
AmyP53 Prevents the Formation of Neurotoxic β-Amyloid Oligomers through an Unprecedent Mechanism of Interaction with Gangliosides: Insights for Alzheimer’s Disease Therapy




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
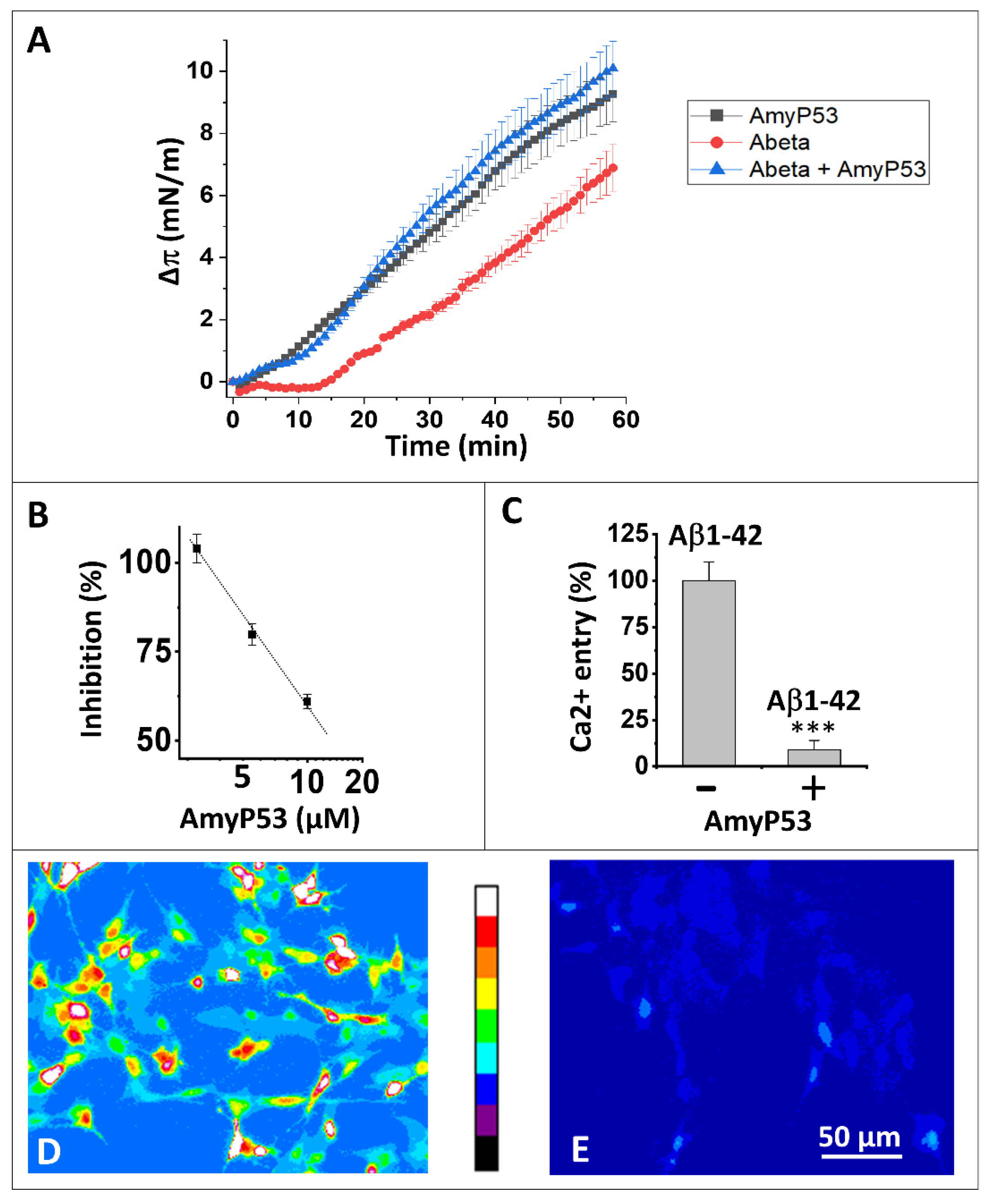
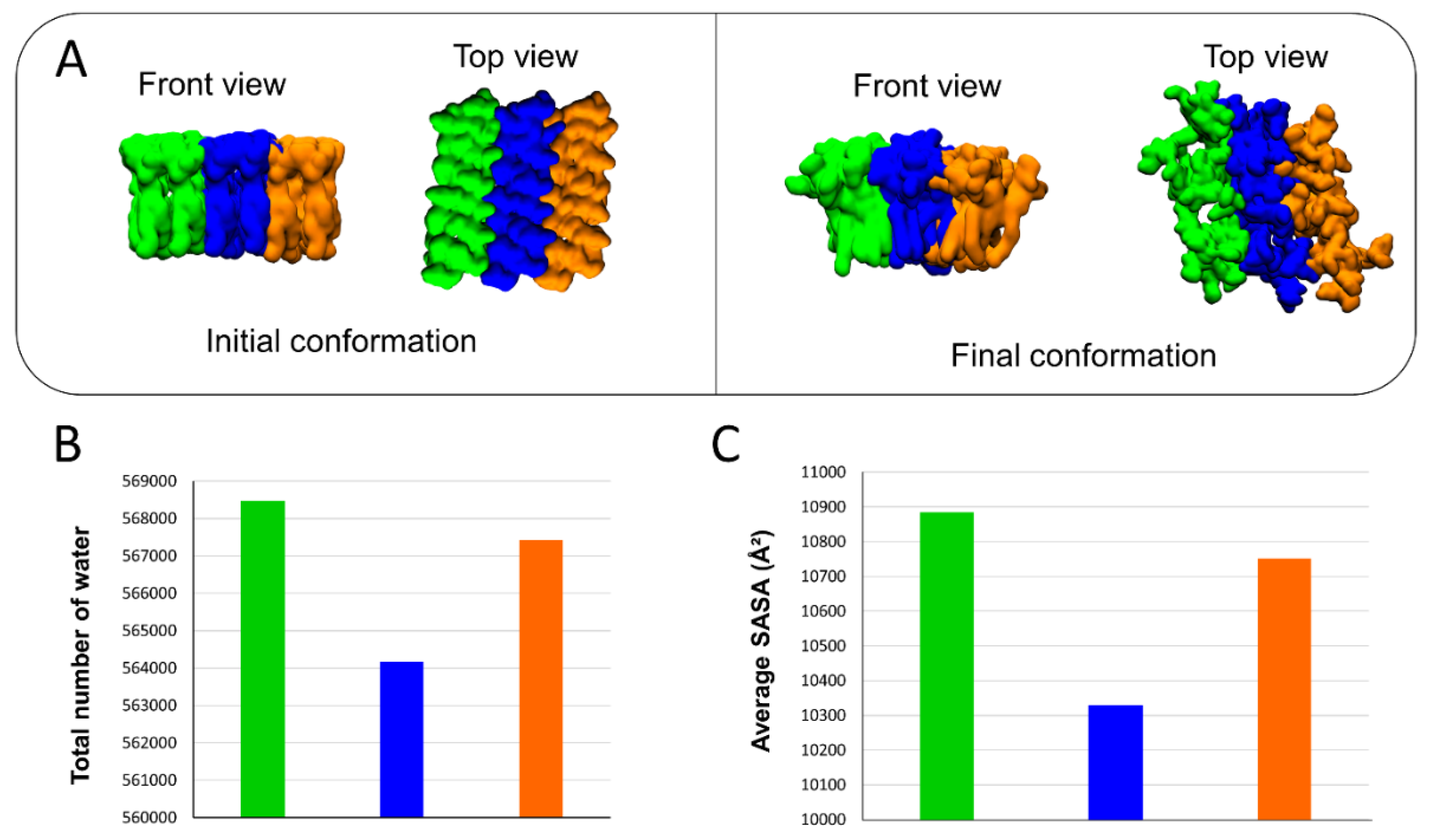
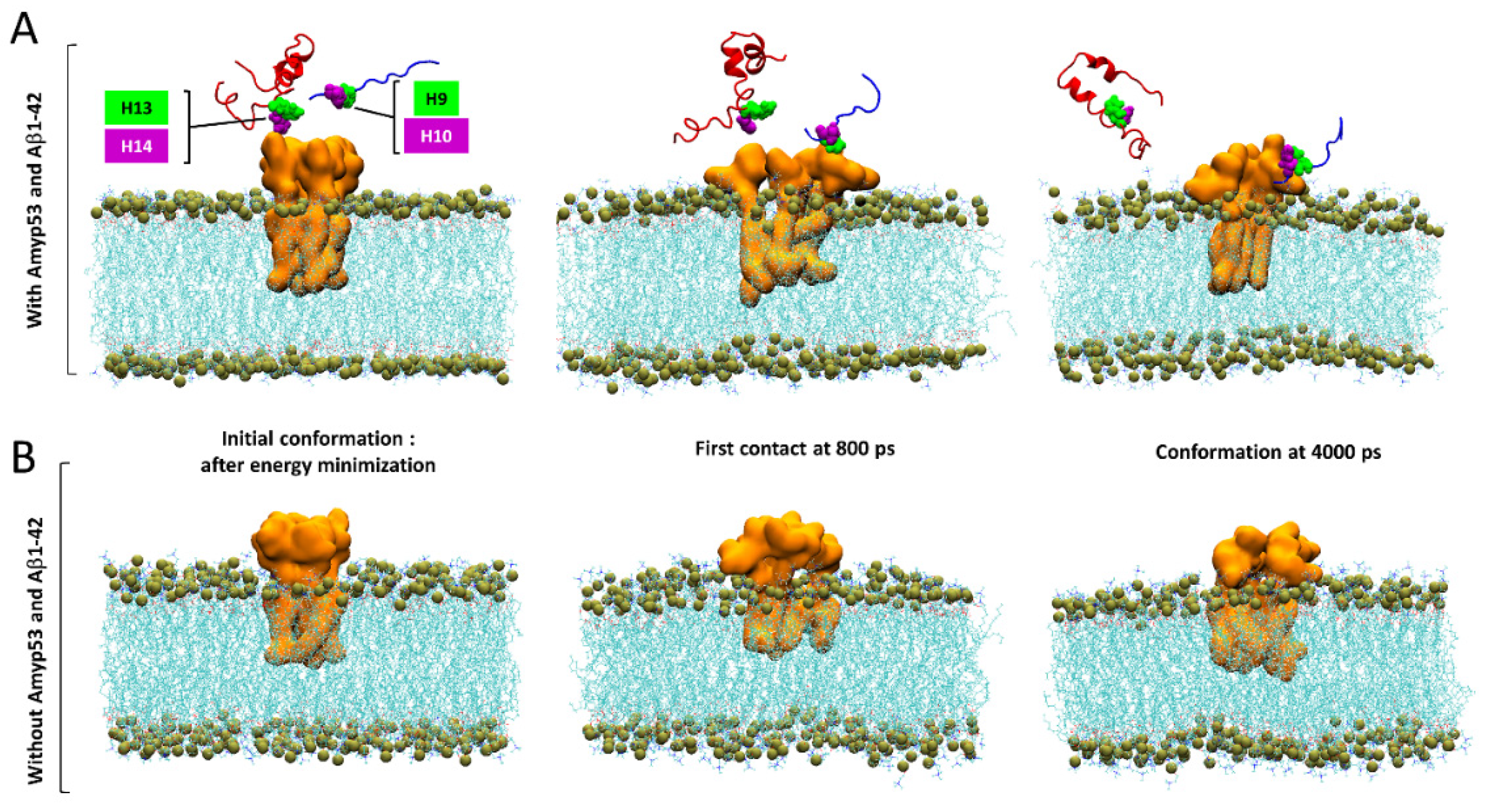
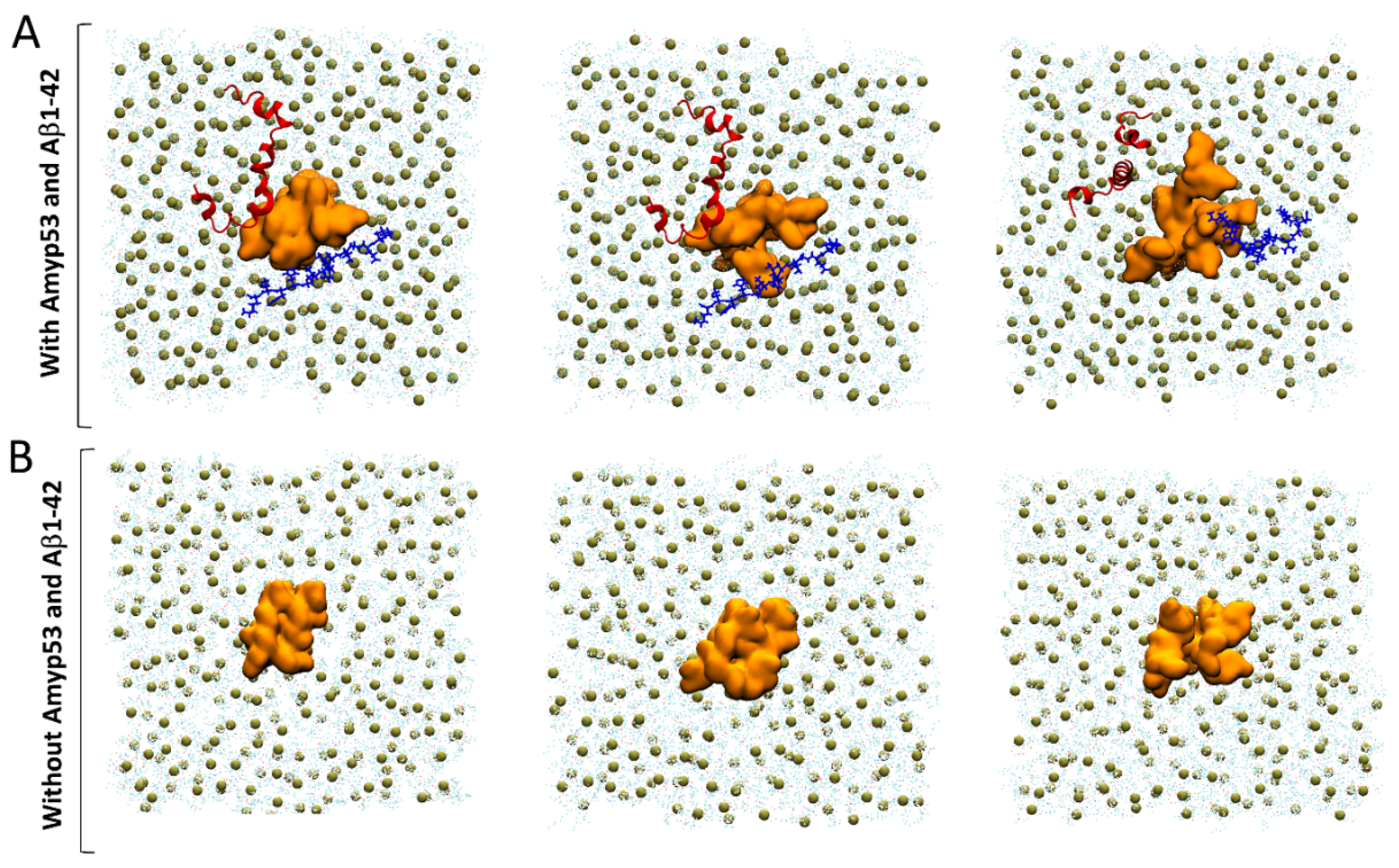
2.1. Interaction of AmyP53 and Aβ1–42 with GM1 in Lipid Rafts
2.2. AmyP53 Is a Competitive Inhibitor of Aβ1–42 Binding to Aged Neural Cells
2.3. AmyP53 Blocks the Formation of Ca2+-Permeable Amyloid Pores
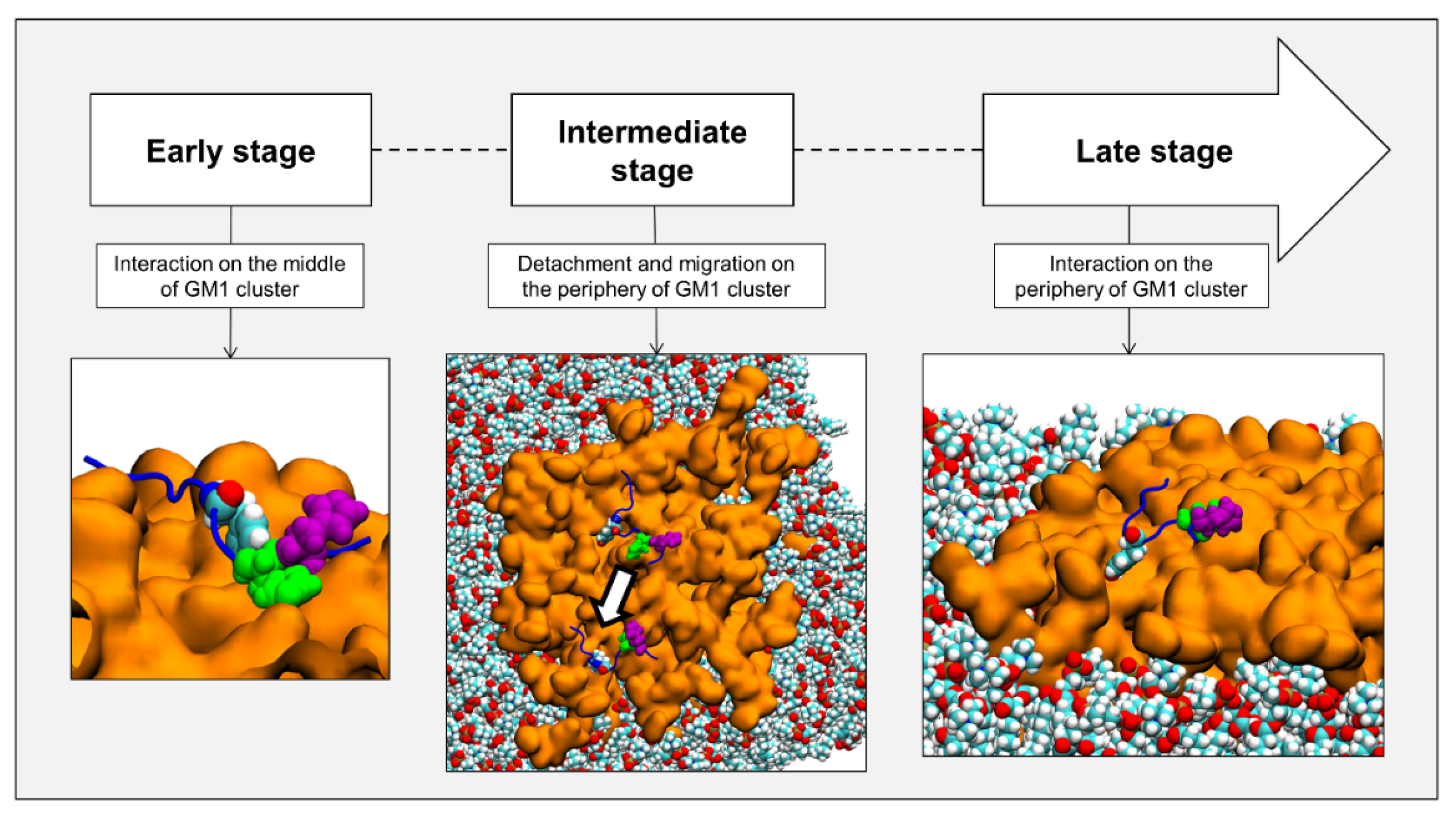
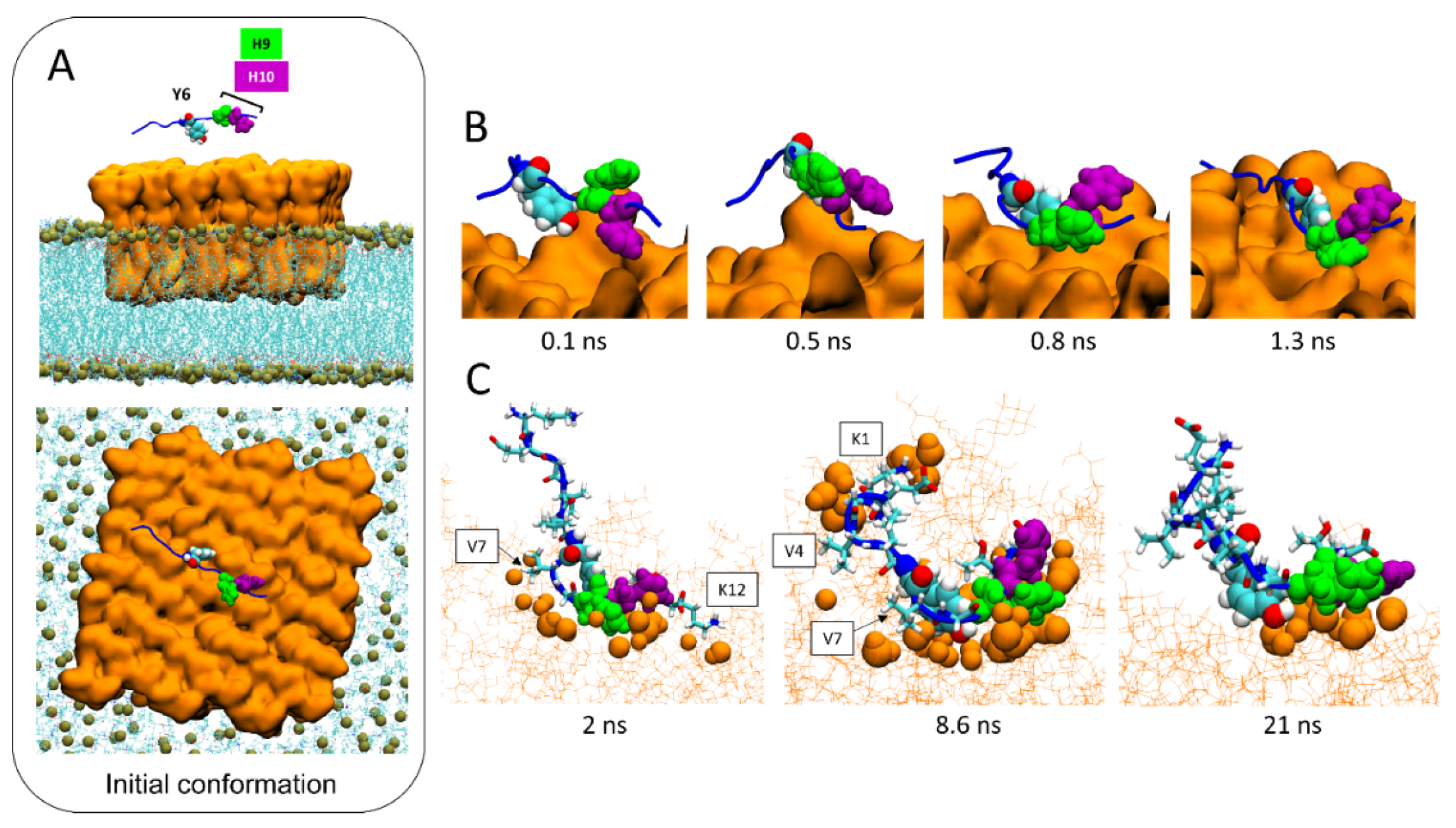
2.4. Timeline of the Journey of AmyP53 on the Surface of a GM1 Lipid Raft
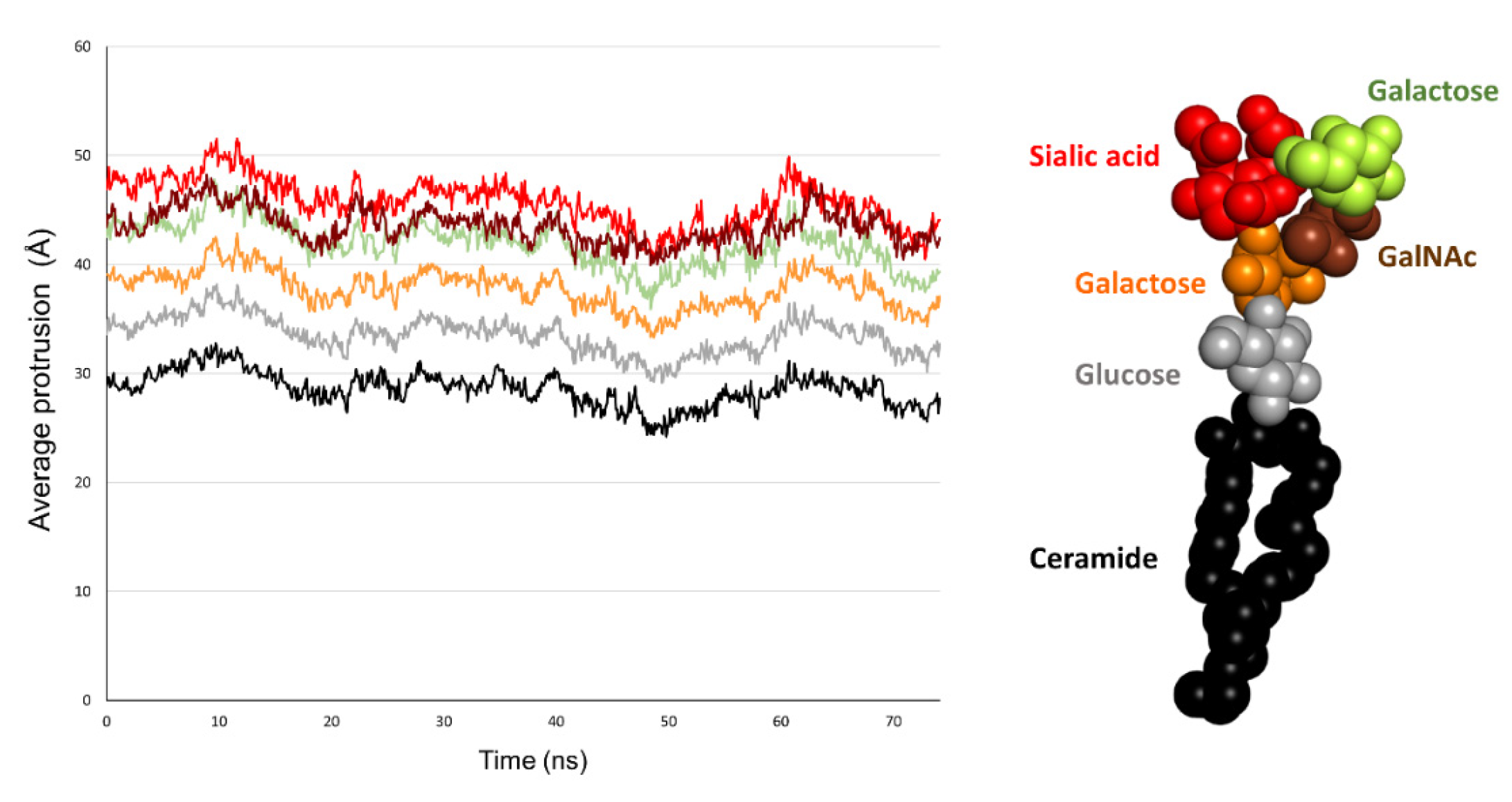
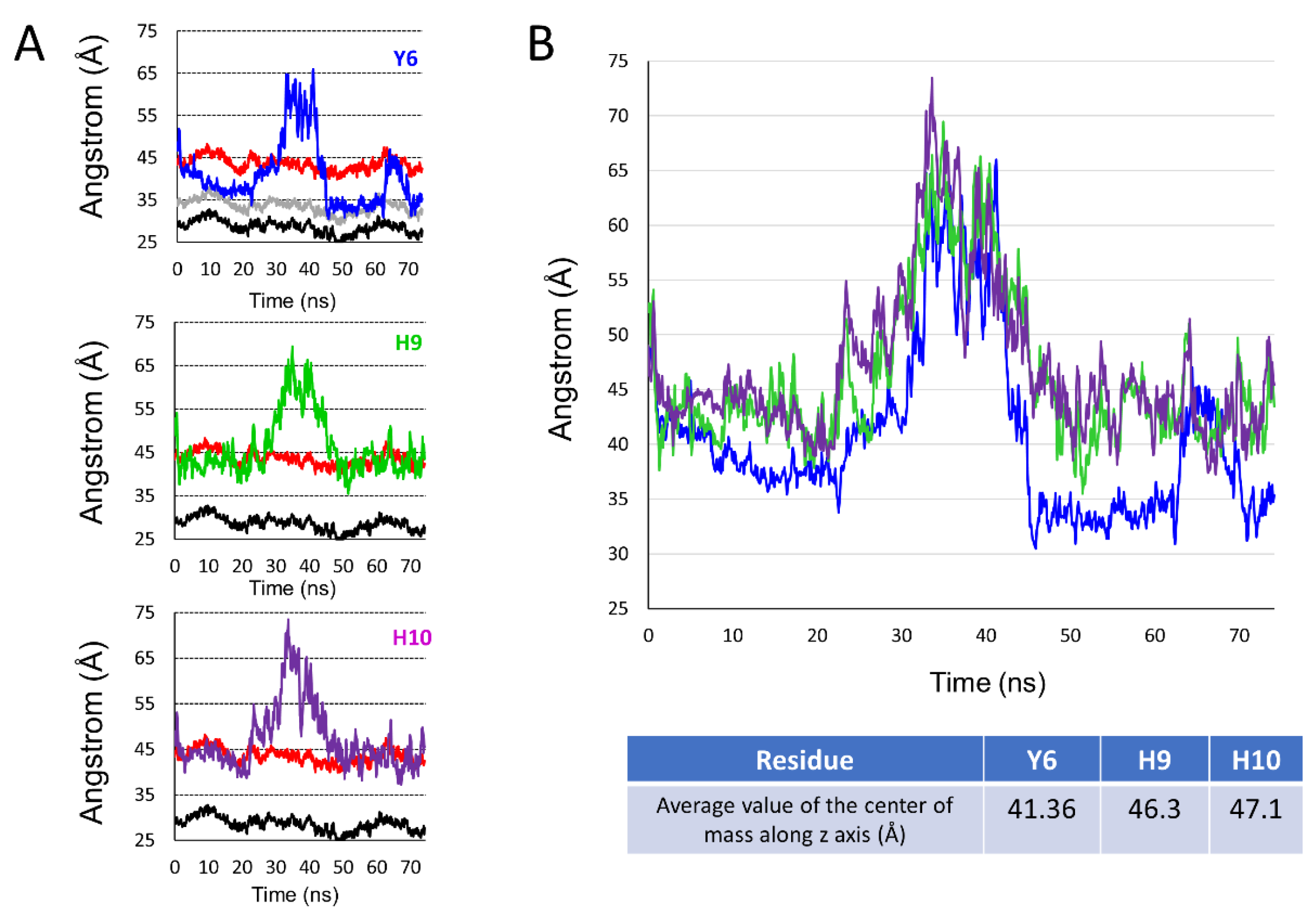
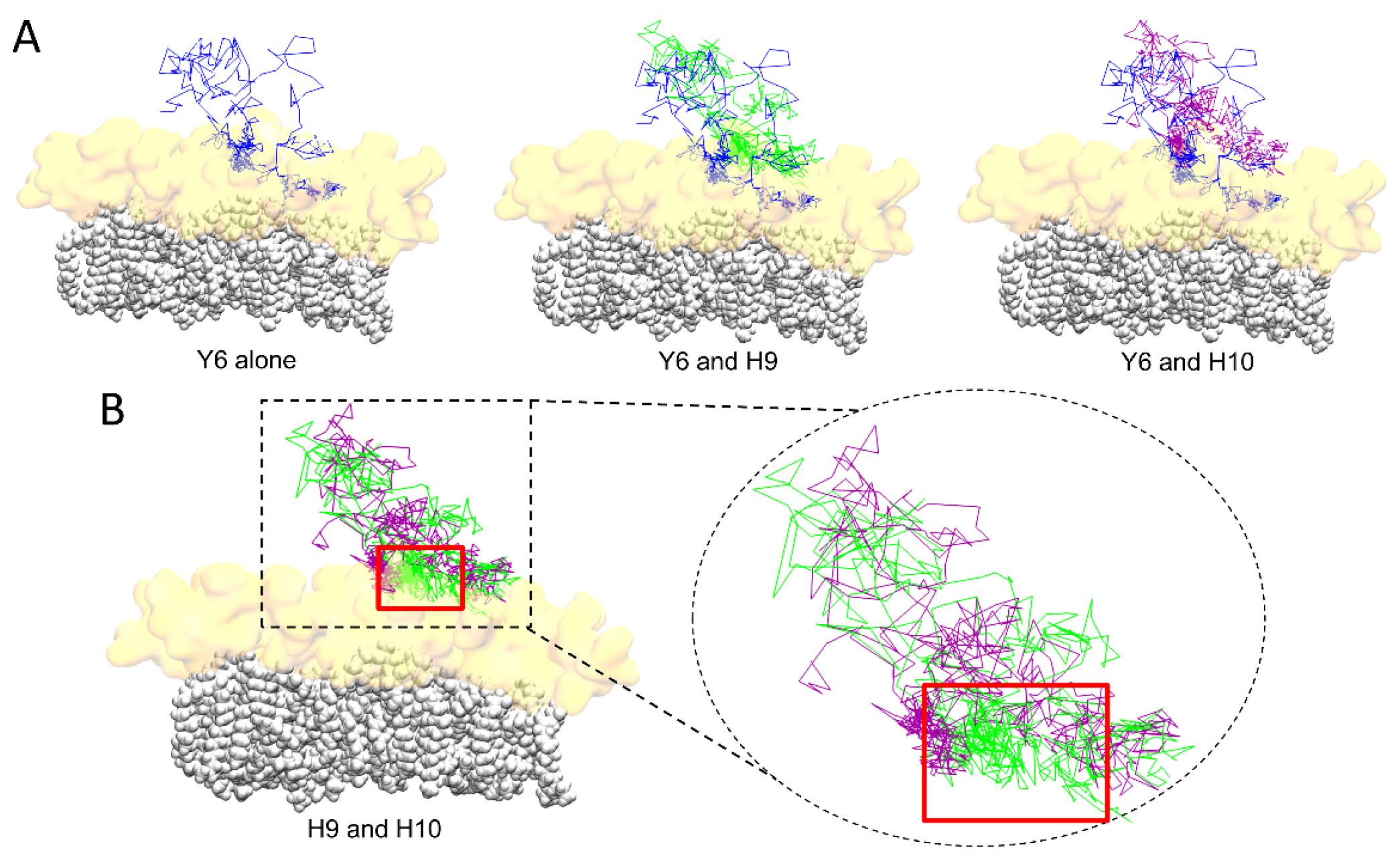
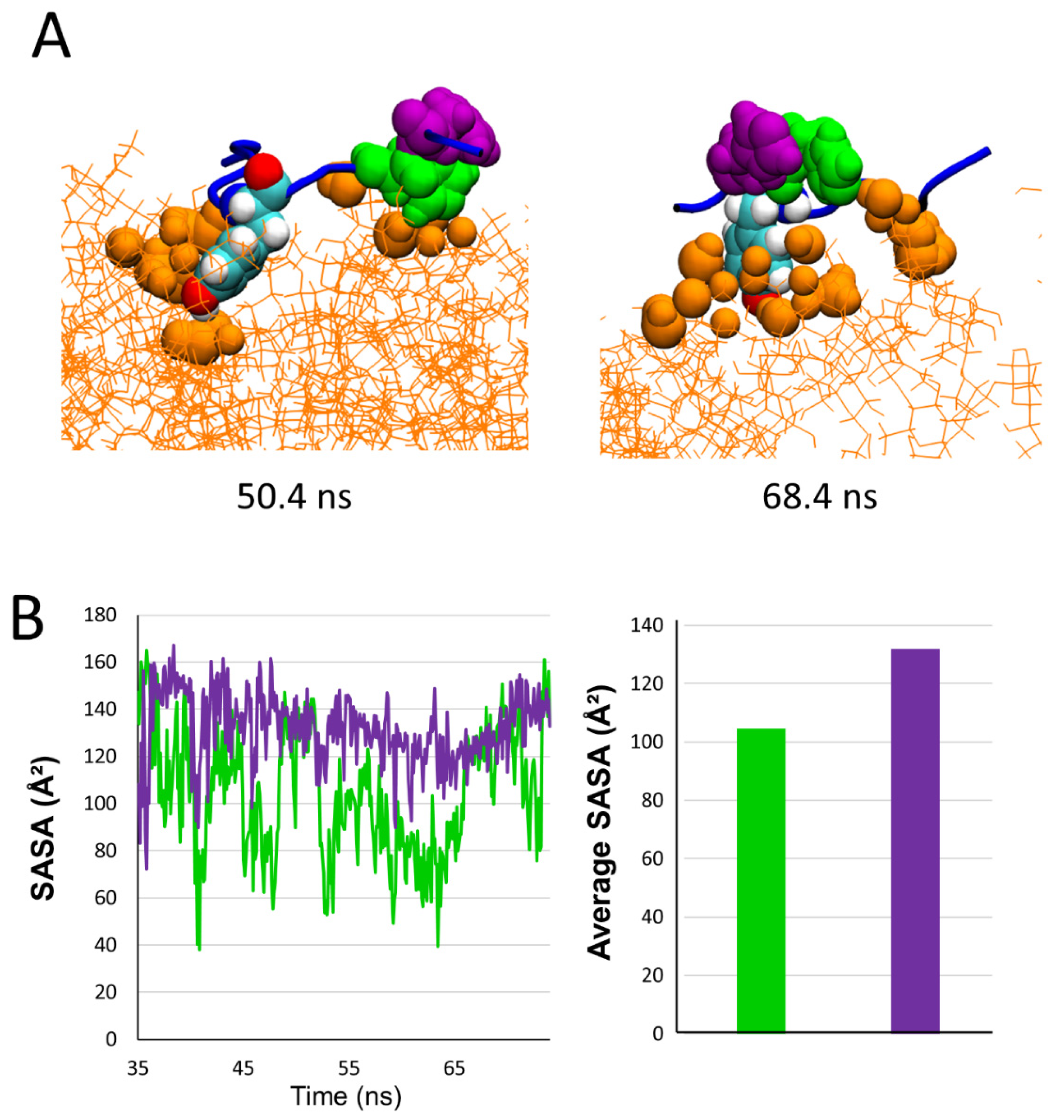
2.5. Comparison of the Burial of Aromatic Residues of AmyP53 in the Lipid Raft Surface
2.6. Molecular Details of Raft-AmyP53 Interactions at the Early Stage
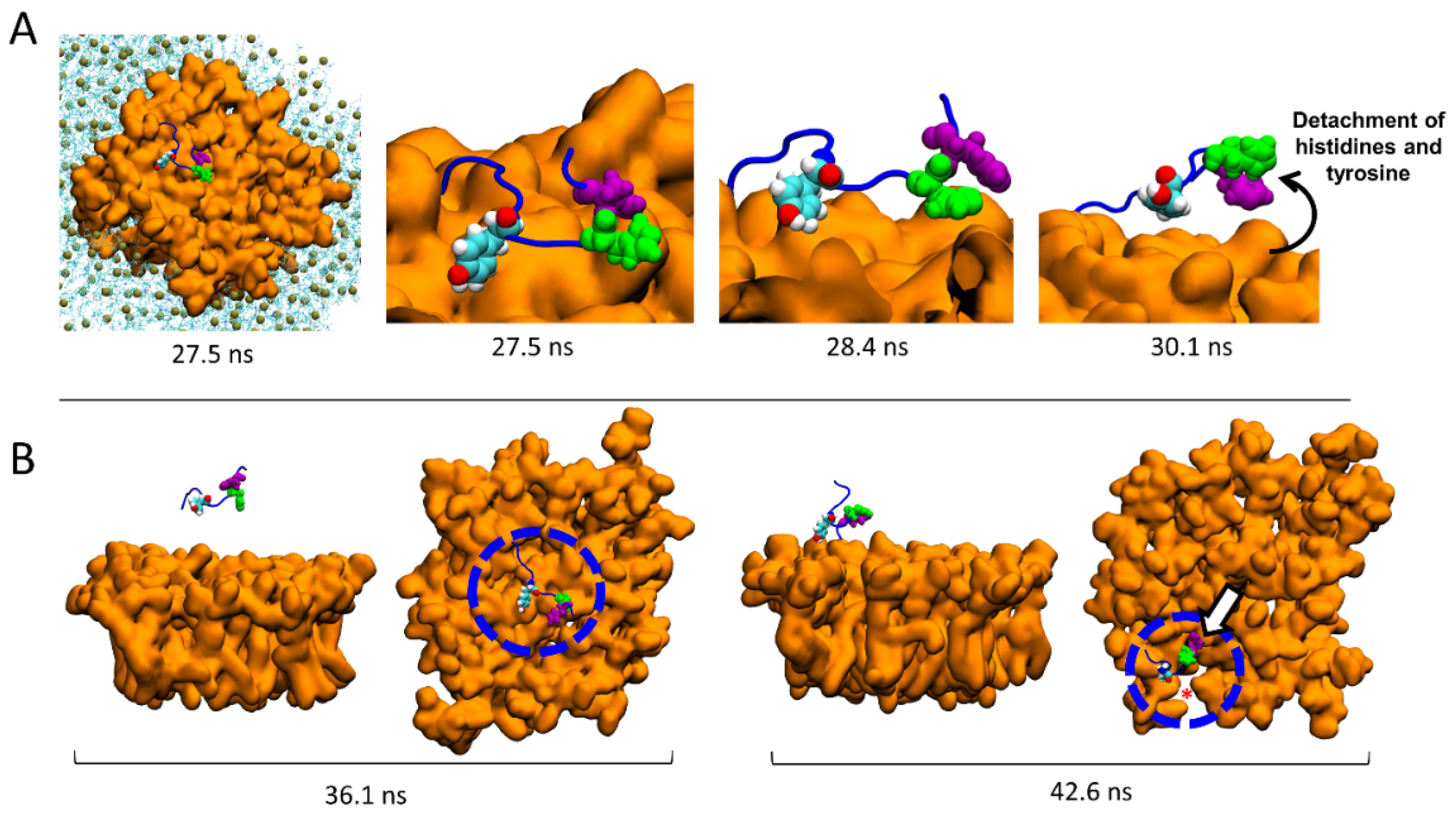
2.7. Molecular Details of the Detachment and Migration of AmyP53 towards the Periphery of the GM1 Lipid Raft
2.8. Conformational Analysis of AmyP53 at the Periphery of the Lipid Raft
2.9. Competition between Aβ1–42 and AmyP53 for GM1 Binding
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Monolayers Studies
4.3. Cell Culture
4.4. Aβ1–42 Binding to SH-SY5Y Cells
4.5. Ca2+ Flux Experiments
4.6. Statistical Analysis
4.7. Molecular Modeling
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nandi, A.; Counts, N.; Chen, S.; Seligman, B.; Tortorice, D.; Vigo, D.; Bloom, D.E. Global and regional projections of the economic burden of Alzheimer’s disease and related dementias from 2019 to 2050: A value of statistical life approach. EClinicalMedicine 2022, 51, 101580. [Google Scholar] [CrossRef] [PubMed]
- Rajan, K.B.; Weuve, J.; Barnes, L.L.; McAninch, E.A.; Wilson, R.S.; Evans, D.A. Population estimate of people with clinical Alzheimer’s disease and mild cognitive impairment in the United States (2020–2060). Alzheimer’s Dement. 2021, 17, 1966–1975. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Nahed, P.; Kambar, M.E.Z.N.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s disease drug development pipeline: 2022. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2022, 8, e12295. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Yahi, N. Brain Lipids in Synaptic Function and Neurological Disease: Clues to Innovative Therapeutic Strategies for Brain Disorders; Academic Press: San Diego, CA, USA, 2015. [Google Scholar]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [Green Version]
- Santoro, A.; Grimaldi, M.; Buonocore, M.; Stillitano, I.; D’Ursi, A.M. Exploring the early stages of the amyloid Aβ (1–42) peptide aggregation process: An NMR study. Pharmaceuticals 2021, 14, 732. [Google Scholar] [CrossRef]
- Burdick, D.; Soreghan, B.; Kwon, M.; Kosmoski, J.; Knauer, M.; Henschen, A.; Yates, J.; Cotman, C.; Glabe, C. Assembly and aggregation properties of synthetic Alzheimer’s A4/beta amyloid peptide analogs. J. Biol. Chem. 1992, 267, 546–554. [Google Scholar] [CrossRef]
- Jeremic, D.; Jiménez-Díaz, L.; Navarro-López, J.D. Past, present and future of therapeutic strategies against amyloid-β peptides in Alzheimer’s disease: A systematic review. Ageing Res. Rev. 2021, 72, 101496. [Google Scholar] [CrossRef]
- Valiukas, Z.; Ephraim, R.; Tangalakis, K.; Davidson, M.; Apostolopoulos, V.; Feehan, J. Immunotherapies for Alzheimer’s Disease—A Review. Vaccines 2022, 10, 1527. [Google Scholar] [CrossRef]
- Makin, S. The amyloid hypothesis on trial. Nature 2018, 559, S4–S7. [Google Scholar] [CrossRef] [Green Version]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimer’s Res. Ther. 2021, 13, 1–14. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S. Lecanemab in early Alzheimer’s disease. N. Engl. J. Med. 2022, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Chahinian, H.; Yahi, N. Progress toward Alzheimer’s disease treatment: Leveraging the Achilles’ heel of Aβ oligomers? Protein Sci. A Publ. Protein Soc. 2020, 29, 1748–1759. [Google Scholar] [CrossRef] [PubMed]
- Arispe, N.; Rojas, E.; Pollard, H.B. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: Blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. USA 1993, 90, 567–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quist, A.; Doudevski, I.; Lin, H.; Azimova, R.; Ng, D.; Frangione, B.; Kagan, B.; Ghiso, J.; Lal, R. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA 2005, 102, 10427–10432. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.I.; Yi, J.S.; Ko, Y.G. Amyloid β oligomerization is induced by brain lipid rafts. J. Cell. Biochem. 2006, 99, 878–889. [Google Scholar] [CrossRef]
- Yu, X.; Wang, Q.; Pan, Q.; Zhou, F.; Zheng, J. Molecular interactions of Alzheimer amyloid-β oligomers with neutral and negatively charged lipid bilayers. Phys. Chem. Chem. Phys. 2013, 15, 8878–8889. [Google Scholar] [CrossRef] [Green Version]
- Meleleo, D.; Galliani, A.; Notarachille, G. AβP1-42 incorporation and channel formation in planar lipid membranes: The role of cholesterol and its oxidation products. J. Bioenerg. Biomembr. 2013, 45, 369–381. [Google Scholar] [CrossRef]
- Ashley, R.H.; Harroun, T.A.; Hauss, T.; Breen, K.C.; Bradshaw, J.P. Autoinsertion of soluble oligomers of Alzheimer’s Aβ (1–42) peptide into cholesterol-containing membranes is accompanied by relocation of the sterol towards the bilayer surface. BMC Struct. Biol. 2006, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kandel, N.; Matos, J.O.; Tatulian, S.A. Structure of amyloid β25–35 in lipid environment and cholesterol-dependent membrane pore formation. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Venko, K.; Novič, M.; Stoka, V.; Žerovnik, E. Prediction of Transmembrane Regions, Cholesterol, and Ganglioside Binding Sites in Amyloid-Forming Proteins Indicate Potential for Amyloid Pore Formation. Front. Mol. Neurosci. 2021, 14, 619496. [Google Scholar] [CrossRef]
- Morgado, I.; Garvey, M. Lipids in amyloid-β processing, aggregation, and toxicity. Lipids Protein Misfolding 2015, 67–94. [Google Scholar]
- Tempra, C.; Scollo, F.; Pannuzzo, M.; Lolicato, F.; La Rosa, C. A unifying framework for amyloid-mediated membrane damage: The lipid-chaperone hypothesis. Biochim. Et Biophys. Acta (BBA) Proteins Proteom. 2022, 1870, 140767. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, Y.H.; Arce, F.T.; Gillman, A.L.; Jang, H.; Kagan, B.L.; Nussinov, R.; Yang, J.; Lal, R. Amyloid β ion channels in a membrane comprising brain total lipid extracts. ACS Chem. Neurosci. 2017, 8, 1348–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Scala, C.; Yahi, N.; Boutemeur, S.; Flores, A.; Rodriguez, L.; Chahinian, H.; Fantini, J. Common molecular mechanism of amyloid pore formation by Alzheimer’s β-amyloid peptide and α-synuclein. Sci. Rep. 2016, 6, 28781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Ostaszewski, B.L.; Yang, T.; O’Malley, T.T.; Jin, M.; Yanagisawa, K.; Li, S.; Bartels, T.; Selkoe, D.J. Soluble Aβ oligomers are rapidly sequestered from brain ISF in vivo and bind GM1 ganglioside on cellular membranes. Neuron 2014, 82, 308–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Scala, C.; Yahi, N.; Flores, A.; Boutemeur, S.; Kourdougli, N.; Chahinian, H.; Fantini, J. Broad neutralization of calcium-permeable amyloid pore channels with a chimeric Alzheimer/Parkinson peptide targeting brain gangliosides. Biochim. Et Biophys. Acta 2016, 1862, 213–222. [Google Scholar] [CrossRef]
- Fantini, J.; Yahi, N. Molecular insights into amyloid regulation by membrane cholesterol and sphingolipids: Common mechanisms in neurodegenerative diseases. Expert Rev. Mol. Med. 2010, 12, e27. [Google Scholar] [CrossRef] [Green Version]
- Fantini, J.; Yahi, N. Molecular basis for the glycosphingolipid-binding specificity of α-synuclein: Key role of tyrosine 39 in membrane insertion. J. Mol. Biol. 2011, 408, 654–669. [Google Scholar] [CrossRef]
- Yahi, N.; Fantini, J. Deciphering the glycolipid code of Alzheimer’s and Parkinson’s amyloid proteins allowed the creation of a universal ganglioside-binding peptide. PLoS ONE 2014, 9, e104751. [Google Scholar] [CrossRef] [Green Version]
- Di Scala, C.; Armstrong, N.; Chahinian, H.; Chabrière, E.; Fantini, J.; Yahi, N. AmyP53, a Therapeutic Peptide Candidate for the Treatment of Alzheimer’s and Parkinson’s Disease: Safety, Stability, Pharmacokinetics Parameters and Nose-to Brain Delivery. Int. J. Mol. Sci. 2022, 23, 13383. [Google Scholar] [CrossRef]
- Fantini, J.; Yahi, N.; Garmy, N. Cholesterol accelerates the binding of Alzheimer’s β-amyloid peptide to ganglioside GM1 through a universal hydrogen-bond-dependent sterol tuning of glycolipid conformation. Front. Physiol. 2013, 4, 120. [Google Scholar] [CrossRef] [PubMed]
- Cameron, M.; Kékesi, O.; Morley, J.W.; Tapson, J.; Breen, P.P.; Van Schaik, A.; Buskila, Y. Calcium imaging of AM dyes following prolonged incubation in acute neuronal tissue. PLoS ONE 2016, 11, e0155468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yahi, N.; Di Scala, C.; Chahinian, H.; Fantini, J. Innovative treatment targeting gangliosides aimed at blocking the formation of neurotoxic α-synuclein oligomers in Parkinson’s disease. Glycoconj. J. 2022, 39, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury, P.T. Amyloid pores from pathogenic mutations. Nature 2002, 418, 291. [Google Scholar] [CrossRef] [Green Version]
- Di Scala, C.; Chahinian, H.; Yahi, N.; Garmy, N.; Fantini, J. Interaction of Alzheimer’s β-amyloid peptides with cholesterol: Mechanistic insights into amyloid pore formation. Biochemistry 2014, 53, 4489–4502. [Google Scholar] [CrossRef]
- Di Scala, C.; Troadec, J.D.; Lelièvre, C.; Garmy, N.; Fantini, J.; Chahinian, H. Mechanism of cholesterol-assisted oligomeric channel formation by a short Alzheimer β-amyloid peptide. J. Neurochem. 2014, 128, 186–195. [Google Scholar] [CrossRef]
- Yu, X.; Zheng, J. Cholesterol promotes the interaction of Alzheimer β-amyloid monomer with lipid bilayer. J. Mol. Biol. 2012, 421, 561–571. [Google Scholar] [CrossRef]
- Liguori, N.; Nerenberg, P.S.; Head-Gordon, T. Embedding Aβ42 in Heterogeneous Membranes Depends on Cholesterol Asymmetries. Biophys. J. 2013, 105, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. A protein-chameleon: Conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J. Biomol. Struct. Dyn. 2003, 21, 211–234. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically disordered proteins and their environment: Effects of strong denaturants, temperature, pH, counter ions, membranes, binding partners, osmolytes, and macromolecular crowding. Protein J. 2009, 28, 305–325. [Google Scholar] [CrossRef]
- Uversky, V.N. The mysterious unfoldome: Structureless, underappreciated, yet vital part of any given proteome. J. Biomed. Biotechnol. 2010, 2010, 568068. [Google Scholar] [CrossRef]
- Uversky, V.N. Dancing Protein Clouds: The Strange Biology and Chaotic Physics of Intrinsically Disordered Proteins. J. Biol. Chem. 2016, 291, 6681–6688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, S.-H. Cyclic peptides as therapeutic agents and biochemical tools. Biomol. Ther. 2012, 20, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Y.S.; Lane, D.P.; Verma, C.S. Stapled peptide design: Principles and roles of computation. Drug Discov. Today 2016, 21, 1642–1653. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, K. GM1 ganglioside and Alzheimer’s disease. Glycoconj. J. 2015, 32, 87–91. [Google Scholar] [CrossRef]
- Steponkus, P.L.; Lynch, D.V. Freeze/thaw-induced destabilization of the plasma membrane and the effects of cold acclimation. J. Bioenerg. Biomembr. 1989, 21, 21–41. [Google Scholar] [CrossRef]
- Zhang, S.; Zhu, N.; Gu, J.; Li, H.-F.; Qiu, Y.; Liao, D.-F.; Qin, L. Crosstalk between lipid rafts and aging: New frontiers for delaying aging. Aging Dis. 2022, 13, 1042. [Google Scholar] [CrossRef]
- Colin, J.; Gregory-Pauron, L.; Lanhers, M.-C.; Claudepierre, T.; Corbier, C.; Yen, F.T.; Malaplate-Armand, C.; Oster, T. Membrane raft domains and remodeling in aging brain. Biochimie 2016, 130, 178–187. [Google Scholar] [CrossRef]
- Prinetti, A.; Chigorno, V.; Prioni, S.; Loberto, N.; Marano, N.; Tettamanti, G.; Sonnino, S. Changes in the lipid turnover, composition, and organization, as sphingolipid-enriched membrane domains, in rat cerebellar granule cells developing in vitro. J. Biol. Chem. 2001, 276, 21136–21145. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Chen, Y.; Wang, H.; Bai, Y.; Zhao, J.; Zhang, X.; Liang, L.; Chen, Y.; Ye, C.; Li, Y. Integrated lipidomics and transcriptomics characterization upon aging-related changes of lipid species and pathways in human bone marrow mesenchymal stem cells. J. Proteome Res. 2019, 18, 2065–2077. [Google Scholar] [CrossRef]
- Lesuisse, C.; Martin, L.J. Long-term culture of mouse cortical neurons as a model for neuronal development, aging, and death. J. Neurobiol. 2002, 51, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Cheng, S.; Huang, F.; Fan, W.; Chen, Y.; Shi, H.; He, H. Mitochondrial dysfunction in long-term neuronal cultures mimics changes with aging. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2011, 17, BR91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzaz, F.; Yahi, N.; Di Scala, C.; Chahinian, H.; Fantini, J. Ganglioside binding domains in proteins: Physiological and pathological mechanisms. Adv. Protein Chem. Struct. Biol. 2022, 128, 289–324. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Yahi, N. The driving force of alpha-synuclein insertion and amyloid channel formation in the plasma membrane of neural cells: Key role of ganglioside- and cholesterol-binding domains. Adv. Exp. Med. Biol. 2013, 991, 15–26. [Google Scholar] [CrossRef] [PubMed]
- El-Battari, A.; Rodriguez, L.; Chahinian, H.; Delézay, O.; Fantini, J.; Yahi, N.; Di Scala, C. Gene Therapy Strategy for Alzheimer’s and Parkinson’s Diseases Aimed at Preventing the Formation of Neurotoxic Oligomers in SH-SY5Y Cells. Int. J. Mol. Sci. 2021, 22, 11550. [Google Scholar] [CrossRef]
- Di Scala, C.; Fantini, J. Hybrid In Silico/In Vitro Approaches for the Identification of Functional Cholesterol-Binding Domains in Membrane Proteins. Methods Mol. Biol. 2017, 1583, 7–19. [Google Scholar] [CrossRef]
- Nelson, T.J.; Alkon, D.L. Protection against β-amyloid-induced apoptosis by peptides interacting with β-amyloid. J. Biol. Chem. 2007, 282, 31238–31249. [Google Scholar] [CrossRef] [Green Version]
- Froimowitz, M. HyperChem: A software package for computational chemistry and molecular modeling. BioTechniques 1993, 14, 1010–1013. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Lee, J.; Patel, D.S.; Ståhle, J.; Park, S.J.; Kern, N.R.; Kim, S.; Lee, J.; Cheng, X.; Valvano, M.A.; Holst, O.; et al. CHARMM-GUI Membrane Builder for Complex Biological Membrane Simulations with Glycolipids and Lipoglycans. J. Chem. Theory Comput. 2019, 15, 775–786. [Google Scholar] [CrossRef] [Green Version]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Davila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azzaz, F.; Chahinian, H.; Yahi, N.; Fantini, J.; Di Scala, C. AmyP53 Prevents the Formation of Neurotoxic β-Amyloid Oligomers through an Unprecedent Mechanism of Interaction with Gangliosides: Insights for Alzheimer’s Disease Therapy. Int. J. Mol. Sci. 2023, 24, 1760. https://doi.org/10.3390/ijms24021760
Azzaz F, Chahinian H, Yahi N, Fantini J, Di Scala C. AmyP53 Prevents the Formation of Neurotoxic β-Amyloid Oligomers through an Unprecedent Mechanism of Interaction with Gangliosides: Insights for Alzheimer’s Disease Therapy. International Journal of Molecular Sciences. 2023; 24(2):1760. https://doi.org/10.3390/ijms24021760
Chicago/Turabian StyleAzzaz, Fodil, Henri Chahinian, Nouara Yahi, Jacques Fantini, and Coralie Di Scala. 2023. "AmyP53 Prevents the Formation of Neurotoxic β-Amyloid Oligomers through an Unprecedent Mechanism of Interaction with Gangliosides: Insights for Alzheimer’s Disease Therapy" International Journal of Molecular Sciences 24, no. 2: 1760. https://doi.org/10.3390/ijms24021760
APA StyleAzzaz, F., Chahinian, H., Yahi, N., Fantini, J., & Di Scala, C. (2023). AmyP53 Prevents the Formation of Neurotoxic β-Amyloid Oligomers through an Unprecedent Mechanism of Interaction with Gangliosides: Insights for Alzheimer’s Disease Therapy. International Journal of Molecular Sciences, 24(2), 1760. https://doi.org/10.3390/ijms24021760