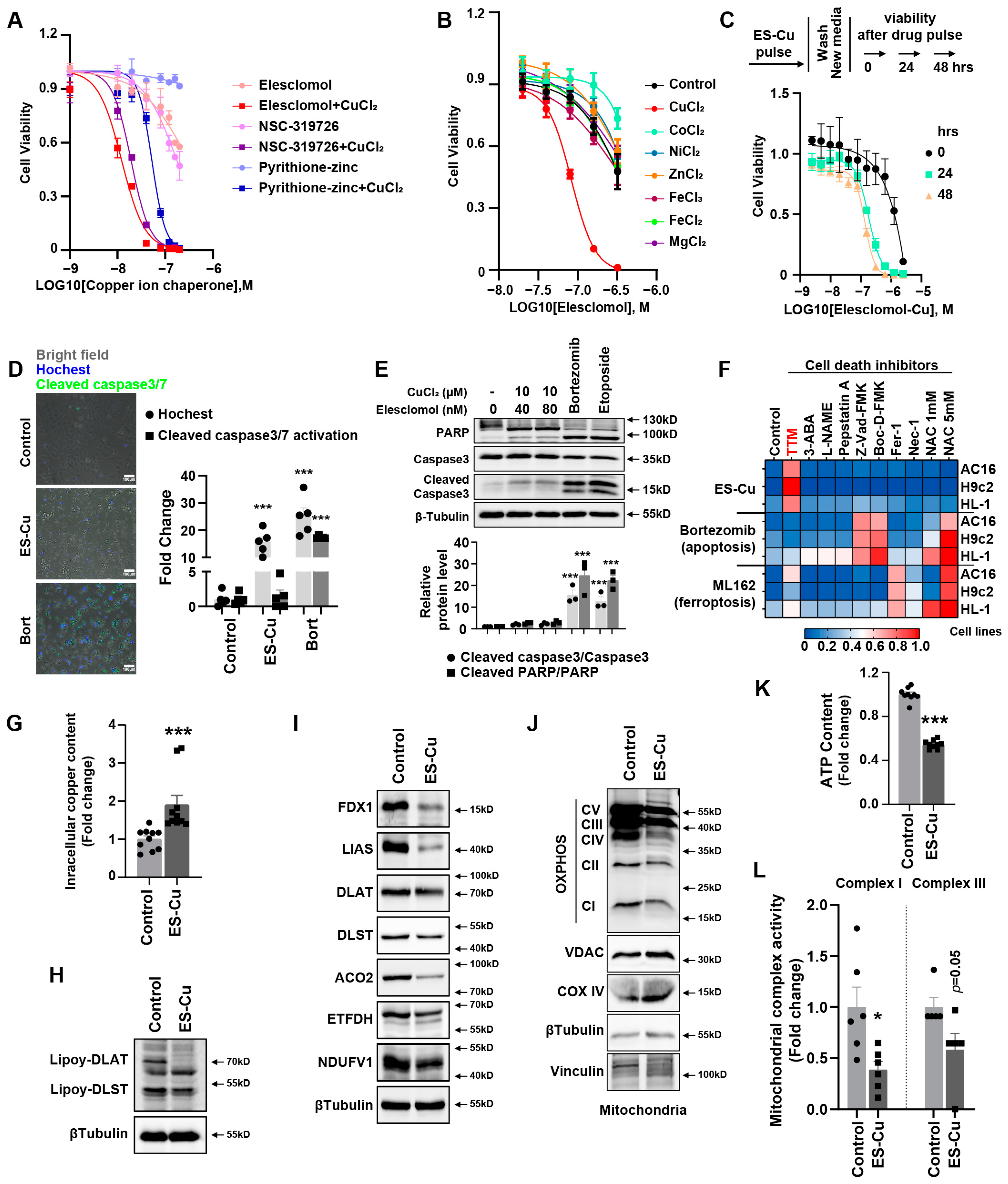
Cuproptosis as the novel and unexpected programmed cell death triggered by targeting Cu to mitochondria has been demonstrated [
16]. Copper ionophore elesclomol induced cuproptosis in a variety of tumor cells [
16], but cuproptosis has not been investigated in cardiomyocytes. Our data suggest that copper overload also promotes cuproptosis in cardiomyocytes. We found elesclomol showed a higher affinity for copper than for other metal ions in cardiomyocytes. Compared with other copper ionophores, it exhibited stronger copper toxicity to cardiomyocytes. This copper-induced cuproptosis is also a unique form of irreversible damage and is only prevented by copper chelator TTM. Whole-genome and metabolism-focused CRISPR screens revealed that FDX1 functions as a direct binder of elesclomol. FDX1 donates an electron to lipoate synthase and reduces Cu
2+ to Cu
+, releasing it within the mitochondrial matrix [
16,
35]. Cuproptosis is elicited by the adventitious nonspecific binding of Cu to lipoylated proteins in mitochondria. FDX1 loss promotes accumulation and depletion of TCA cycle intermediates that correspond to specific reductions in metabolic enzyme lipoylation and accumulation of lipoylation DLAT [
16]. We found similar characteristic changes in cardiomyocytes. Elesclomol–CuCl
2 significantly induced a decrease in Fe–S cluster protein and decrease in lipoyl–DLAT and lipoyl–DLST and a significant abundance of HSP70 protein in cardiomyocytes. These factors contributed to decreased mitochondrial oxidative respiratory chain protein expression, depressed mitochondrial complex I and III activities and decreased ATP content. Depressed mitochondrial oxidative respiration is the main outcome of cuproptosis in cardiomyocytes, further suggesting the inability of the cells to respond to an energetic demand, which eventually leads to cardiomyocyte death.
AGEs are produced by glycation. AGEs are risk factors for multiple diabetes complications. Although the formation reaction of AGEs is very slow under physiological internal environment, this protein modification by glucose is significantly accelerated under the hyperglycemic conditions of diabetes [
21,
22,
23]. The toxicity of AGEs on cardiomyocytes is mainly induced by calcium overload, oxidative stress or excessive autophagy [
26,
27,
28]. Notably, catalytically-active Cu(II), probably binding AGEs and localizing themselves to blood vessels, contributes to pathogenic damage of atherosclerosis [
10]. Meanwhile, a positive-feedback loop exists in Alzheimer’s disease: oxidative reactions catalyzed by Cu(II) are involved in the glycation of AGEs formation, and copper metalloproteins binding copper would undergo conformational changes and fragment after glycation, causing the release of catalytically-active Cu(II) [
24,
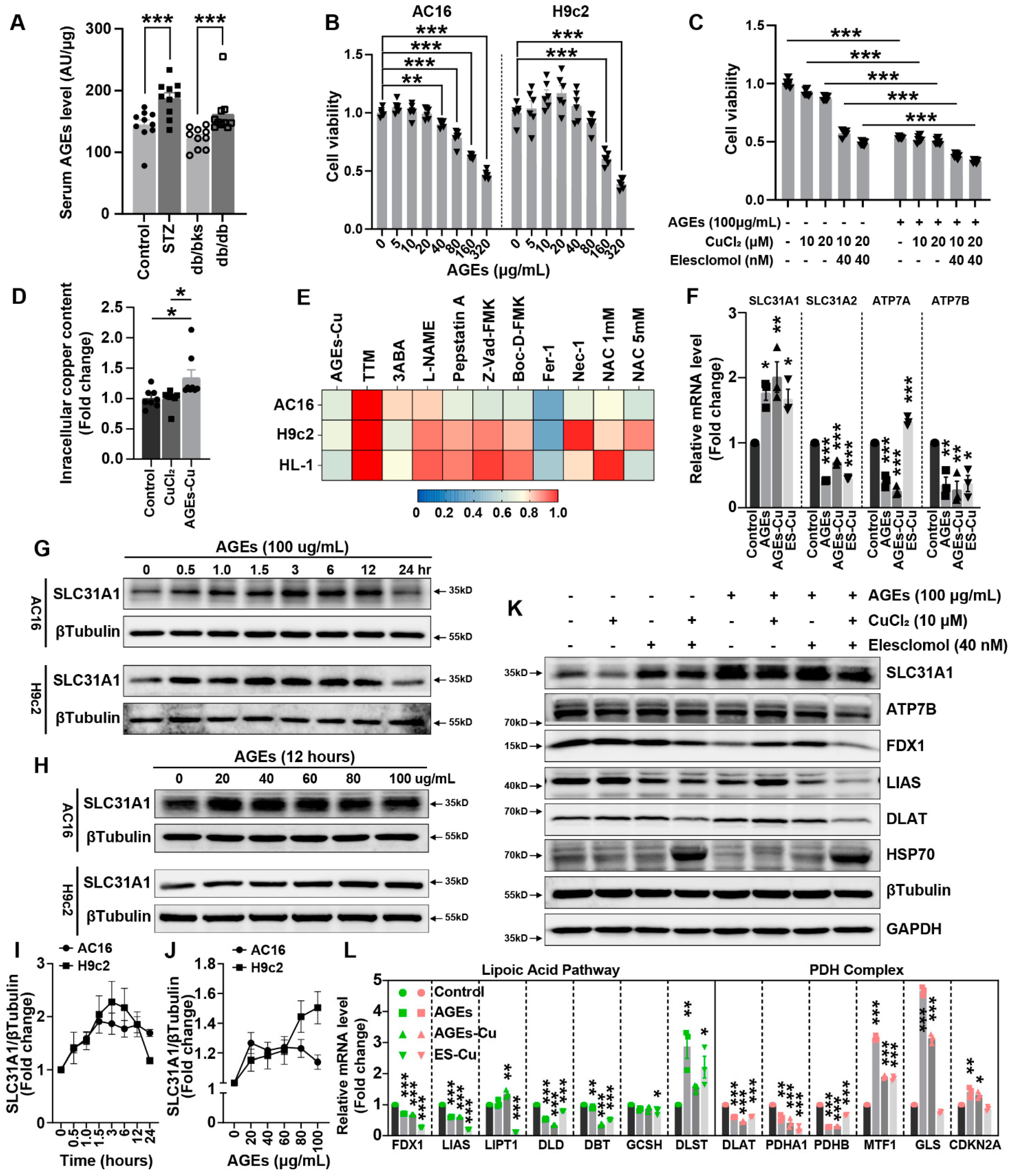
25]. However, the relationship between AGEs and cuproptosis has not been reported. We found that the concentrations of AGEs in the blood and myocardium were significantly upregulated in diabetic mice. Our data also illustrate that AGEs significantly induced cardiomyocyte death. More importantly, synergistic toxicity of AGEs and Cu was more obvious and could be alleviated by copper chelators to a certain extent, indicating that AGEs might promote copper overload-induced cardiotoxicity and participate in cuproptosis. We further analyzed the changes in copper transporters after AGEs treatment, and found that the mRNA and protein levels of copper importer SLC31A1 were significantly upregulated, while the exporters ATP7A and ATP7B were downregulated, indicating that AGEs might increase copper accumulation and thereby induce copper overload in cardiomyocytes. AGEs–CuCl2 similarly changed the cuproptosis-related protein expression and suppressed mitochondrial oxidative respiration. We also observed that SLC31A1 upregulation, cuproptosis-related protein expression changes and mitochondrial oxidative respiration decreases in diabetic murine hearts induced by STZ or db/db. To date, only the ATP7A protein is markedly downregulated in vessels isolated from T2DM patients and diabetic mice [
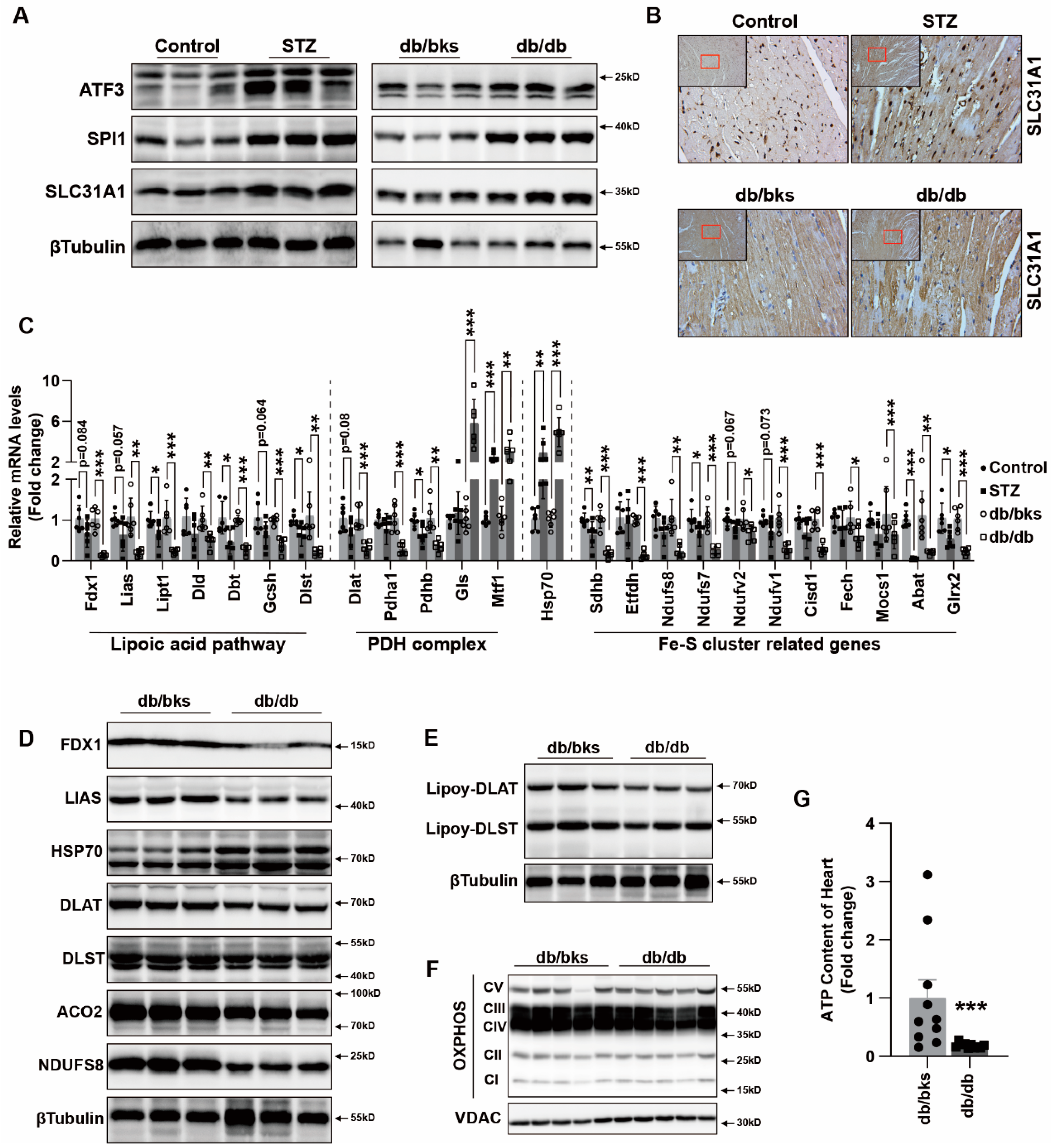
36], whereas how the other copper transporters, ATP7B and SLC31A1, change in diabetes was uncertain. Our results indicated SLC31A1 was upregulated in DCM, which might be the reason for copper overload and cuproptosis in DCM. The upregulation of SLC31A1 might be an alternative illustration of the mechanism of DCM [
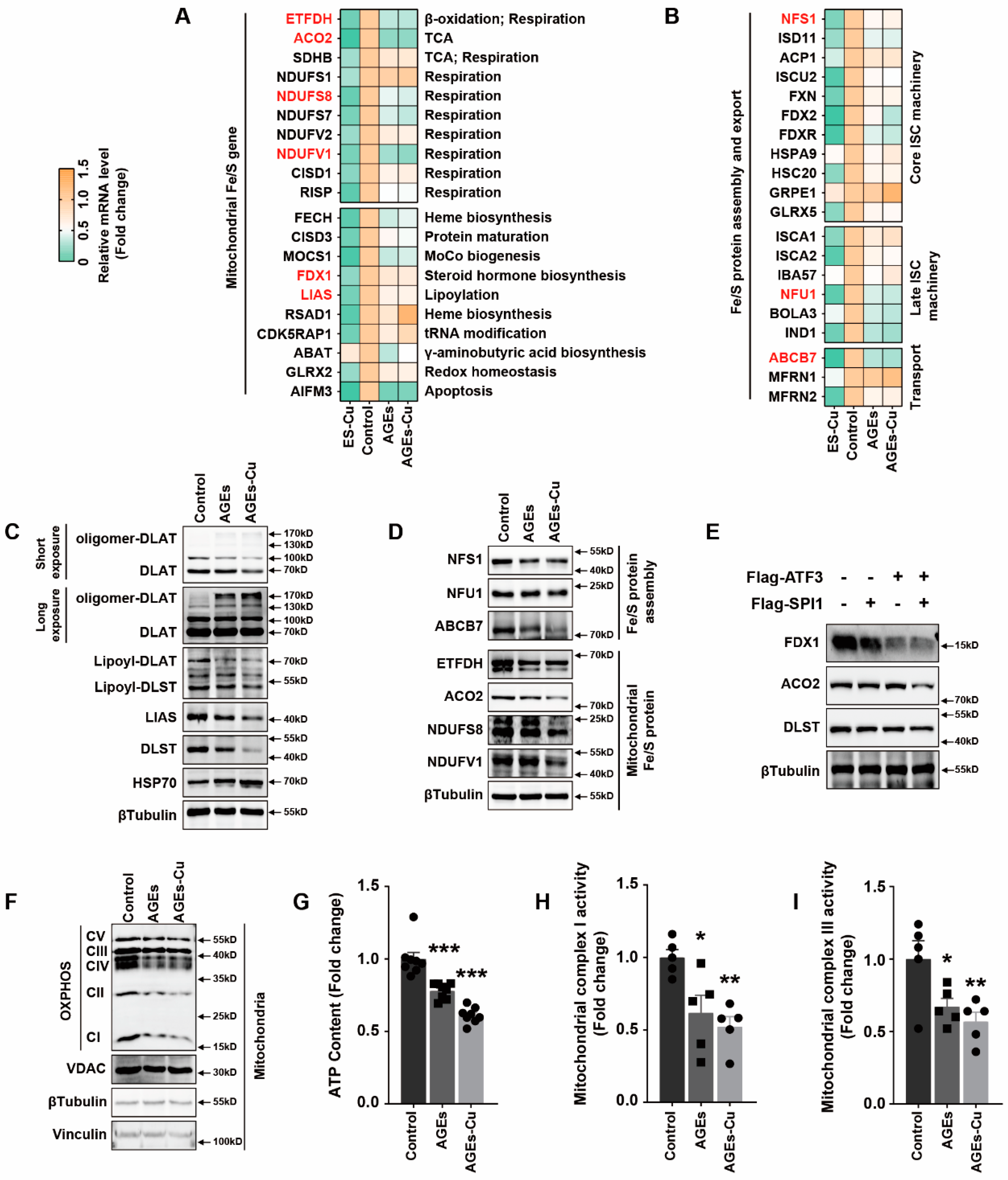
16].
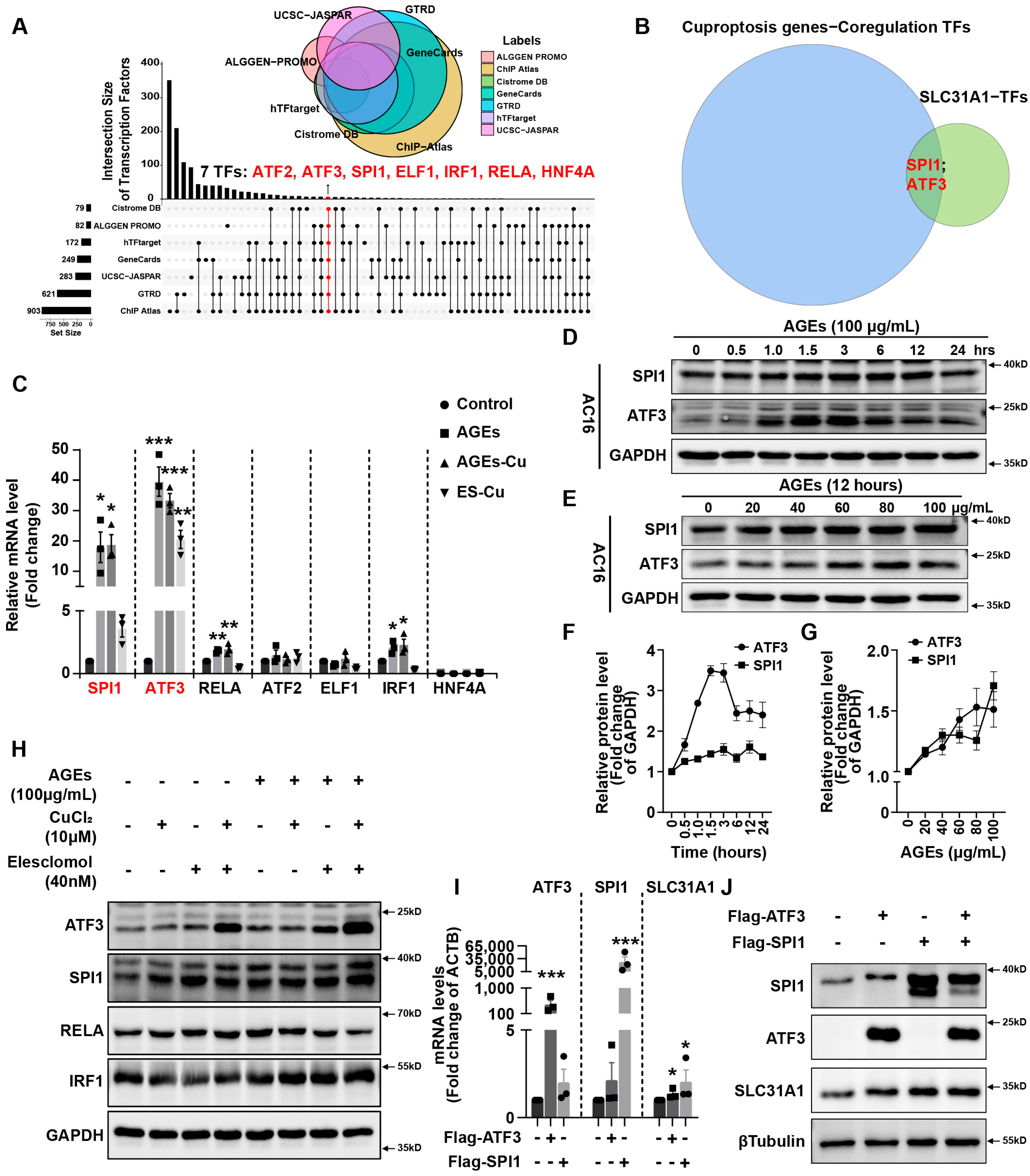
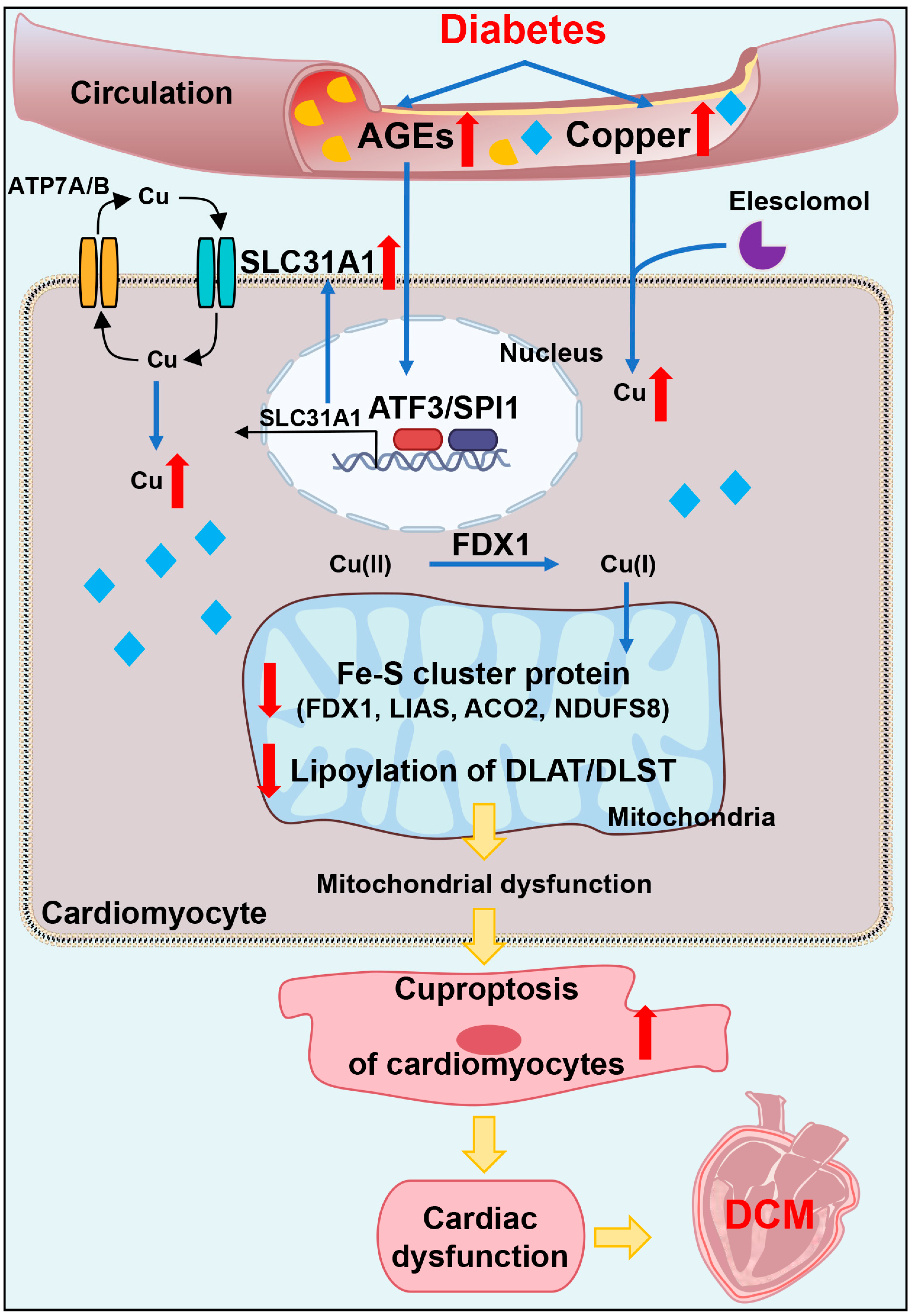
In addition, we explored the underlying mechanism of how AGEs upregulated the expression of SLC31A1 in AC16 cardiomyocytes. Multiple databases, includingALGGEN PROMO, ChIP Atlas, Cistrome DB, GeneCards, GTRD, hTFtarget and UCSC JASPAR, were integrated to predict possible transcription factors of SLC31A1. Meanwhile, the co-transcription factors of the cuproptosis gene set, including 10 genes of FDX1, LIAS, LIPT1, DLD, DLAT, PDHA1, PDHB, MTF1, GLS and CDKN2A, were predicted by the hTFtarget database. We found that ATF3 and SPI1 were the overlapping genes of the two predicted TF sets. The overexpression of ATF3 and/or SPI1 upregulated SLC31A1 and promoted the loss of Fe–S cluster protein expression. Our data indicated that ATF3 and/or SPI1 might be upstream of SLC31A1 and regulate cuproptosis of cardiomyocytes in diabetes (
Figure 7). The transcriptional regulation of SLC31A1 by ATF3 and SPI1 has not been reported, not only in cardiovascular but also in diabetic disease. Activating transcription factor 3 (ATF3) is an adaptive-response gene. ATF3 plays a dual role in cardiac remodeling, and both ATF3 knockdown and overexpression in adult mice resulted in cardiac hypertrophy and dysfunction in response to pathological stimulation [
37,
38]. In diabetes, ATF3 also promoted β-cell dysfunction, accelerated STZ- induced diabetic liver injury and aggravated podocyte injury of db/db mice. This indicated the pathological effect of ATF3 in diabetes [
39,
40,
41]. There is a positive feedback loop between ATF3 and oxidative stress [
42,
43]. Moreover, ATF3 could promote erastin-induced ferroptosis by repressing SLC7A11 expression and suppressing system Xc [
44]. This indicated that ATF3 might be involved in pathogenicity by affecting redox homeostasis and regulating the expression of metal ion transporters. Our findings also proved that ATF3 might influence copper homeostasis and promote cuproptosis by regulating the expression of the copper importer SLC31A1. Spi-1 proto-oncogene (SPI1) is a multifaceted TF that involves numerous normal and pathogenic functions within the hematopoietic system [
45]. However, there is less evidence investigating the relationship between SP1 and diabetes or cardiovascular diseases. A previous study has shown that SPI1 upregulation activated the TLR4/NFκB axis and aggravated myocardial infarction [
46]. SPI1 also promoted the toxicity of nickel ions in THP-1 cells, indicating its important role in the homeostasis of metal ions [
47]. Our findings revealed the potential regulatory effect of SPI1 on copper homeostasis and cuproptosis in diabetic cardiomyopathy.
This experiment indeed contained some limitations. Firstly, we performed most experiments in cell lines, but there still existed subtle differences between cell lines and primary cells. Hence, further investigation should be performed for the probable mechanism in primary cells. Next, further research is needed to determine whether intervention in the ATF3/SPI1/SLC31A1 signaling pathway could alleviate diabetic cardiovascular injury in diabetic animals. The exploration of inhibitors to related signaling pathways might also be a potential therapeutic target for the treatment of diabetic cardiomyopathy. Finally, ATF3/SPI1/SLC31A1 signaling might not be the only and the most important molecular target of AGEs–CuCl2 treatment. Whole-genome CRIPSR-Cas9 positive selection screen or Genome siRNA Library screen would find more underlying targets for AGEs–CuCl2 treatment.
In conclusion, our data showed, for the first time, that AGEs-induced cuproptosis might be a novel mechanism of DCM. The excessive increase in AGEs and copper in diabetes induced the upregulation of copper importer SLC31A1 through ATF3/SPI1, thereby mediating the accumulation of copper in cardiomyocytes, disturbing copper homeostasis and promoting cuproptosis. The decline in the Fe–S cluster protein and lipoylation of DLAT and DLST aggravated mitochondrial dysfunction in cardiomyocytes and resulted in myocardial dysfunction. Collectively, AGEs–CuCl2-induced cuproptosis via the ATF3/SPI1/SLC31A1 pathway might be an alternative potential therapeutic target for DCM.
4. Materials and Methods
4.1. Cell Culture and Compounds
The myocardial cell line H9c2 was obtained from the American Type Culture Collection. AC16 and HL-1 were donated by Hesong Zeng group of Division of Cardiology, Department of Internal Medicine, Tongji Hospital, Tongji Medical College and Huazhong University of Science and Technology (Wuhan, China). AC16 cardiomyocytes were validated by the STR profiling method. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Keygen Biotech, Nanjing, China) supplemented with 10% (v/v) fetal bovine serum (FBS, Gibico, Thermo Fisher Scientific, Grand Island, NY, USA) and 1% (v/v) penicillin/streptomycin (Sangon, Shanghai, China) in a humidified atmosphere of 5% CO2 at 37 °C.
Metal ion chloride: Copper(II) chloride (CuCl2, 751944, Sigma-Aldrich, Darmstadt, Germany), cobalt(II) chloride (CoCl2, 232696, Sigma-Aldrich, Darmstadt, Germany), zinc chloride (ZnCl2, 208086, Sigma-Aldrich, Darmstadt, Germany), iron(III) chloride (FeCl3, 157740, Sigma-Aldrich, Darmstadt, Germany) and magnesium chloride (MgCl2, 208337, Sigma-Aldrich, Darmstadt, Germany) were purchased from Sigma-Aldrich Technology (Darmstadt, Germany). Nickel(II) chloride (NiCl2, N829833, Macklin, Shanghai, China) was purchased from Macklin Biochemical Technology (Shanghai, China). Iron(II) chloride (FeCl2, I106504, Aladdin, Shanghai, China) were purchased from Aladdin Biochemical Technology (Shanghai, China).
Copper ionophores: Elesclomol (S1052, Selleck, Shanghai, China) was purchased from Selleck (Shanghai, China). Thiram (45689, Sigma-Aldrich, Darmstadt, Germany), 2-Methyl-8-quinolinol (8-HQ, H57602, Sigma-Aldrich, Darmstadt, Germany) and tetramethylthiuram monosulfide (TMT, 567205, Sigma-Aldrich, Darmstadt, Germany) were purchased from Sigma-Aldrich. NSC-319726 (HY-18634, MCE, Shanghai, China), pyrithione-zinc (HY-B0572, MCE, Shanghai, China) and disulfiram (HY-B0240, MCE, Shanghai, China) were purchased from MedChemExpress Technology (MCE, Shanghai, China).
Cell death inhibitors: Ammonium tetrathiomolybdate (TTM, 323446, Sigma-Aldrich, Germany) was purchased from Sigma-Aldrich. L-NAME (HY-18729A, MCE, Shanghai, China), Boc-D-FMK (HY-13229, MCE, Shanghai, China) and Ferrostatin-1 (Fer-1, HY-100579, MCE, Shanghai, China) were purchased from MCE. Pepstatin A (S7381, Selleck, Shanghai, China), 3-Aminobenzamide (3-ABA, S1132, Selleck, Shanghai, China), N-Acetyl-L-cysteine (NAC, S1623, Selleck, Shanghai, China), necrostatin-1 (Nec-1, S8037, Selleck, Shanghai, China) and Z-VAD-FMK (S7023, Selleck, Shanghai, China) were purchased from Selleck.
Cell death inducers: Bortezomib (S1013, Selleck, Shanghai, China), Etoposide (S1225, Selleck, Shanghai, China) and ML162 (S4452, Selleck, Shanghai, China) were purchased from Selleck (Shanghai, China). Advanced glycation end products (AGEs, bs-1158P, bioss, Shanghai, China) were purchased from bioss Biochemical Technology (Shanghai, China).
4.2. Cell Viability Assay
Cellular viability was analyzed using the cell counting kit-8 (B34304, Selleck, Shanghai, China). The 5 × 103 cells were seeded in 96-well plates and treated with indicated reagents for indicated time-points. A 10 μL CCK-8 solution was added to each well and the cultures were incubated at 37 °C for 45 min to 1 hour. Absorbance at 450 nm was measured using a microplate reader (Synergy 2, Bio-Tek Instruments, Winooski, VT, USA). The cell viability quantification in the heatmap was based on the absorbance at 450 nm. Four to six replicate wells for each group were established in every single experiment, and at least two independent times were carried out for each experiment. The absorbance of each group was normalized by the blank group (the indicated cells without any treatment). Given that the copper chelator TTM pretreatment significantly improved the cell viability, the absorbance of the TTM group was defined as 1, and the heatmap was plotted according to the average value of two independent experiments.
4.3. Caspase 3/7 Activity Analysis
Caspase 3/7 activation of caspase 3/7 cleavage in AC16 cells was detected by CellEvent Caspase-3/7 Green ReadyProbes (R37111, Invitrogen, Life Technologies Corporation, Eugene, OR, USA). AC16 cells were grown in a six-well in duplicate. Cells were subjected to elesclomol–CuCl2, Bortezomib and etoposide for 24 h and then incubated with caspase-3/7 green regent (2 drops/mL) for at least 30 min. The fluorescence image is detected through standard FITC filters by the MShot fluorescence microscope (Wuhan, China).
4.4. Intracellular Content of Copper
Intracellular content of copper was measured by copper assay kit (ab272528, Abcam, Boston, MA, USA). Then, 1 × 107 AC16 cardiomyocytes after elesclomol–CuCl2, AGEs or AGEs–CuCl2 treatment were collected, and they were homogenized after adding 1 mL distilled water. Next, 100 µL samples per well were transferred to a flat-bottom 96-well UV plate. For each assay well, 35 µL Reagent, 5 µL Reagent B and 150 µL Reagent C were thoroughly mixed with samples, incubated for 5 min at room temperature and the optical density read at 359 nm using a microplate reader (Synergy 2, Bio-Tek Instruments, Winooski, VT, USA). Copper content was normalized by protein concentration.
4.5. ATP Content Detection
ATP content in cells and heart tissue was measured using a commercially available intracellular ATP assay kit (S0026, Beyotime, Shanghai, China). Previously, 100 μL ATP detection reagent was added to each well of 96-well plate and incubated at room temperature for 5 min to minimize the background. In addition, 20 μL cell or tissue lysate per well was then mixed with ATP detection reagent, and luminescence (RLU) was measured by microplate reader (Synergy 2, Bio-Tek Instruments, Winooski, VT, USA). ATP concentration was normalized by protein concentration.
4.6. Mitochondrial Fractionation
The mitochondrial extracts were isolated using the cell mitochondria isolation kit (C3601, Beyotime Biotechnology, Shanghai, China) according to the manufacturer’s protocol. Briefly, 5 × 107 cells were washed with cold PBS twice. Ice-cold isolation reagent (800 μL, plus protease inhibitor) was added to the pellet and resuspended at 4 °C for 15 min. The suspension was homogenized about 10–30 times at 4 °C. After centrifuging at 600× g at 4 °C for 10 min, the supernatant was collected and transferred to a new Eppendorf tube. After centrifuging at 600× g at 4 °C for 10 min, the pellet of isolated mitochondria was ready for subsequent experiments.
4.7. Mitochondrial Complex I and III Activity
The extracted mitochondria of AC16 cardiomyocytes were used for mitochondrial complex activity measurement by CheKine micro mitochondrial complex I and III activity assay kits (KTB1850 and KTB1870, Abbkine, Wuhan, China). Mitochondria precipitation was resuspended by indicated reagents in steps. The samples and working reagents were mixed in a 96-well UV plate. The absorbance value was read at 0 min and 2 min after mixture and then was calculated according to the corresponding formula. The optimal absorbance of mitochondrial complex I and III was detected at 340 nm and 550 nm, respectively, by microplate reader (Synergy 2, Bio-Tek Instruments, Winooski, VT, USA).
4.8. Transcription Factor Prediction of Target Genes
ALGGEN PROMO (
http://alggen.lsi.upc.es/ (accessed on 1 June 2022)), ChIP Atlas (
https://chip-atlas.org/ (accessed on 1 June 2022)), Cistrome DB (
http://cistrome.org/db/ (accessed on 1 June 2022)), GeneCards (
https://www.genecards.org/ (accessed on 1 June 2022)), GTRD (
http://gtrd.biouml.org/ (accessed on 1 June 2022)), hTFtarget (
http://bioinfo.life.hust.edu.cn/hTFtarget/ (accessed on 1 June 2022)) and UCSC JASPAR (
https://genome.ucsc.edu/ (accessed on 1 June 2022)) are seven databases used for predicting the possible transcription factors of target genes. We predicted the possible transcription factors of copper importer SLC31A1 (solute carrier family 31 member 1, Homo sapiens, Gene ID: 1317) online according to the detailed steps of each website. The cuproptosis gene set was obtained from the previously published article (PMID: 35298263). This gene set includes the seven genes of FDX1, LIAS, LIPT1, DLD, DLAT, PDHA1 and PDHB, positively associating with cuproptosis, and negatively cuproptosis-regulated genes of MTF1, GLS and CDKN2A. The co-transcription factors of the cuproptosis gene set were predicted by hTFtarget database. Overlapping TFs of the two predicted TF sets were identified by Venn diagram.
4.9. ATF3 and SPI1 Overexpression
Cells were transfected by seeding at a density of 100,000 cells/mL in 6-well plates in 2 mL of DMEM. After adherence, overexpression plasmid pcDNA3.1(+)-Flag-ATF3 and/or pcDNA3.1(+)-Flag-SPI1 (PAIWEI Technology, Wuhan, China) was transfected with Lipofectamine 3000 transfection reagent (L3000008, ThermoFisher) in Opti-MEM I reduced serum medium (31985070, ThermoFisher). After a 6-h incubation period, the medium was replaced with DMEM and cells were cultured for 48 h.
4.10. Whole Cell Protein Extraction and Western Blotting
Homogenate myocardial tissue and the collected cultured cells were lysed in RIPA lysis buffer with 1 mM protease inhibitor and phosphatase inhibitor for 40 min and then centrifuged at 12,000 rpm for 20 min at 4 °C. The supernatant was collected and quantified by BCA protein assay (P5026, Sangon, Shanghai, China). Proteins (15–30 μg) were loaded onto 8–15% Bis-Tris SDS-polyacrylamide gels and underwent electrophoresis at 60 V for 30 min and then 120 V for 1 hour. Then, separated proteins in gels were transferred to 0.45 μm PVDF membranes (Millipore, Tullagreen, Carrigtwohill, Ireland) at 230 mA for 100 min. After blocking with TBS-T (Tris-buffer saline with 0.1% Tween 20) containing 5% powdered milk for 90 min, the membranes were incubated with primary antibodies overnight. HRP-conjugated secondary antibodies and ultra-high sensitivity ECL (HY-K1005, MCE, Shanghai, China) were used for immunoblot imaging by ChemiScope 6100 Imager (QinXiang Products Ltd., Shanghai, China).
4.11. Primary Antibodies
PARP1 (#9532), cleaved caspase3 (#9664), VDAC (#4661), COX IV (#4844), ACO2 (#6571), DLST (#11954) and RELA (#8242) were obtained from Cell Signaling Technology (Beverly, MA, USA).
Lipoic acid (ab58724), total OXPHOS cocktail (ab110411) and FDX1 (ab108257) were obtained from Abcam Technology (Boston, MA, USA).
βTubulin (10068-1-AP), Vinculin (66305-1-Ig), LIAS (11577-1-AP) and GAPDH (10494-1-AP) were obtained from Proteintech Technology (Wuhan, China).
SLC31A1 (T510261), ATP7B (T58616) and HSP70 (M20033) were obtained from Abmart Technology (Shanghai, China).
ATF3 (A13469), SPI1 (A20461), IRF1 (A7692), DLAT (A8814), NDUFS8 (A13034), NDUFV1 (A8014), NFS1 (A13385), NFU1 (A7097) and ABCB7 (A14699) were obtained from ABclonal Technology (Wuhan, China).
4.12. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA was isolated from frozen heart tissues or collected cultured cells using the FastPure Cell/Tissue Total RNA Isolation Kit (RC112, Vazyme, Nanjing, China). The RNA quality and concentration were determined spectrophotometrically (NanoDrop 2000 spectrophotometer, Thermo scientific, USA). Reverse transcription for cDNA synthesis was performed using HiScipt III RT SuperMix (R323, Vazyme, Nanjing, China). Quantitative real-time polymerase chain reaction was performed with SYBR green Fast qPCR mix (RK21203, ABclonal, Wuhan, China) on StepOnePlus (96-well, Life Technologies, Singapore) or LightCycler 480II (384-well, Roche Diagnostics, Switzerland) real-time PCR system. The mRNA expressions were normalized by housekeeping gene ACTB with the ΔΔCt method. The primer sequences are listed in the
Supplementary Table S1.
4.13. Diabetic Mice Model
Male C57BL/6J, db/bks and db/db mice were purchased from the Charles River Laboratory Animal Technology Co., Ltd. (Beijing, China) and maintained on a regular chow diet. Two diabetic mice models were established: STZ-induced diabetic mice (n = 10) and control mice (n = 10); db/bks mice (n = 10) and db/db diabetic mice (n = 10). STZ-induced diabetes in C57BL/6J mice was performed by high-fat diet feeding (D12492, Research Diets, USA) for eight weeks and then an intraperitoneal injection of STZ (50 mg/kg body weight per day, S0130, Sigma-Aldrich Technology, Germany) in acetate phosphate buffer (C1013, pH 4.5, Solarbio, Beijing, China) for five consecutive days. Animals were considered diabetic when their blood glucose levels exceeded a pre-established value of 15 mmol/L (350 mg/dL). The db/db mice would show spontaneous elevated blood glucose and were considered diabetic when their blood glucose levels exceeded a pre-established value of 15 mmol/L (350 mg/dL) after 8 weeks. After 20 weeks of feeding, the mice were sacrificed. All animal experiments were supported by the institutional review board of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology.
4.14. Blood Glucose and OGTT
Contour TS Blood Glucose Monitoring System (Ascensia Diabetes Care, Parsippany, NJ, USA) and blood glucose test strips (Ascensia Diabetes Care, USA) were used to measure blood glucose and oral glucose tolerance test (OGTT). To perform the OGTT, mice were fasted for 16 hours before receiving a gavage of 20% glucose (2 g/Kg) (G8150, Solarbio, Beijing, China), and blood samples were collected from the tail 0, 15, 30, 45, 60, 90 and 120 min later for blood glucose determination.
4.15. Serum AGEs Level
An Advanced Glycation End Products (AGEs) assay kit (ab273298, Abcam, USA) was used for detecting the AGEs’ serum levels. A 10 µL portion of serums and 190 µL AGEs assay buffer were added into wells of a white 96-well plate. The plate was then incubated at room temperature for 5 min and fluorescence (Ex/Em = 360/460 nm) measured at room temperature in end point mode using a fluorescence microplate reader (Synergy 2, Bio-Tek Instruments, Winooski, VT, USA). AGEs concentration was normalized by protein concentration.
4.16. Copper Levels in Serum and Myocardial Tissue
A copper colorimetric assay kit (E-BC-K300-M, Elabscience, Wuhan, China) was used for detecting copper levels in serum and myocardial tissue. Then, 20 µL of serum or heart tissue homogenate was added into wells of a 96-well plate, and 300 µL of detection reagent was mixed with samples. The plate was incubated at 37 °C for 5 min and optical density was read at 580 nm using a microplate reader (Synergy 2, Bio-Tek Instruments, USA). The copper concentration in heart tissue was normalized by protein concentration.
4.17. Transthoracic Echocardiography
Transthoracic echocardiography was performed before the mice were sacrificed (VINNO 6 VET, VINNO technology, Suzhou, China). Mice were anesthetized with isoflurane (3% for induction and 2% for maintenance) mixed in 1 L/min 100% O2 via a facemask. The left ventricular systolic function of the mice was evaluated by left ventricular ejection fraction (LVEF), fractional shortening (LVFS) and left ventricular end-systolic volume (LVESV). Cardiac parameters were measured and averaged from at least three separate cardiac cycles. The echocardiography operator was blinded to the grouping of mice.
4.18. Immunohistochemistry (IHC) Staining
A murine heart was fixed in a 10% formation fixative solution, conventionally dehydrated, embedded and the section was cut to 5μm thick. Tissue sections were deparaffinized and rehydrated through xylene and graded alcohols. Heat induced antigen retrieval was carried out with citrate buffer (pH = 6.0) and a pressure cooker at 122 °C for 45 s. Endogenous peroxidase activity was blocked by incubation, incubating sections for 15 min in 3% hydrogen peroxide and anhydrous ethanol (1:1). Sections were incubated at room temperature for 45 min with SLC31A1 antibody (1:500, T510261, Abmart). Application of the primary antibodies was followed by incubation for 30 min with HRP goat anti-rabbit IgG as a secondary antibody (ab6721, Abcam) and visualized with 3, 3′–diaminobenzidine (DAB) as a chromogen and hematoxylin counterstaining.
4.19. Cardiac Fibrosis Assessment
Cardiac fibrosis was evaluated by a Masson-trichrome staining kit (G1340, Solarbio, Beijing, China) and Sirius red staining kit (G1472, Solarbio, Beijing, China). Tissue sections were deparaffinized and rehydrated through xylene and graded alcohols. These sections were stained with Weigert’s Iron Hematoxylin Solution for 10 min and differentiated with Acid Alcohol Differentiation Solution for 10–15 s. Masson-trichrome staining was performed using the following steps: Blue in bluing Solution for 2–5 min, and rinse in deionized water. Then, stain with Ponceau-Acid Fucshin Solution for 10 min and rinse in deionized water. Differentiate in Phosphomolybic Acid Solution for 1–2 min or until collagen is not red. Without rinsing, add Aniline Blue Solution to the section and stain for 1–2 min. Finally, place sections in Acetic Acid Working Solution (Acetic Acid solution:deionized water = 1:2) for one minute. Sirius red staining was performed as follows: Sections were incubated for 30 min in Sirius Red Staining solution and subsequently washed with running water. The sections were then dehydrated in 95% ethanol, absolute ethanol and transparent xylene.
4.20. Copper Salt Staining
After conventionally dewaxing to water, heart tissue sections were stained with dithiooxamide staining solution (G3040, Solarbio Life Science, China) in a 37 °C water bath for 48 h. The section was rinsed with 70% ethanol and slightly washed with distilled water. Then, the section was dried and stained with a nuclear fast red solution for one minute. Next, it was washed with distilled water and conventionally dehydrated and transparently sealed with resinene. Copper salt was stained blackish green, and the nucleus was light red.
4.21. Reproducibility and Statistical Analysis
All experiments have been carried out at least two independent times. The indicated “n” in figure legends represents biological replicates. Data were expressed as the mean ± SD. A two-tailed student’s t-test or an ordinary one-way ANOVA followed by the Bonferroni post-test with multiple comparisons were used to compare the means among experimental groups and control.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






