
GPR30 Alleviates Pressure Overload-Induced Myocardial Hypertrophy in Ovariectomized Mice by Regulating Autophagy

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
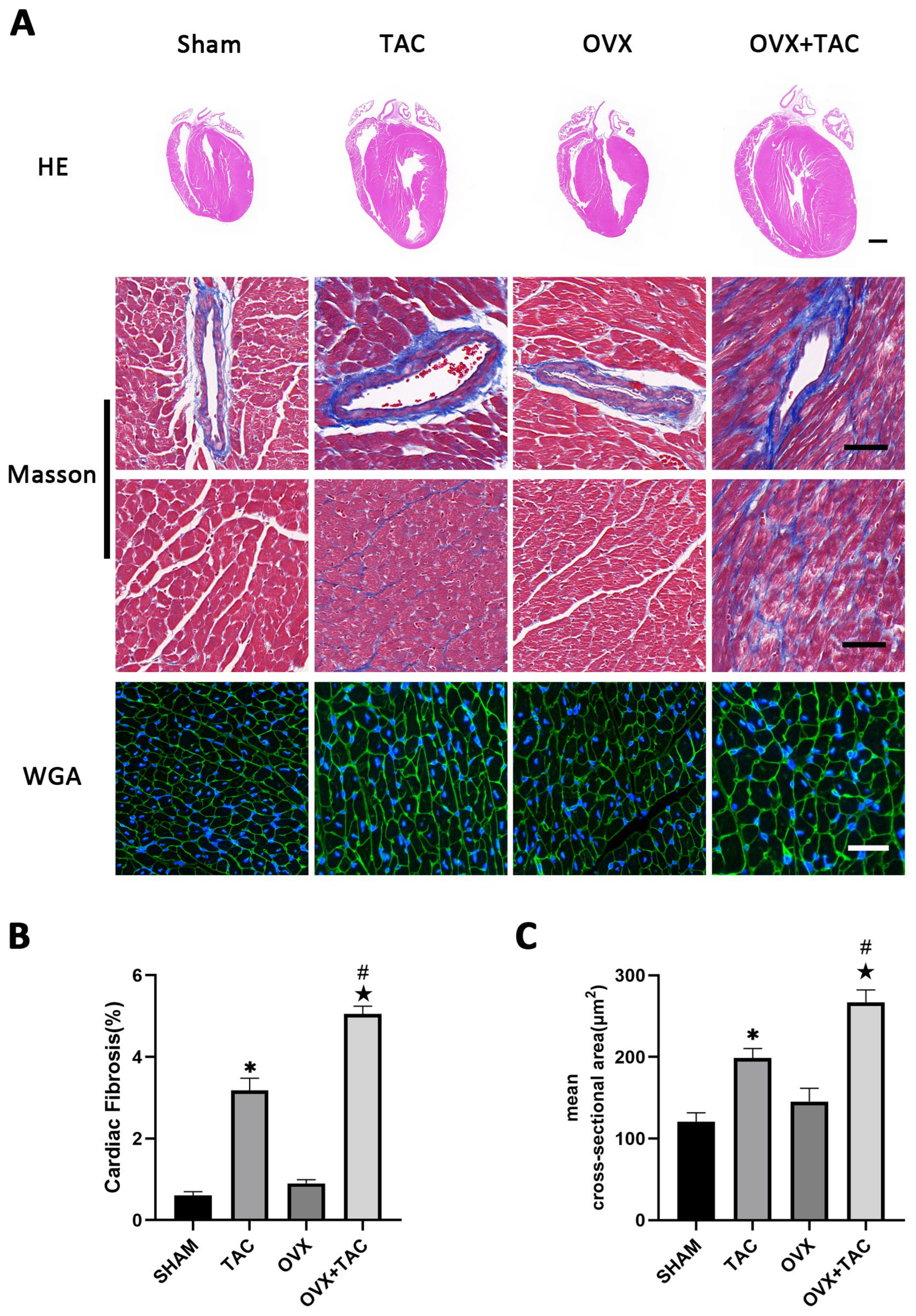
2.1. Effects of Pressure Overload on Cardiac Hypertrophy and Fibrosis in Sham and OVX Mice
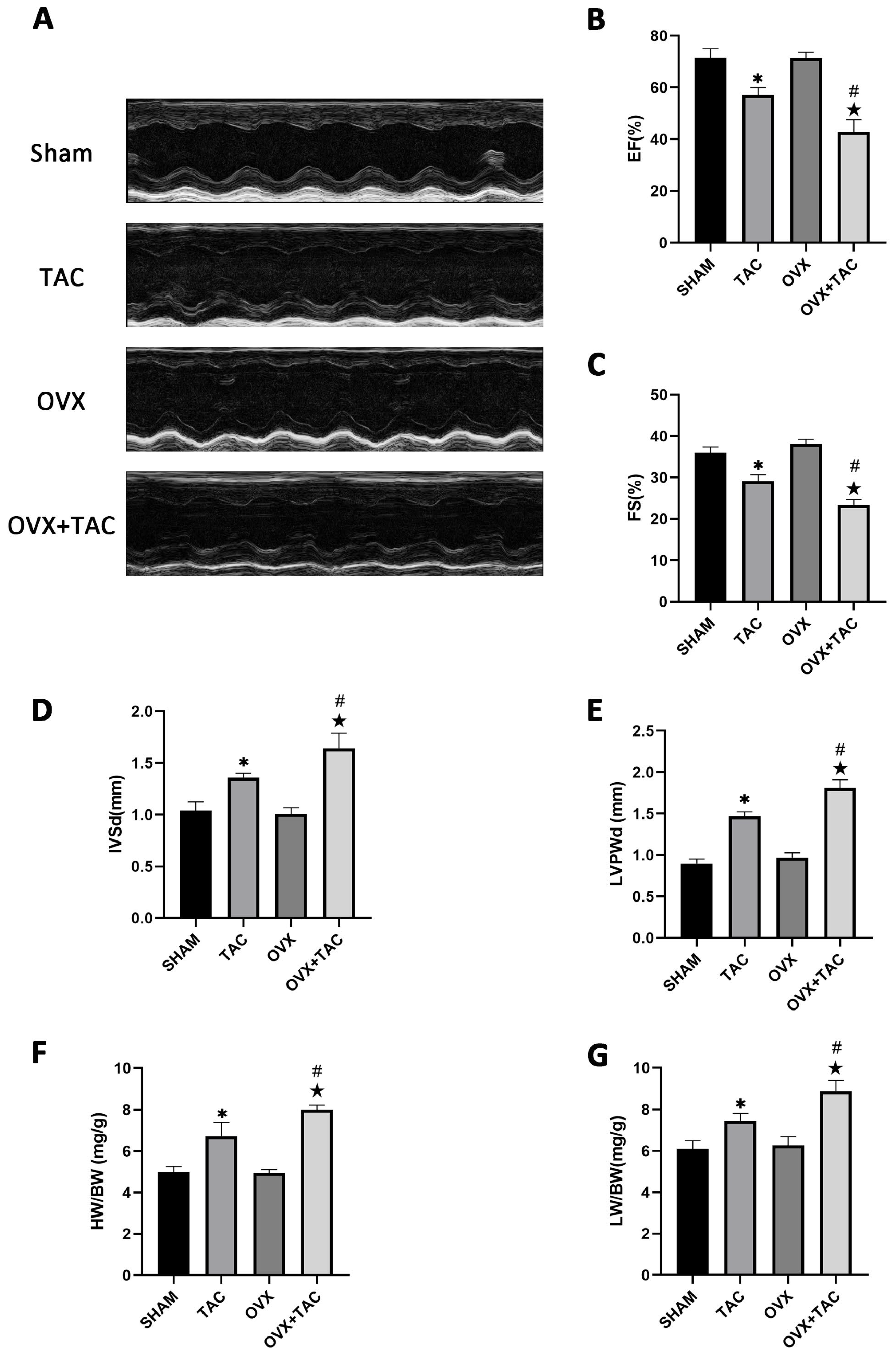
2.2. Effects of Pressure Overload on Cardiac Function, HW/BW and LW/BW after 8 Weeks of TAC
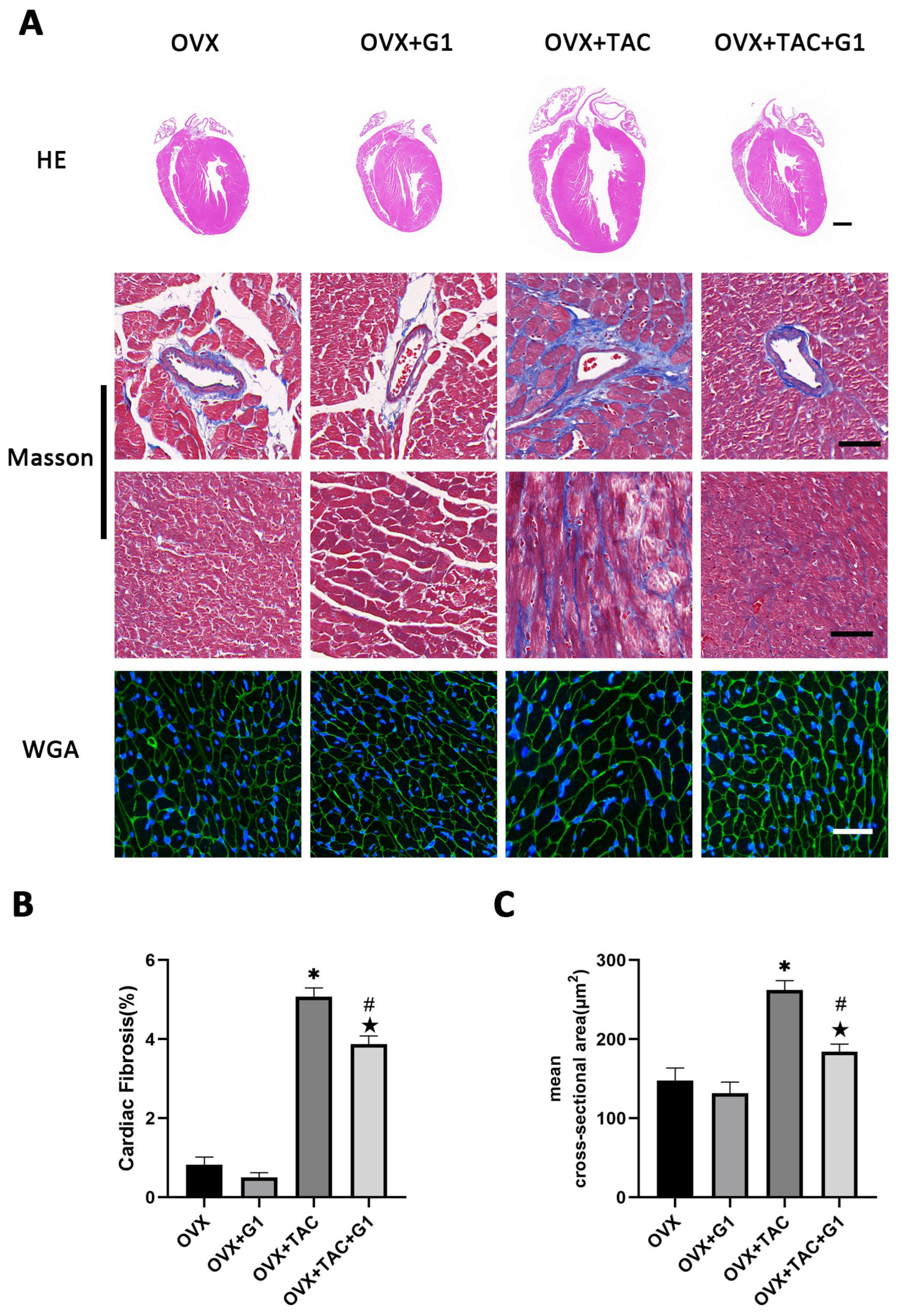
2.3. GPR30 Agonist G1 Alleviated Cardiac Hypertrophy and Fibrosis of Mice in the OVX + TAC Group
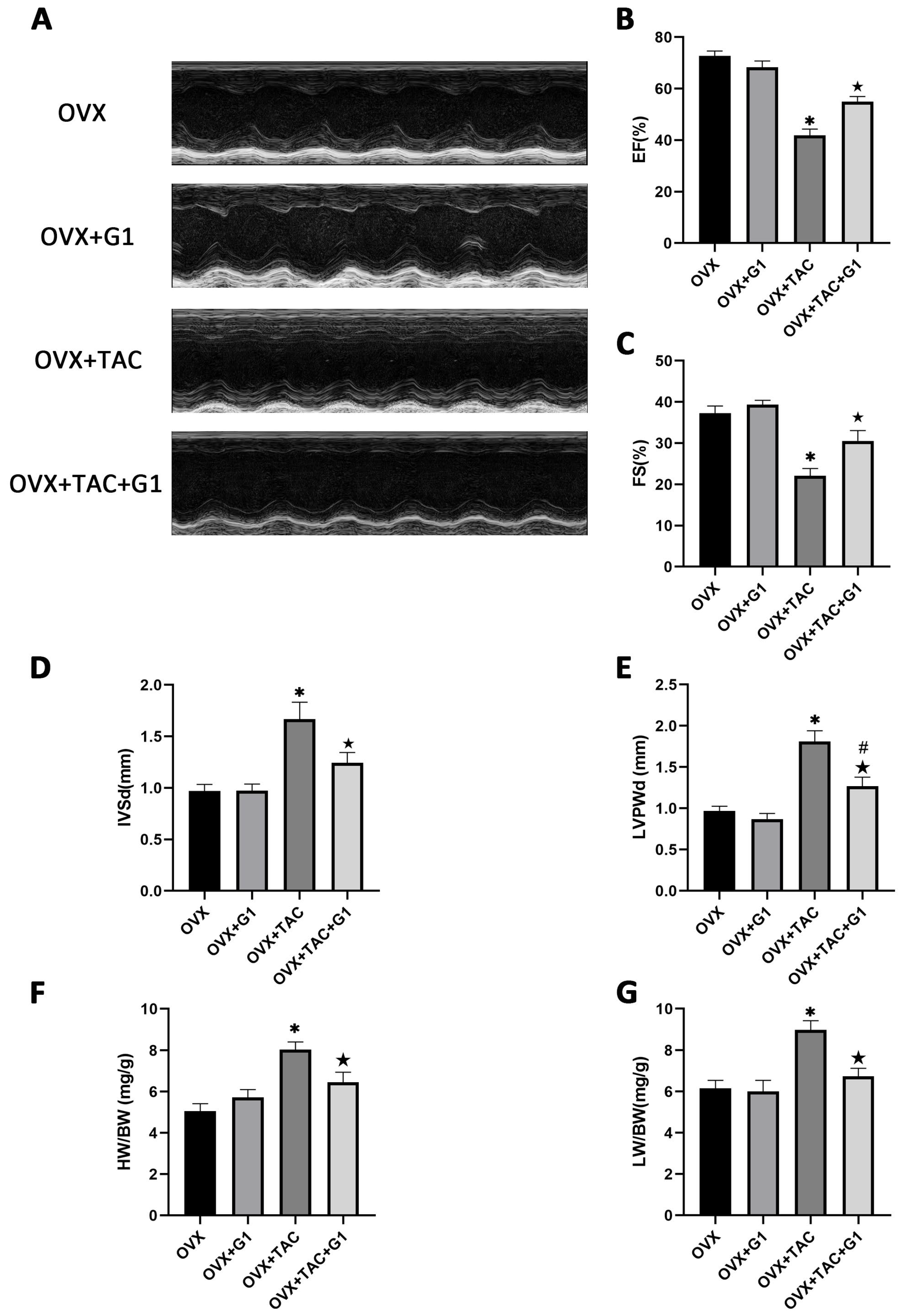
2.4. G1 Treatment Improved Cardiac Function of OVX + TAC Mice
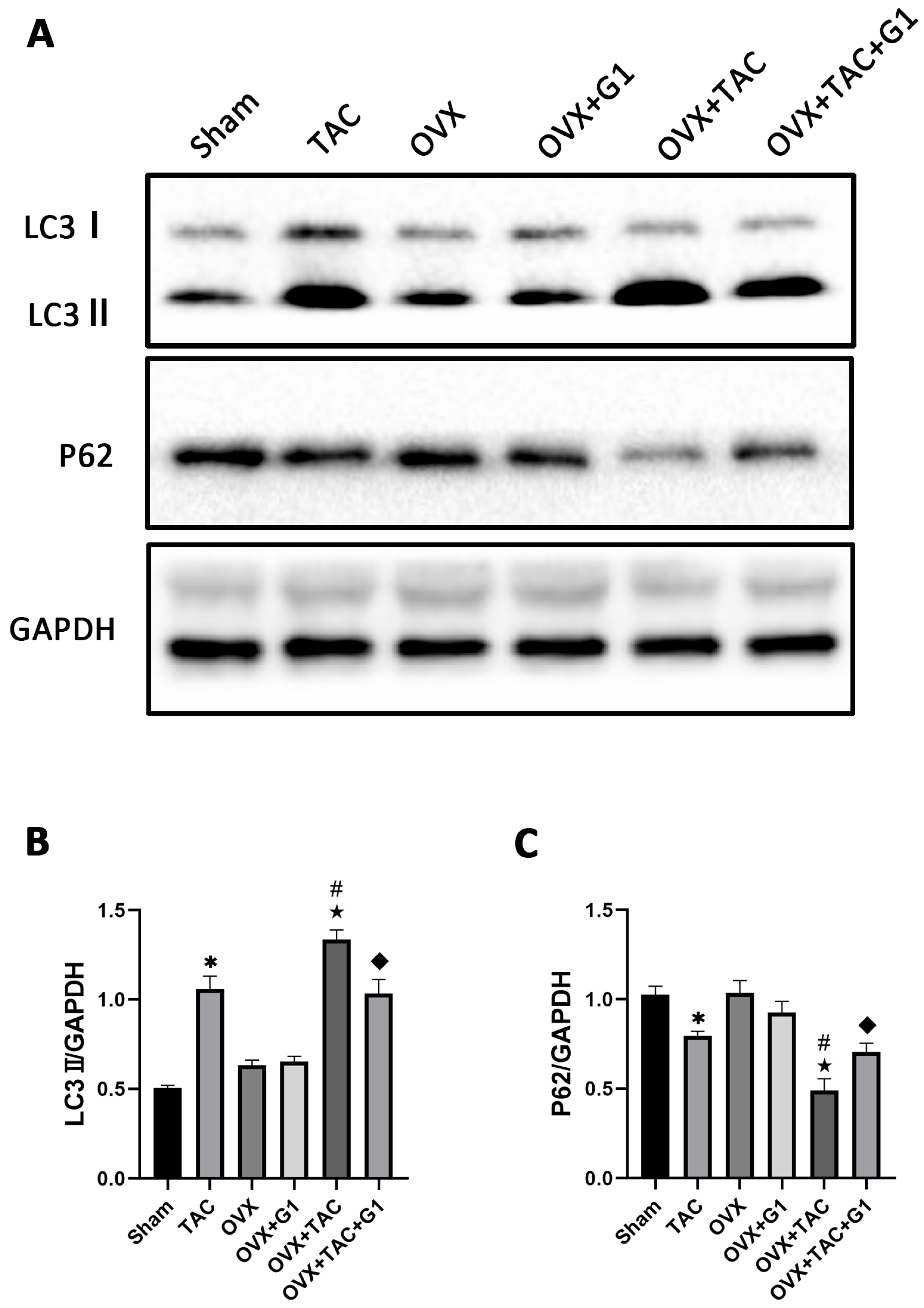
2.5. G1 Decreased the Autophagy Level of Myocardial Tissue in OVX + TAC Mice
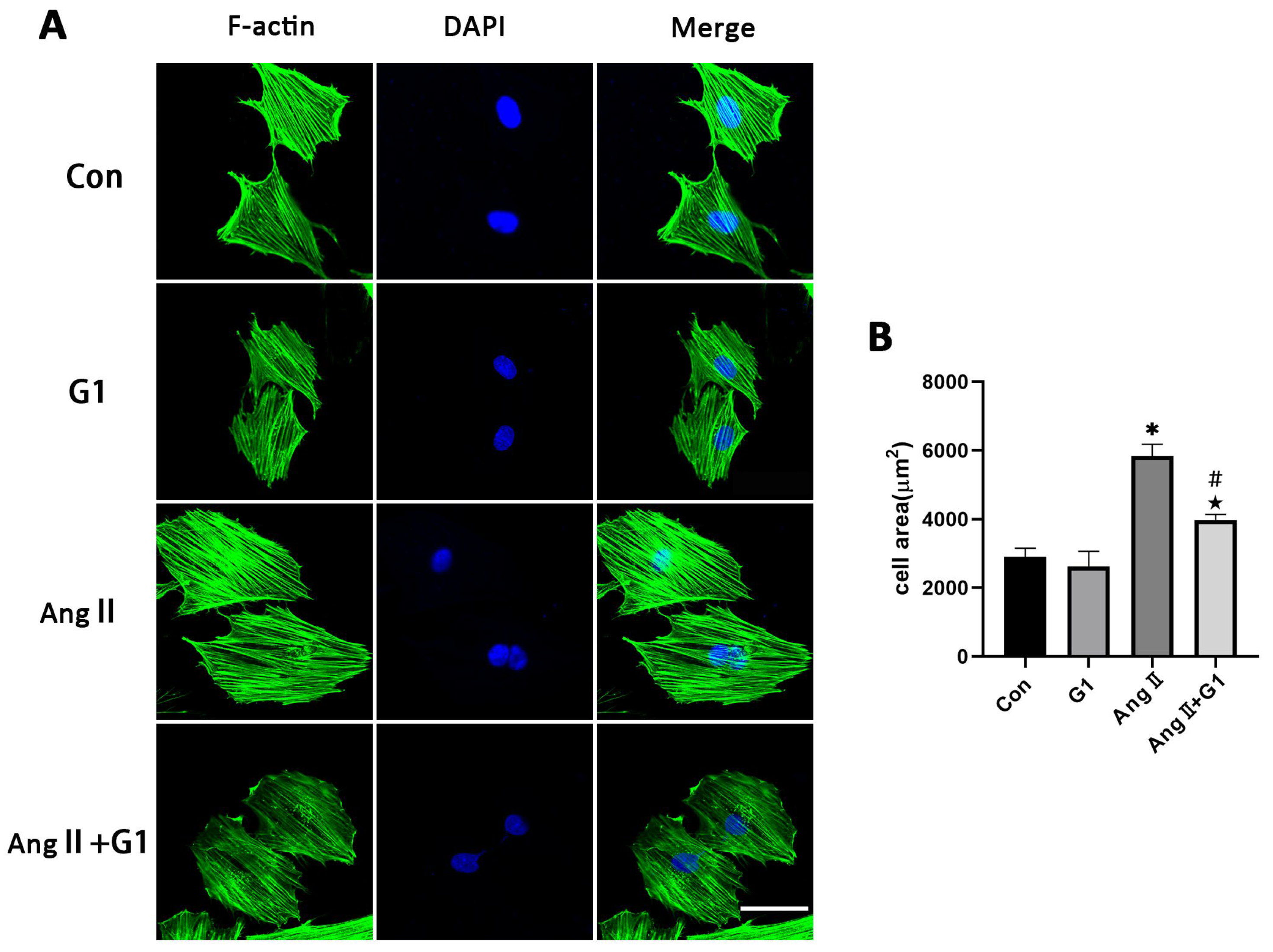
2.6. G1 Attenuated AngII-Induced Hypertrophy of H9c2 Cells In Vitro
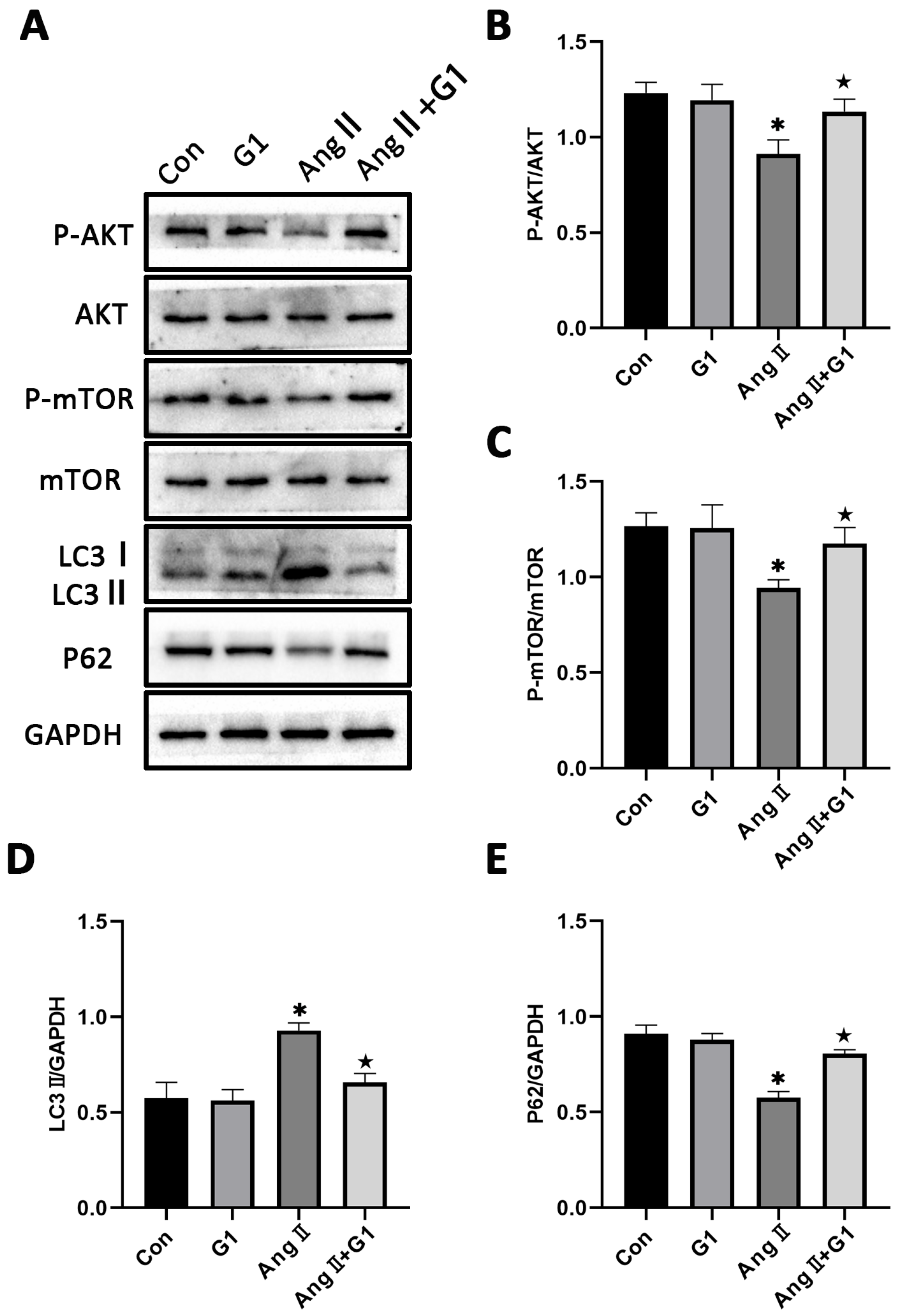
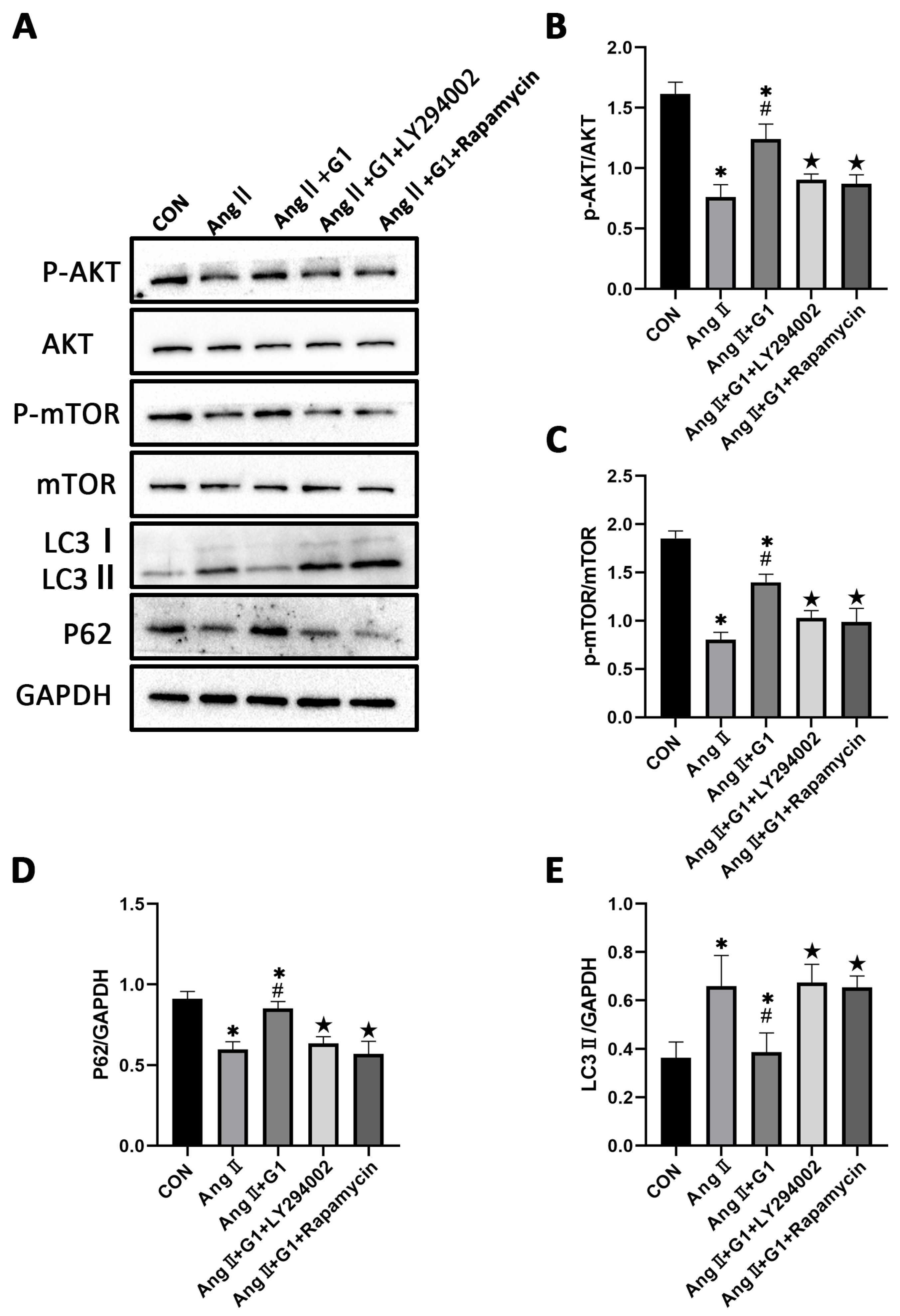
2.7. The Akt/mTOR Pathway and Autophagy Were Involved in G1 Alleviating AngII-Induced Hypertrophy of H9c2 Cells
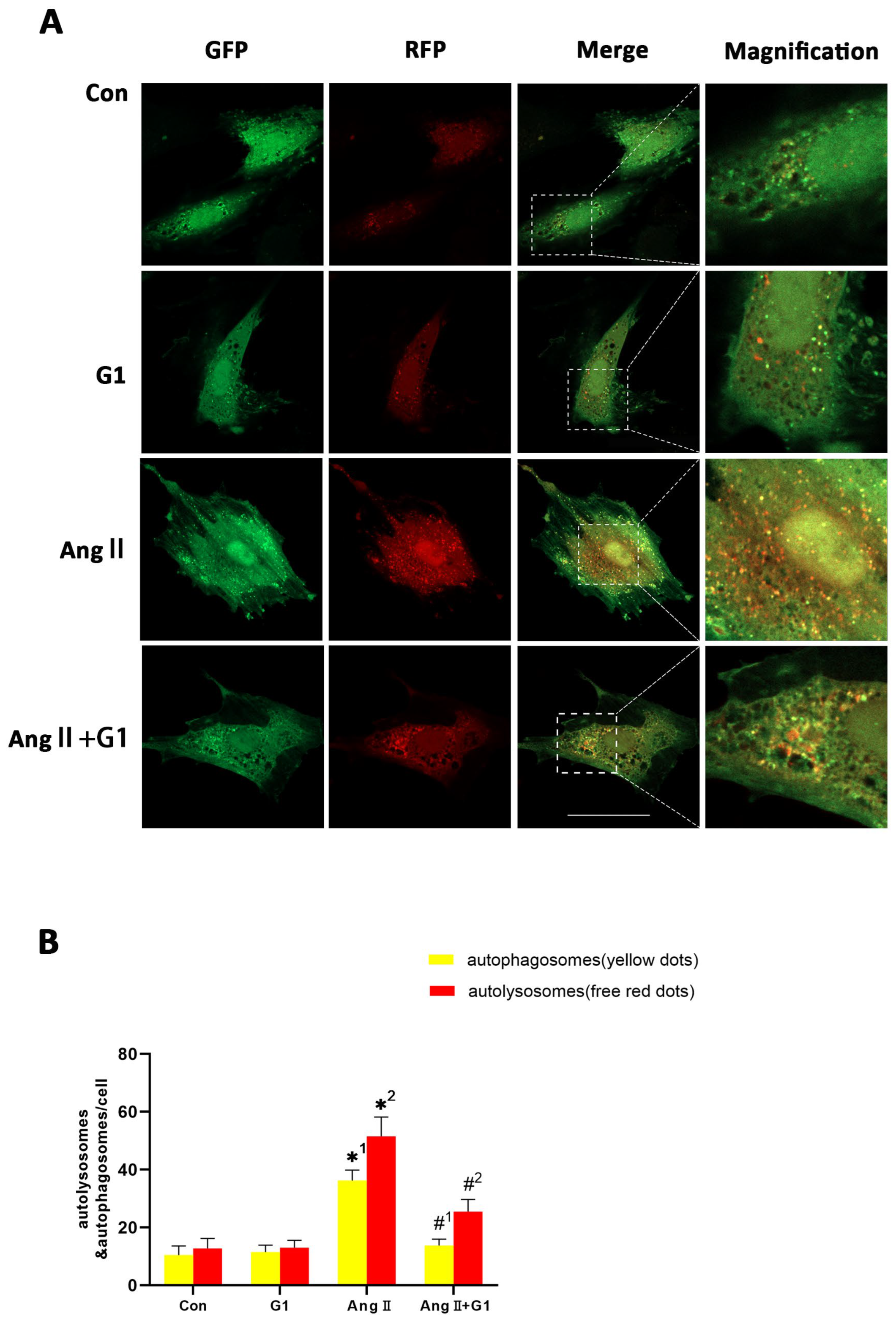
2.8. G1 Decreased the Autophagic Flux of H9c2 Cells Enhanced by AngII
2.9. LY294002 or Rapamycin Blocked the Effect of G1 Decreasing the Elevated Autophagy Induced by AngII in H9c2 Cells
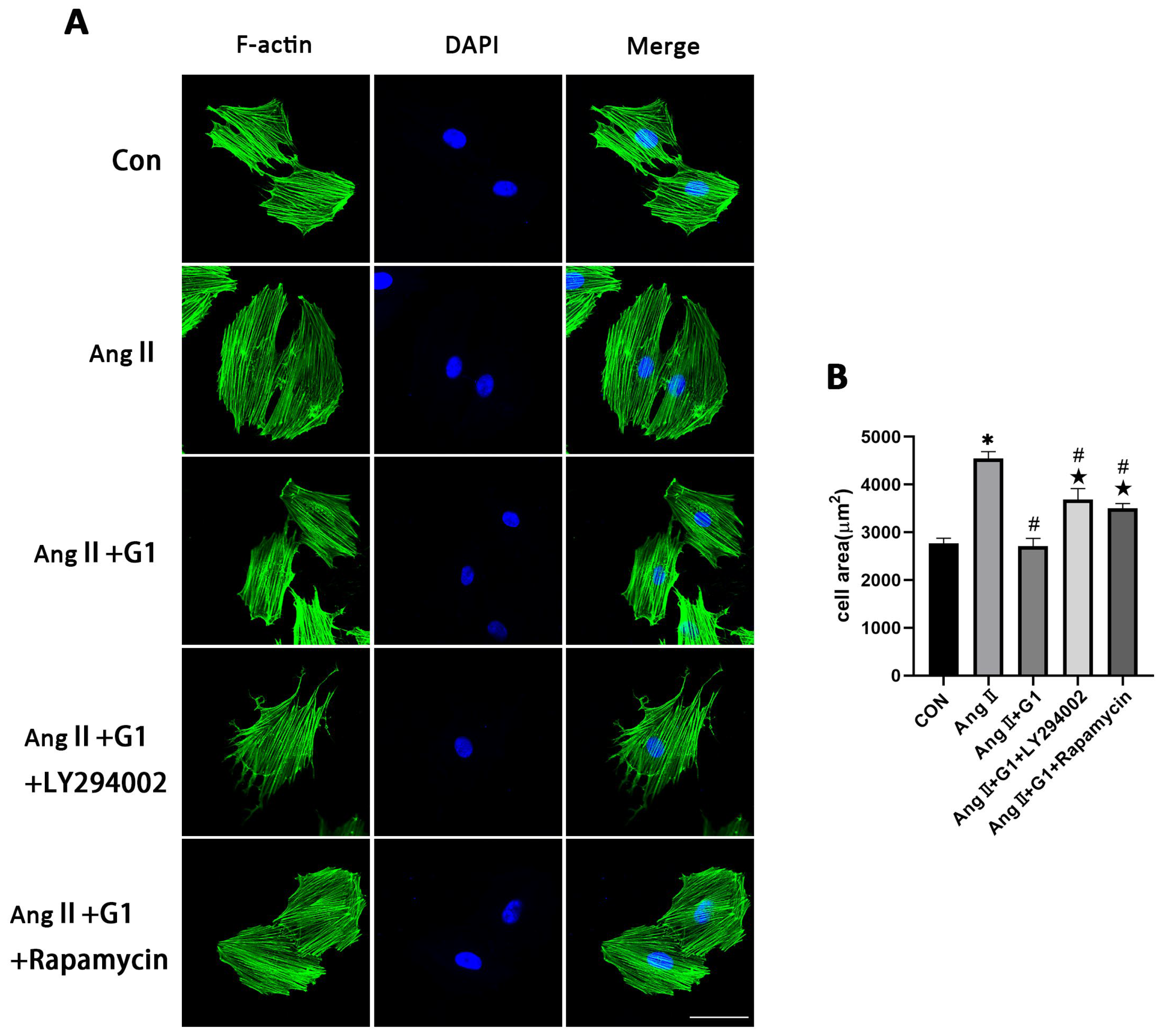
2.10. Effect of LY294002 or Rapamycin on Cardiac Hypertrophy of H9c2 Cells in AngII + G1 Group
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Experimental Animals
4.3. Ovariectomy and Transverse Aortic Constriction(TAC) Surgery
4.4. Experimental Design and Treatment
4.5. H9c2 Cells Culture and In Vitro Study Design
4.6. Echocardiographic Assessment of Cardiac Function
4.7. Histological Examination
4.8. Phalloidin Staining
4.9. Western Blotting
4.10. Tandem mRFP-GFP-LC3 Fluorescence Microscopy
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ziaeian, B.; Fonarow, G.C. Epidemiology and aetiology of heart failure. Nat. Rev. Cardiol. 2016, 13, 368–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roger, V.L. Epidemiology of Heart Failure: A Contemporary Perspective. Circ. Res. 2021, 128, 1421–1434. [Google Scholar] [CrossRef] [PubMed]
- Regitz-Zagrosek, V.; Oertelt-Prigione, S.; Seeland, U.; Hetzer, R. Sex and gender differences in myocardial hypertrophy and heart failure. Circ. J. 2010, 74, 1265–1273. [Google Scholar] [CrossRef] [Green Version]
- Regitz-Zagrosek, V.; Brokat, S.; Tschope, C. Role of gender in heart failure with normal left ventricular ejection fraction. Prog. Cardiovasc Dis. 2007, 49, 241–251. [Google Scholar] [CrossRef]
- Redfield, M.M. Heart Failure with Preserved Ejection Fraction. New Engl. J. Med. 2016, 375, 1868–1877. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, M.A.; Shah, A.M.; Borlaug, B.A. Heart Failure With Preserved Ejection Fraction In Perspective. Circ. Res. 2019, 124, 1598–1617. [Google Scholar] [CrossRef] [PubMed]
- Borlaug, B.A. The pathophysiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2014, 11, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: C omorbidities drive myocardial dysfunction and remodeling through coron ary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubey, R.K.; Imthurn, B.; Zacharia, L.C.; Jackson, E.K. Hormone replacement therapy and cardiovascular disease: What went wron g and where do we go from here? Hypertension 2004, 44, 789–795. [Google Scholar] [CrossRef]
- Humphrey, L.L.; Chan, B.K.S.; Sox, H.C. Postmenopausal hormone replacement therapy and the primary prevention of cardiovascular disease. Ann. Intern. Med. 2002, 137, 273–284. [Google Scholar] [CrossRef]
- Zhang, G.-Q.; Chen, J.-L.; Luo, Y.; Mathur, M.B.; Anagnostis, P.; Nurmatov, U.; Talibov, M.; Zhang, J.; Hawrylowicz, C.M.; Lumsden, M.A.; et al. Menopausal hormone therapy and women’s health: An umbrella review. PLoS Med. 2021, 18, e1003731. [Google Scholar] [CrossRef] [PubMed]
- El Khoudary, S.R.; Aggarwal, B.; Beckie, T.M.; Hodis, H.N.; Johnson, A.E.; Langer, R.D.; Limacher, M.C.; Manson, J.E.; Stefanick, M.L.; Allison, M.A.; et al. Menopause Transition and Cardiovascular Disease Risk: Implications for Timing of Early Prevention: A Scientific Statement From the American Heart Association. Circulation 2020, 142, e506–e532. [Google Scholar] [CrossRef] [PubMed]
- Feldman, R.D.; Limbird, L.E. GPER (GPR30): A Nongenomic Receptor (GPCR) for Steroid Hormones with I mplications for Cardiovascular Disease and Cancer. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 567–584. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Arterburn, J.B. International Union of Basic and Clinical Pharmacology. XCVII. G Prote in-Coupled Estrogen Receptor and Its Pharmacologic Modulators. Pharmacol. Rev. 2015, 67, 505–540. [Google Scholar] [CrossRef] [Green Version]
- Prossnitz, E.R.; Barton, M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat. Rev. Endocrinol. 2011, 7, 715–726. [Google Scholar] [CrossRef] [Green Version]
- Sharma, G.; Hu, C.; Staquicini, D.; Brigman, J.; Liu, M.; Mauvais-Jarvis, F.; Pasqualini, R.; Arap, W.; Arterburn, J.; Hathaway, H.; et al. Preclinical efficacy of the GPER-selective agonist G-1 in mouse models of obesity and diabetes. Sci. Transl. Med. 2020, 12, eaau5956. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jessup, J.A.; Lin, M.S.; Chagas, C.; Lindsey, S.H.; Groban, L. Activation of GPR30 attenuates diastolic dysfunction and left ventricl e remodelling in oophorectomized mRen2.Lewis rats. Cardiovasc. Res. 2012, 94, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lu, L.; Tan, Y.; Jiang, L.; Zhao, M.; Gao, E.; Yu, S.; Liu, J. GPR 30 reduces myocardial infarct area and fibrosis in female ovariect omized mice by activating the PI3K/AKT pathway. Life Sci. 2019, 226, 22–32. [Google Scholar] [CrossRef]
- Wang, X.; Ma, J.; Zhang, S.; Li, Z.; Hong, Z.; Jiang, L.; Duan, W.; Liu, J. G Protein–Coupled Estrogen Receptor 30 Reduces Transverse Aortic Const riction–Induced Myocardial Fibrosis in Aged Female Mice by Inhibiting the ERK1/2 -MMP-9 Signaling Pathway. Front. Pharmacol. 2021, 12, 731609. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosi s. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B. Autophagy in Human Diseases. New Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef] [PubMed]
- Del Re, D.P.; Amgalan, D.; Linkermann, A.; Liu, Q.; Kitsis, R.N. Fundamental Mechanisms of Regulated Cell Death and Implications for He art Disease. Physiol. Rev. 2019, 99, 1765–1817. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, Z.; Yu, X.Y.; Xu, S.; Luo, J. Autophagy and cardiac diseases: Therapeutic potential of natural produ cts. Med. Res. Rev. 2020, 41, 314–341. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2016, 97, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Ren, J.; Ba, L.; Song, C.; Zhang, Q.; Cao, Y.; Shi, P.; Fu, B.; Liu, Y.; Sun, H. MSTN Attenuates Cardiac Hypertrophy through Inhibition of Excessive Ca rdiac Autophagy by Blocking AMPK/mTOR and miR-128/PPARγ/NF-κB. Mol. Ther. Nucleic. Acids 2020, 19, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.J.; Wang, Z.V.; Battiprolu, P.K.; Jiang, N.; Morales, C.R.; Kong, Y.; Rothermel, B.A.; Gillette, T.G.; Hill, J.A. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc. Natl. Acad. Sci. USA 2011, 108, 4123–4128. [Google Scholar] [CrossRef] [Green Version]
- Shirakabe, A.; Ikeda, Y.; Sciarretta, S.; Zablocki, D.K.; Sadoshima, J. Aging and Autophagy in the Heart. Circ. Res. 2016, 118, 1563–1576. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S. Autophagy and cardiac aging. Cell Death Differ. 2019, 26, 653–664. [Google Scholar] [CrossRef] [Green Version]
- Yue, J.; Wang, X.-S.; Feng, B.; Hu, L.-N.; Yang, L.-K.; Lu, L.; Zhang, K.; Wang, Y.-T.; Liu, S.-B. Activation of G-Protein-Coupled Receptor 30 Protects Neurons against E xcitotoxicity through Inhibiting Excessive Autophagy Induced by Glutam ate. ACS Chem. Neurosci. 2019, 10, 4227–4236. [Google Scholar] [CrossRef]
- Wang, X.-S.; Yue, J.; Hu, L.-N.; Tian, Z.; Zhang, K.; Yang, L.; Zhang, H.-N.; Guo, Y.-Y.; Feng, B.; Liu, H.-Y.; et al. Activation of G protein-coupled receptor 30 protects neurons by regula ting autophagy in astrocytes. Glia 2020, 68, 27–43. [Google Scholar] [CrossRef]
- Zhang, S.; Li, J.; Jiang, H.; Gao, Y.; Cheng, P.; Cao, T.; Li, D.; Wang, J.; Song, Y.; Liu, B.; et al. Dorsal Root Ganglion Maintains Stemness of Bone Marrow Mesenchymal Stem Cells by Enhancing Autophagy through the AMPK/mTOR Pathway in a Coculture System. Stem Cells Int. 2018, 2018, 8478953. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Tang, R.; Li, B.; Cheng, L.; Ye, S.; Yang, T.; Han, Y.C.; Liu, C.; Dong, Y.; Qu, L.H.; et al. The cardiac translational landscape reveals that micropeptides are new players involved in cardiomyocyte hypertrophy. Mol. Ther. J. Am. Soc. Gene Ther. 2021, 29, 2253–2267. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Gao, Z.; Liu, W.; Wang, C.; Luo, D.; Chao, S.; Li, S.; Li, Z.; Wang, C.; Zhou, J. Promoting maturation and contractile function of neonatal rat cardiomyocytes by self-powered implantable triboelectric nanogenerator. Nano Energy 2022, 103, 107798. [Google Scholar] [CrossRef]
- Lerchenmüller, C.; Rabolli, C.P.; Yeri, A.; Kitchen, R.; Salvador, A.M.; Liu, L.X.; Ziegler, O.; Danielson, K.; Platt, C.; Shah, R.; et al. CITED4 Protects Against Adverse Remodeling in Response to Physiologica l and Pathological Stress. Circ. Res. 2020, 127, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef]
- Bhuiyan, M.S.; Shioda, N.; Fukunaga, K. Ovariectomy augments pressure overload-induced hypertrophy associated with changes in Akt and nitric oxide synthase signaling pathways in fe male rats. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1606–E1614. [Google Scholar] [CrossRef] [Green Version]
- Rothermel, B.A.; Hill, J.A. Autophagy in load-induced heart disease. Circ. Res. 2008, 103, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Ornatowski, W.; Lu, Q.; Yegambaram, M.; Garcia, A.E.; Zemskov, E.A.; Maltepe, E.; Fineman, J.R.; Wang, T.; Black, S.M. Complex interplay between autophagy and oxidative stress in the develo pment of pulmonary disease. Redox Biol. 2020, 36, 101679. [Google Scholar] [CrossRef]
- Yin, Y.; Sun, G.; Li, E.; Kiselyov, K.; Sun, D. ER stress and impaired autophagy flux in neuronal degeneration and bra in injury. Ageing Res. Rev. 2017, 34, 3–14. [Google Scholar] [CrossRef]
- Schneider, J.; Lother, A.; Hein, L.; Gilsbach, R. Chronic cardiac pressure overload induces adrenal medulla hypertrophy and increased catecholamine synthesis. Basic Res. Cardiol. 2011, 106, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.-N.; Kong, L.-H.; Ding, P.; Liu, Y.; Fan, Z.-G.; Gao, E.-H.; Yang, J.; Yang, L.-F. Melatonin ameliorates pressure overload-induced cardiac hypertrophy by attenuating Atg5-dependent autophagy and activating the Akt/mTOR path way. Biochim. Et Biophys. Acta. Mol. Basis Dis. 2020, 1866, 165848. [Google Scholar] [CrossRef]
- Zhu, H.; Tannous, P.; Johnstone, J.L.; Kong, Y.; Shelton, J.M.; Richardson, J.A.; Le, V.; Levine, B.; Rothermel, B.A.; Hill, J.A. Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Investig. 2007, 117, 1782–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, G.; Mauvais-Jarvis, F.; Prossnitz, E.R. Roles of G protein-coupled estrogen receptor GPER in metabolic regulation. J. Steroid. Biochem. 2018, 176, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Brunsing, R.L.; Prossnitz, E.R. Induction of interleukin-10 in the T helper type 17 effector populatio n by the G protein coupled estrogen receptor (GPER) agonist G-1. Immunology 2011, 134, 93–106. [Google Scholar] [CrossRef]
- Chan, Q.K.Y.; Lam, H.M.; Ng, C.F.; Lee, A.Y.Y.; Chan, E.S.Y.; Ng, H.K.; Ho, S.M.; Lau, K.M. Activation of GPR30 inhibits the growth of prostate cancer cells throu gh sustained activation of Erk1/2, c-jun/c-fos-dependent upregulation of p21, and induction of G(2) cell-cycle arrest. Cell Death Differ. 2010, 17, 1511–1523. [Google Scholar] [CrossRef]
- Zucchetti, A.E.; Barosso, I.R.; Boaglio, A.C.; Basiglio, C.L.; Miszczuk, G.; Larocca, M.C.; Ruiz, M.L.; Davio, C.A.; Roma, M.G.; Crocenzi, F.A.; et al. G-protein-coupled receptor 30/adenylyl cyclase/protein kinase A pathwa y is involved in estradiol 17ß-D-glucuronide-induced cholestasis. Hepatology 2014, 59, 1016–1029. [Google Scholar] [CrossRef]
- Cheng, L.; Poulsen, S.B.; Wu, Q.; Esteva-Font, C.; Olesen, E.T.B.; Peng, L.; Olde, B.; Leeb-Lundberg, L.M.F.; Pisitkun, T.; Rieg, T.; et al. Rapid Aldosterone-Mediated Signaling in the DCT Increases Activity of the Thiazide-Sensitive NaCl Cotransporter. J. Am. Soc. Nephrol. 2019, 30, 1454–1470. [Google Scholar] [CrossRef]
- Rowlands, D.J.; Chapple, S.; Siow, R.C.M.; Mann, G.E. Equol-stimulated mitochondrial reactive oxygen species activate endoth elial nitric oxide synthase and redox signaling in endothelial cells: Roles for F-actin and GPR30. Hypertension 2011, 57, 833–840. [Google Scholar] [CrossRef]
- Fredette, N.C.; Meyer, M.R.; Prossnitz, E.R. Role of GPER in estrogen-dependent nitric oxide formation and vasodila tion. J. Steroid. Biochem. Mol. Biol. 2018, 176, 65–72. [Google Scholar] [CrossRef]
- Wani, A.; Gupta, M.; Ahmad, M.; Shah, A.M.; Ahsan, A.U.; Qazi, P.H.; Malik, F.; Singh, G.; Sharma, P.R.; Kaddoumi, A.; et al. Alborixin clears amyloid-β by inducing autophagy through PTEN-mediated inhibition of the AKT pathway. Autophagy 2019, 15, 1810–1828. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gao, E.; Chan, T.O.; Zhang, X.Q.; Song, J.; Shang, X.; Koch, W.J.; Feldman, A.M.; Cheung, J.Y. Induced overexpression of Na(+)/Ca(2+) exchanger does not aggravate myocardial dysfunction induced by transverse aortic constriction. J. Card Fail. 2013, 19, 60–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Wang, X.; Li, C.; Li, Q.; An, Y.A.; Luo, X.; Deng, Y.; Gillette, T.G.; Scherer, P.E.; Wang, Z.V. Integrated Stress Response Couples Mitochondrial Protein Translation With Oxidative Stress Control. Circulation 2021, 144, 1500–1515. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Ma, J.; Wang, X.; Zhao, D.; Zhang, J.; Jiang, L.; Duan, W.; Wang, X.; Hong, Z.; Li, Z.; et al. GPR30 Alleviates Pressure Overload-Induced Myocardial Hypertrophy in Ovariectomized Mice by Regulating Autophagy. Int. J. Mol. Sci. 2023, 24, 904. https://doi.org/10.3390/ijms24020904
Zhang S, Ma J, Wang X, Zhao D, Zhang J, Jiang L, Duan W, Wang X, Hong Z, Li Z, et al. GPR30 Alleviates Pressure Overload-Induced Myocardial Hypertrophy in Ovariectomized Mice by Regulating Autophagy. International Journal of Molecular Sciences. 2023; 24(2):904. https://doi.org/10.3390/ijms24020904
Chicago/Turabian StyleZhang, Shuaishuai, Jipeng Ma, Xiaowu Wang, Diancai Zhao, Jinglong Zhang, Liqing Jiang, Weixun Duan, Xiaoya Wang, Ziwei Hong, Zilin Li, and et al. 2023. "GPR30 Alleviates Pressure Overload-Induced Myocardial Hypertrophy in Ovariectomized Mice by Regulating Autophagy" International Journal of Molecular Sciences 24, no. 2: 904. https://doi.org/10.3390/ijms24020904
APA StyleZhang, S., Ma, J., Wang, X., Zhao, D., Zhang, J., Jiang, L., Duan, W., Wang, X., Hong, Z., Li, Z., & Liu, J. (2023). GPR30 Alleviates Pressure Overload-Induced Myocardial Hypertrophy in Ovariectomized Mice by Regulating Autophagy. International Journal of Molecular Sciences, 24(2), 904. https://doi.org/10.3390/ijms24020904