
A Rare Skeletal Disorder, Fibrous Dysplasia: A Review of Its Pathogenesis and Therapeutic Prospects


Abstract
:1. Introduction
2. Fibrous Dysplasia: Clinical Heterogeneity
3. Somatic Mosaicism and Its Clinical Relevance
4. Histopathology and Cytopathology
5. Pathogenesis
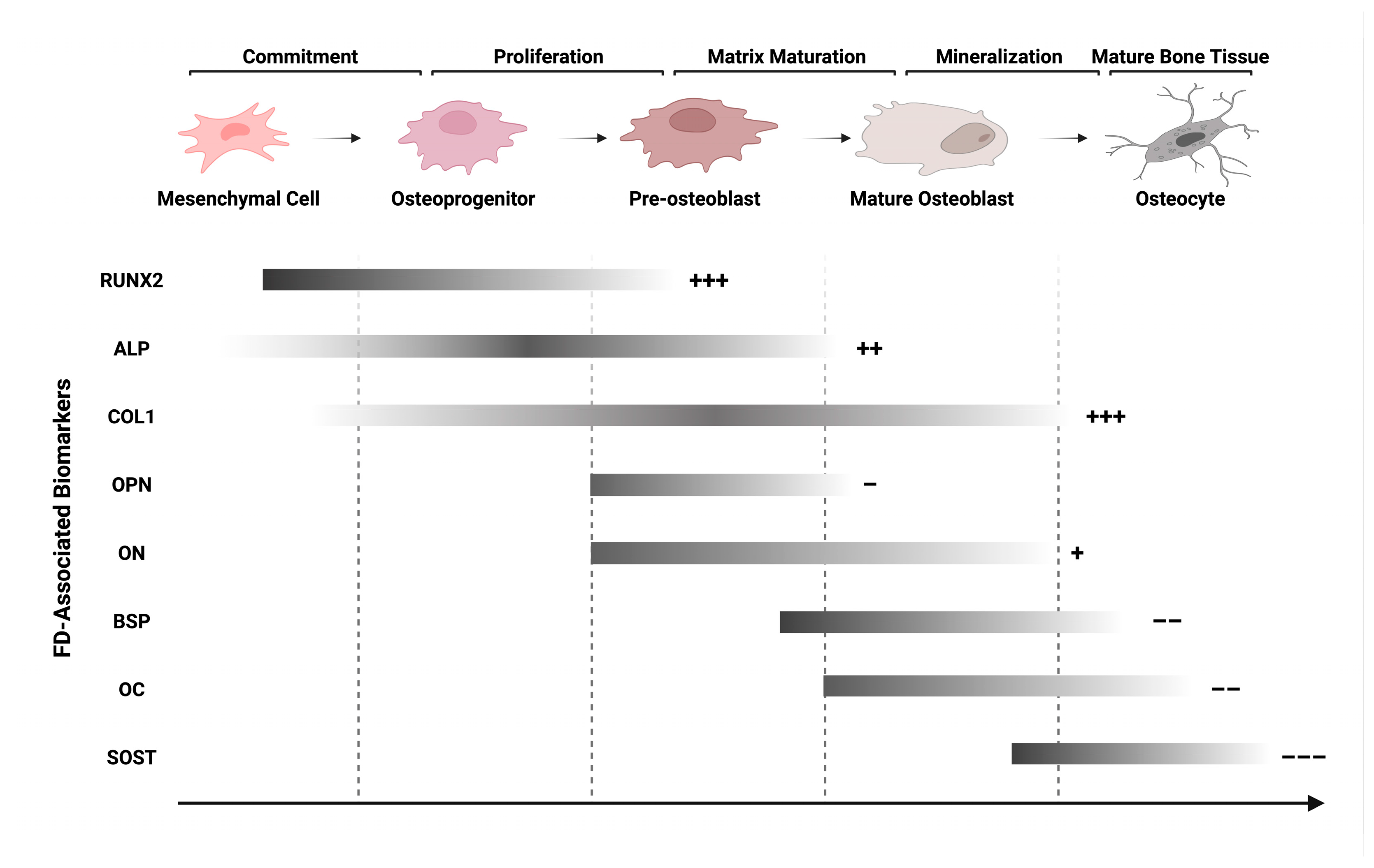
5.1. Aberrant Bone Formation
5.1.1. Enhanced Proliferation and Osteoblast Differentiation
5.1.2. Impaired Osteoblast Maturation
5.1.3. Insufficient Mineralization
5.2. Fibrous Matrix Deposition
5.2.1. Delineating Fibroblastic Cell Population
5.2.2. Role of TGF-β
5.2.3. Sharpey’s Fiber Formation
5.3. Bone Remodeling Imbalance
5.3.1. IL6 and Osteoclastogenesis
5.3.2. Other Cytokines and Chemokines
5.4. Bone Pain
5.4.1. Nociceptive Factors
5.4.2. Neuropathic Factors
6. Current Treatment Prospects
6.1. Modulation of Osteoclastogenesis
6.1.1. Bisphosphonates
6.1.2. RANKL Inhibitors
6.1.3. IL6 Inhibitors
6.1.4. CSF1R Inhibitors
6.2. Blockade of Neural Sensitization
6.2.1. NGF/TrKA Inhibitors
6.2.2. TRPV1 Antagonists
6.3. Inhibition of Gsα
6.3.1. G Protein Antagonists
6.3.2. GNAS Gene Editors/Silencers

{kind=link}
{kind=link}
| Agent | Target | Action | Type of Study | Efficacy | Potential Complications | Refs. | ||
|---|---|---|---|---|---|---|---|---|
| Bone Turnover Decrease | Bone Pain Reduction | Radiographic Improvement | ||||||
| Current therapies | ||||||||
| Bisphosphonates 1. Alendronate | Bone resorption/bone formation | Osteoclast function and proliferation inhibition, osteoclast apoptosis, osteoblast differentiation, and activity promotion | CR (n = 1) | + | + | + | No adverse effects | [181] |
| CR (n = 1) | + | + | No adverse effects | [182] | ||||
| Phase II (n = 40) | + | - | - | Nausea and vomit (pediatric) | [142] | |||
| 2. Pamidronate | CS (n = 9) | + | + | + | Transient mineralization defect | [183] | ||
| CS (n = 9) | + | + | No adverse effects | [184] | ||||
| CS (n = 13) | + | + | + | No adverse effects | [185] | |||
| Phase II (n = 58) | + | + | + | Transient fever | [143] | |||
| 3. Zoledronic acid | CS (n = 11) | + | + | No adverse effects | [186] | |||
| CS (n = 10) peds. | + | + | + | Hypocalcemia | [144] | |||
| CS (n = 7) | + | + | - | Transient fever and myalgia | [187] | |||
| Anti-RANKL (denosumab) | Bone resorption | Osteoclast activation suppression | CR (n = 1) | + | + | + | Hypophosphatemia and secondary hyperparathyroidism | [127] |
| CS (n = 12) | + | + | No adverse events | [128] | ||||
| CR (n = 2) | + | + | + | No adverse events | [188] | |||
| Phase II (n = 8) | + | + | + | Hypercalcemia post-discontinuation | [189] | |||
| Phase II (n = 15 estimated) | NA | NA | NA | NA | * | |||
| Phase IV (n = 82 estimated) | NA | NA | NA | NA | * | |||
| Anti-IL6 (tocilizumab) | Bone resorption | RANKL production suppression | CR (n = 1) | + | No adverse events | [190] | ||
| Phase II (n = 16) | - | - | - | No adverse events | [151] | |||
| Potential therapies | ||||||||
| CSF1R inhibitors | Bone resorption | Macrophage formation inhibition | [191] | |||||
| Anti-NGF/TrKA (tanezumab) | Bone pain | Nerve sprouting and sensitization blockade | [192] | |||||
| TRPV1 antagonists | pH-sensitive neurons’ blockade | [193] | ||||||
| Gsα inhibitor (suramin sodium) | G protein | Mutant Gsα inhibition | [194] | |||||
| Gsα gene editor | Gsα mutation correction | [195] | ||||||
7. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schoenau, E.; Rauch, F. Fibrous Dysplasia. Horm. Res. Paediatr. 2002, 57 (Suppl. S2), 79–82. [Google Scholar] [CrossRef]
- Pai, B.; Ferdinand, D. Fibrous Dysplasia Causing Safeguarding Concerns. Arch. Dis. Child. 2013, 98, 1003. [Google Scholar] [CrossRef] [PubMed]
- Lail, R.A.; Majeed, A. Clinical Presentation and Outcome of Fibrous Dysplasia in Patients Attending Sahiwal Teaching Hospital, Punjab. J. Univ. Coll. Med. Dent. 2022, 1, 20–23. [Google Scholar] [CrossRef]
- Kushchayeva, Y.S.; Kushchayev, S.V.; Glushko, T.Y.; Tella, S.H.; Teytelboym, O.M.; Collins, M.T.; Boyce, A.M. Fibrous Dysplasia for Radiologists: Beyond Ground Glass Bone Matrix. Insights Imaging 2018, 9, 1035–1056. [Google Scholar] [CrossRef] [PubMed]
- Dumitrescu, C.E.; Collins, M.T. McCune-Albright Syndrome. Orphanet J. Rare Dis. 2008, 3, 12. [Google Scholar] [CrossRef]
- Mancini, F.; Corsi, A.; De Maio, F.; Riminucci, M.; Ippolito, E. Scoliosis and Spine Involvement in Fibrous Dysplasia of Bone. Eur. Spine J. Off. Publ. Eur. Spine Soc. Eur. Spinal Deform. Soc. Eur. Sect. Cerv. Spine Res. Soc. 2009, 18, 196–202. [Google Scholar] [CrossRef]
- Lee, J.S.; FitzGibbon, E.J.; Chen, Y.R.; Kim, H.J.; Lustig, L.R.; Akintoye, S.O.; Collins, M.T.; Kaban, L.B. Clinical Guidelines for the Management of Craniofacial Fibrous Dysplasia. Orphanet J. Rare Dis. 2012, 7 (Suppl. S1), S2. [Google Scholar] [CrossRef]
- Li, Z.; Raynald; Wang, Z.; Qian, H. Malignant Transformation of Craniofacial Fibrous Dysplasia: A Systematic Review of Overall Survival. Neurosurg. Rev. 2020, 43, 911–921. [Google Scholar] [CrossRef]
- Stanton, R.P.; Ippolito, E.; Springfield, D.; Lindaman, L.; Wientroub, S.; Leet, A. The Surgical Management of Fibrous Dysplasia of Bone. Orphanet J. Rare Dis. 2012, 7 (Suppl. S1), S1. [Google Scholar] [CrossRef]
- Robinson, C.; Collins, M.T.; Boyce, A.M. Fibrous Dysplasia/McCune-Albright Syndrome: Clinical and Translational Perspectives. Curr. Osteoporos. Rep. 2016, 14, 178–186. [Google Scholar] [CrossRef]
- Hopkins, C.; De Castro, L.F.; Corsi, A.; Boyce, A.; Collins, M.T.; Riminucci, M.; Heegaard, A.-M. Fibrous Dysplasia Animal Models: A Systematic Review. Bone 2022, 155, 116270. [Google Scholar] [CrossRef] [PubMed]
- Saggio, I.; Remoli, C.; Spica, E.; Cersosimo, S.; Sacchetti, B.; Robey, P.G.; Holmbeck, K.; Cumano, A.; Boyde, A.; Bianco, P.; et al. Constitutive Expression of Gsα R201C in Mice Produces a Heritable, Direct Replica of Human Fibrous Dysplasia Bone Pathology and Demonstrates Its Natural History: Mouse Model of Human Fibrous Dysplasia. J. Bone Miner. Res. 2014, 29, 2357–2368. [Google Scholar] [CrossRef] [PubMed]
- Charoenlarp, P.; Cholitgul, W.; Sinpitaksakul, P.; Dhanuthai, K.; Sessirisombat, S. Successive Generations with Inherited Craniofacial Fibrous Dysplasia. Oral Radiol. 2012, 28, 121–128. [Google Scholar] [CrossRef]
- Kuznetsov, S.A.; Cherman, N.; Riminucci, M.; Collins, M.T.; Robey, P.G.; Bianco, P. Age-Dependent Demise of GNAS -Mutated Skeletal Stem Cells and “Normalization” of Fibrous Dysplasia of Bone. J. Bone Miner. Res. 2008, 23, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Ringel, M.D.; Schwindinger, W.F.; Levine, M.A. Clinical Implications of Genetic Defects in G Proteins: The Molecular Basis of McCune-Albright Syndrome and Albright Hereditary Osteodystrophy. Medicine 1996, 75, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Bianco, P.; Robey, P.G. Diseases of Bone and the Stromal Cell Lineage. J. Bone Miner. Res. 1999, 14, 336–341. [Google Scholar] [CrossRef]
- Bianco, P.; Kuznetsov, S.A.; Riminucci, M.; Fisher, L.W.; Spiegel, A.M.; Robey, P.G. Reproduction of Human Fibrous Dysplasia of Bone in Immunocompromised Mice by Transplanted Mosaics of Normal and Gsalpha-Mutated Skeletal Progenitor Cells. J. Clin. Investig. 1998, 101, 1737–1744. [Google Scholar] [CrossRef]
- Pereira, T.D.S.F.; Gomes, C.C.; Brennan, P.A.; Fonseca, F.P.; Gomez, R.S. Fibrous Dysplasia of the Jaws: Integrating Molecular Pathogenesis with Clinical, Radiological, and Histopathological Features. J. Oral Pathol. Med. 2019, 48, 3–9. [Google Scholar] [CrossRef]
- Tabareau-Delalande, F.; Collin, C.; Gomez-Brouchet, A.; Decouvelaere, A.-V.; Bouvier, C.; Larousserie, F.; Marie, B.; Delfour, C.; Aubert, S.; Rosset, P.; et al. Diagnostic Value of Investigating GNAS Mutations in Fibro-Osseous Lesions: A Retrospective Study of 91 Cases of Fibrous Dysplasia and 40 Other Fibro-Osseous Lesions. Mod. Pathol. 2013, 26, 911–921. [Google Scholar] [CrossRef]
- Liang, Q.; Wei, M.; Hodge, L.; Fanburg-Smith, J.C.; Nelson, A.; Miettinen, M.; Foss, R.D.; Wang, G. Quantitative Analysis of Activating Alpha Subunit of the G Protein (Gsα) Mutation by Pyrosequencing in Fibrous Dysplasia and Other Bone Lesions. J. Mol. Diagn. 2011, 13, 137–142. [Google Scholar] [CrossRef]
- Cuttler, L.; Jackson, J.A.; uz-Zafar, M.S.; Levitsky, L.L.; Mellinger, R.C.; Frohman, L.A. Hypersecretion of Growth Hormone and Prolactin in McCune-Albright Syndrome. J. Clin. Endocrinol. Metab. 1989, 68, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Shenker, A.; Laue, L.; Kosugi, S.; Merendino, J.J.; Minegishi, T.; Cutler, G.B. A Constitutively Activating Mutation of the Luteinizing Hormone Receptor in Familial Male Precocious Puberty. Nature 1993, 365, 652–654. [Google Scholar] [CrossRef] [PubMed]
- Malchoff, C.D.; Reardon, G.; MacGillivray, D.C.; Yamase, H.; Rogol, A.D.; Malchoff, D.M. An Unusual Presentation of McCune-Albright Syndrome Confirmed by an Activating Mutation of the Gs Alpha-Subunit from a Bone Lesion. J. Clin. Endocrinol. Metab. 1994, 78, 803–806. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.A. Clinical Implications of Genetic Defects in G Proteins. Arch. Med. Res. 1999, 30, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Idowu, B.D.; Al-Adnani, M.; O’Donnell, P.; Yu, L.; Odell, E.; Diss, T.; Gale, R.E.; Flanagan, A.M. A Sensitive Mutation-Specific Screening Technique for GNAS1 Mutations in Cases of Fibrous Dysplasia: The First Report of a Codon 227 Mutation in Bone. Histopathology 2007, 50, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Elli, F.M.; De Sanctis, L.; Bergallo, M.; Maffini, M.A.; Pirelli, A.; Galliano, I.; Bordogna, P.; Arosio, M.; Mantovani, G. Improved Molecular Diagnosis of McCune–Albright Syndrome and Bone Fibrous Dysplasia by Digital PCR. Front. Genet. 2019, 10, 862. [Google Scholar] [CrossRef]
- Clarke, B. Normal Bone Anatomy and Physiology. Clin. J. Am. Soc. Nephrol. CJASN 2008, 3 (Suppl. S3), S131–S139. [Google Scholar] [CrossRef]
- Riminucci, M.; Robey, P.G.; Saggio, I.; Bianco, P. Skeletal Progenitors and the GNAS Gene: Fibrous Dysplasia of Bone Read through Stem Cells. J. Mol. Endocrinol. 2010, 45, 355–364. [Google Scholar] [CrossRef]
- Riminucci, M.; Liu, B.; Corsi, A.; Shenker, A.; Spiegel, A.M.; Robey, P.G.; Bianco, P. The Histopathology of Fibrous Dysplasia of Bone in Patients with Activating Mutations of the Gs? Gene: Site-Specific Patterns and Recurrent Histological Hallmarks. J. Pathol. 1999, 187, 249–258. [Google Scholar] [CrossRef]
- Raimondo, D.; Remoli, C.; Astrologo, L.; Burla, R.; La Torre, M.; Vernì, F.; Tagliafico, E.; Corsi, A.; Del Giudice, S.; Persichetti, A.; et al. Changes in Gene Expression in Human Skeletal Stem Cells Transduced with Constitutively Active Gsα Correlates with Hallmark Histopathological Changes Seen in Fibrous Dysplastic Bone. PLoS ONE 2020, 15, e0227279. [Google Scholar] [CrossRef]
- Stewart, M.J.; Gilmer, W.S.; Edmonson, A.S. Fibrous Dysplasia of Bone. J. Bone Joint Surg. Br. 1962, 44, 302–318. [Google Scholar] [CrossRef] [PubMed]
- De Castro, L.F.; Burke, A.B.; Wang, H.D.; Tsai, J.; Florenzano, P.; Pan, K.S.; Bhattacharyya, N.; Boyce, A.M.; Gafni, R.I.; Molinolo, A.A.; et al. Activation of RANK/RANKL/OPG Pathway Is Involved in the Pathophysiology of Fibrous Dysplasia and Associated with Disease Burden. J. Bone Miner. Res. 2019, 34, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Riminucci, M.; Fisher, L.W.; Shenker, A.; Spiegel, A.M.; Bianco, P.; Gehron Robey, P. Fibrous Dysplasia of Bone in the McCune-Albright Syndrome: Abnormalities in Bone Formation. Am. J. Pathol. 1997, 151, 1587–1600. [Google Scholar] [PubMed]
- Latham, P.D.; Athanasouz, N.A.; Woods, C.G. Fibrous Dysplasia with Locally Aggressive Malignant Change. Arch. Orthop. Trauma Surg. 1992, 111, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Corsi, A.; Collins, M.T.; Riminucci, M.; Howell, P.G.; Boyde, A.; Robey, P.G.; Bianco, P. Osteomalacic and Hyperparathyroid Changes in Fibrous Dysplasia of Bone: Core Biopsy Studies and Clinical Correlations. J. Bone Miner. Res. 2003, 18, 1235–1246. [Google Scholar] [CrossRef]
- Legrand, M.A.; Millet, M.; Merle, B.; Rousseau, J.; Hemmendinger, A.; Gineyts, E.; Sornay-Rendu, E.; Szulc, P.; Borel, O.; Croset, M.; et al. A Signature of Circulating miRNAs Associated with Fibrous Dysplasia of Bone: The mirDys Study. J. Bone Miner. Res. 2020, 35, 1881–1892. [Google Scholar] [CrossRef]
- Kini, U.; Nandeesh, B.N. Physiology of Bone Formation, Remodeling, and Metabolism. In Radionuclide and Hybrid Bone Imaging; Fogelman, I., Gnanasegaran, G., Van Der Wall, H., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 29–57. [Google Scholar] [CrossRef]
- Donsante, S.; Palmisano, B.; Serafini, M.; Robey, P.G.; Corsi, A.; Riminucci, M. From Stem Cells to Bone-Forming Cells. Int. J. Mol. Sci. 2021, 22, 3989. [Google Scholar] [CrossRef]
- Komori, T. Regulation of Osteoblast Differentiation by Transcription Factors. J. Cell. Biochem. 2006, 99, 1233–1239. [Google Scholar] [CrossRef]
- Raisz, L.G.; Kream, B.E. Regulation of Bone Formation. N. Engl. J. Med. 1983, 309, 83–89. [Google Scholar] [CrossRef]
- Caetano-Lopes, J.; Canhão, H.; Fonseca, J.E. Osteoblasts and Bone Formation. Acta Reumatol. Port. 2007, 32, 103–110. [Google Scholar]
- Lin, X.; Patil, S.; Gao, Y.-G.; Qian, A. The Bone Extracellular Matrix in Bone Formation and Regeneration. Front. Pharmacol. 2020, 11, 757. [Google Scholar] [CrossRef] [PubMed]
- Amarasekara, D.S.; Kim, S.; Rho, J. Regulation of Osteoblast Differentiation by Cytokine Networks. Int. J. Mol. Sci. 2021, 22, 2851. [Google Scholar] [CrossRef]
- Rosen, C.J. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Jilka, R.L. Biology of the Basic Multicellular Unit and the Pathophysiology of Osteoporosis. Med. Pediatr. Oncol. 2003, 41, 182–185. [Google Scholar] [CrossRef]
- Marie, P.J.; Fromigué, O.; Modrowski, D. Deregulation of Osteoblast Differentiation in Primary Bone Cancers. In Bone Cancer; Elsevier: Amsterdam, The Netherlands, 2015; pp. 39–54. [Google Scholar] [CrossRef]
- Sakamoto, A. A Comparative Study of Fibrous Dysplasia and Osteofibrous Dysplasia with Regard to Expressions of C-Fos and c-Jun Products and Bone Matrix Proteins: A Clinicopathologic Review and Immunohistochemical Study of c-Fos, c-Jun, Type I Collagen, Osteonectin, Osteopontin, and Osteocalcin. Hum. Pathol. 1999, 30, 1418–1426. [Google Scholar] [CrossRef] [PubMed]
- Avery, S.J.; Ayre, W.N.; Sloan, A.J.; Waddington, R.J. Interrogating the Osteogenic Potential of Implant Surfaces In Vitro : A Review of Current Assays. Tissue Eng. Part B Rev. 2020, 26, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Warner, D.R.; Weng, G.; Yu, S.; Matalon, R.; Weinstein, L.S. A Novel Mutation in the Switch 3 Region of Gsα in a Patient with Albright Hereditary Osteodystrophy Impairs GDP Binding and Receptor Activation. J. Biol. Chem. 1998, 273, 23976–23983. [Google Scholar] [CrossRef]
- Warner, D.R.; Gejman, P.V.; Collins, R.M.; Weinstein, L.S. A Novel Mutation Adjacent to the Switch III Domain of Gsα in a Patient with Pseudohypoparathyroidism. Mol. Endocrinol. 1997, 11, 1718–1727. [Google Scholar] [CrossRef]
- Weinstein, L.S.; Liu, J.; Sakamoto, A.; Xie, T.; Chen, M. Minireview: GNAS: Normal and Abnormal Functions. Endocrinology 2004, 145, 5459–5464. [Google Scholar] [CrossRef]
- Montminy, M.R.; Gonzalez, G.A.; Yamamoto, K.K. Regulation of Camp-Inducible Genes by Creb. Trends Neurosci. 1990, 13, 184–188. [Google Scholar] [CrossRef]
- Shen, L.; He, Y.; Chen, S.; He, L.; Zhang, Y. PTHrP Modulates the Proliferation and Osteogenic Differentiation of Craniofacial Fibrous Dysplasia-Derived BMSCs. Int. J. Mol. Sci. 2023, 24, 7616. [Google Scholar] [CrossRef]
- Yasuoka, T.; Takagi, N.; Hatakeyama, D.; Yokoyama, K. Fibrous Dysplasia in the Maxilla: Possible Mechanism of Bone Remodeling by Calcitonin Treatment. Oral Oncol. 2003, 39, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Persichetti, A.; Milanetti, E.; Palmisano, B.; Di Filippo, A.; Spica, E.; Donsante, S.; Coletta, I.; Venti, M.D.S.; Ippolito, E.; Corsi, A.; et al. Nanostring Technology on Fibrous Dysplasia Bone Biopsies. A Pilot Study Suggesting Different Histology-Related Molecular Profiles. Bone Rep. 2022, 16, 101156. [Google Scholar] [CrossRef] [PubMed]
- Leet, A.I.; Collins, M.T. Current Approach to Fibrous Dysplasia of Bone and McCune–Albright Syndrome. J. Child. Orthop. 2007, 1, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Regard, J.B.; Cherman, N.; Palmer, D.; Kuznetsov, S.A.; Celi, F.S.; Guettier, J.-M.; Chen, M.; Bhattacharyya, N.; Wess, J.; Coughlin, S.R.; et al. Wnt/β-Catenin Signaling Is Differentially Regulated by Gα Proteins and Contributes to Fibrous Dysplasia. Proc. Natl. Acad. Sci. USA 2011, 108, 20101–20106. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, E.; Sakisaka, Y.; Tsuchiya, M.; Tamura, M.; Nakamura, T.; Kanaya, S.; Shimonishi, M.; Shimauchi, H. Wnt3a Signaling Induces Murine Dental Follicle Cells to Differentiate into Cementoblastic/Osteoblastic Cells via an Osterix-Dependent Pathway. J. Periodontal Res. 2016, 51, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Chen, X.; Jia, H.; Meyers, C.A.; Shrestha, S.; Asatrian, G.; Ding, C.; Tsuei, R.; Zhang, X.; Peault, B.; et al. Effects of WNT3A and WNT16 on the Osteogenic and Adipogenic Differentiation of Perivascular Stem/Stromal Cells. Tissue Eng. Part A 2018, 24, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Shi, S.; Deng, M.; Tang, L.; Zhang, G.; Liu, N.; Ding, B.; Liu, W.; Liu, Y.; Shi, H.; et al. High Levels of β-Catenin Signaling Reduce Osteogenic Differentiation of Stem Cells in Inflammatory Microenvironments through Inhibition of the Noncanonical Wnt Pathway. J. Bone Miner. Res. 2011, 26, 2082–2095. [Google Scholar] [CrossRef]
- Boland, G.M.; Perkins, G.; Hall, D.J.; Tuan, R.S. Wnt3a Promotes Proliferation and Suppresses Osteogenic Differentiation of Adult Human Mesenchymal Stem Cells. J. Cell. Biochem. 2004, 93, 1210–1230. [Google Scholar] [CrossRef]
- Fan, Q.-M.; Yue, B.; Bian, Z.-Y.; Xu, W.-T.; Tu, B.; Dai, K.-R.; Li, G.; Tang, T.-T. The CREB-Smad6-Runx2 Axis Contributes to the Impaired Osteogenesis Potential of Bone Marrow Stromal Cells in Fibrous Dysplasia of Bone. J. Pathol. 2012, 228, 45–55. [Google Scholar] [CrossRef]
- Chen, S.; Wu, Z.; He, Y.; Zhu, L.; Wang, J.; Lin, H.; Xie, J.; Zhou, C.; Zou, S. Cyclic Di-Adenosine Monophosphate Regulates the Osteogenic and Adipogenic Differentiation of hPDLSCs via MAPK and NF-κB Signaling. Acta Biochim. Biophys. Sin. 2023, 55, 426–437. [Google Scholar] [CrossRef]
- Zhao, X.; Deng, P.; Iglesias-Bartolome, R.; Amornphimoltham, P.; Steffen, D.J.; Jin, Y.; Molinolo, A.A.; De Castro, L.F.; Ovejero, D.; Yuan, Q.; et al. Expression of an Active Gαs Mutant in Skeletal Stem Cells Is Sufficient and Necessary for Fibrous Dysplasia Initiation and Maintenance. Proc. Natl. Acad. Sci. USA 2018, 115, E428–E437. [Google Scholar] [CrossRef] [PubMed]
- Park, B.Y.; Cheon, Y.W.; Kim, Y.O.; Pae, N.S.; Lee, W.J. Prognosis for Craniofacial Fibrous Dysplasia after Incomplete Resection: Age and Serum Alkaline Phosphatase. Int. J. Oral Maxillofac. Surg. 2010, 39, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Liang, L.; Gu, B.; Zhang, H.; Wen, W.; Liu, H. A Retrospective Study on Craniofacial Fibrous Dysplasia: Preoperative Serum Alkaline Phosphatase as a Prognostic Marker? J. Cranio-Maxillofac. Surg. 2013, 41, 644–647. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Du, Z.; Li, D.; Yang, R.; Tang, X.; Yan, T.; Guo, W. Increasing Serum Alkaline Phosphatase Is Associated with Bone Deformity Progression for Patients with Polyostotic Fibrous Dysplasia. J. Orthop. Surg. 2020, 15, 583. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, Z.; Yoshida, C.A.; Furuichi, T.; Amizuka, N.; Ito, M.; Fukuyama, R.; Miyazaki, T.; Kitaura, H.; Nakamura, K.; Fujita, T.; et al. Runx2 Determines Bone Maturity and Turnover Rate in Postnatal Bone Development and Is Involved in Bone Loss in Estrogen Deficiency. Dev. Dyn. 2007, 236, 1876–1890. [Google Scholar] [CrossRef]
- Xiao, T.; Fu, Y.; Zhu, W.; Xu, R.; Xu, L.; Zhang, P.; Du, Y.; Cheng, J.; Jiang, H. HDAC8, A Potential Therapeutic Target, Regulates Proliferation and Differentiation of Bone Marrow Stromal Cells in Fibrous Dysplasia. Stem Cells Transl. Med. 2019, 8, 148–161. [Google Scholar] [CrossRef]
- Sakamoto, A.; Oda, Y.; Iwamoto, Y.; Tsuneyoshi, M. Frequent Immunoexpression of TGF-Β1, FGF-2 and BMP-2 in Fibroblast-like Cells in Osteofibrous Dysplasia. Oncol. Rep. 2007, 17, 531–535. [Google Scholar] [CrossRef]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β Signaling in Fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef]
- Sobral, A.P.V.; Etges, A.; Bitu Sousa, F.; Soares De Araújo, N.; Daumas Nunes, F. Immunolocalization of Bmp2/4, Tgfβ-1 and Osteonectin in Fibro-Osseous Lesions of the Jaws. Rev. De Cir. E Traumatol. Buco-Maxilo-Facial 2011, 11, 77–84. [Google Scholar]
- Zou, Y.; Zhang, N.; Tang, Y.; Zhan, Z.; Yang, M.; Lu, Y.; Li, G.-S.; Zhang, L. Predictive Markers for Severe Hypocalcemia in Dialysis Patients with Secondary Hyperparathyroidism after Near-Total Parathyroidectomy. Ann. Palliat. Med. 2021, 10, 10712–10719. [Google Scholar] [CrossRef]
- Roach, H. Why Does Bone Matrix Contain Non-Collagenous Proteins? The Possible Roles of Osteocalcin, Osteonectin, Osteopontin and Bone Sialoprotein in Bone Mineralisation and Resorption. Cell Biol. Int. 1994, 18, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Marie, P.J.; de Pollak, C.; Chanson, P.; Lomri, A. Increased Proliferation of Osteoblastic Cells Expressing the Activating Gs Alpha Mutation in Monostotic and Polyostotic Fibrous Dysplasia. Am. J. Pathol. 1997, 150, 1059–1069. [Google Scholar] [PubMed]
- Feller, L.; Wood, N.H.; Khammissa, R.A.; Lemmer, J.; Raubenheimer, E.J. The Nature of Fibrous Dysplasia. Head Face Med. 2009, 5, 22. [Google Scholar] [CrossRef]
- O’Brien, E.R.; Garvin, M.R.; Stewart, D.K.; Hinohara, T.; Simpson, J.B.; Schwartz, S.M.; Giachelli, C.M. Osteopontin Is Synthesized by Macrophage, Smooth Muscle, and Endothelial Cells in Primary and Restenotic Human Coronary Atherosclerotic Plaques. Arterioscler. Thromb. J. Vasc. Biol. 1994, 14, 1648–1656. [Google Scholar] [CrossRef] [PubMed]
- Malyankar, U.M.; Almeida, M.; Johnson, R.J.; Pichler, R.H.; Giachelli, C.M. Osteopontin Regulation in Cultured Rat Renal Epithelial Cells. Kidney Int. 1997, 51, 1766–1773. [Google Scholar] [CrossRef] [PubMed]
- Iline-Vul, T.; Nanda, R.; Mateos, B.; Hazan, S.; Matlahov, I.; Perelshtein, I.; Keinan-Adamsky, K.; Althoff-Ospelt, G.; Konrat, R.; Goobes, G. Osteopontin Regulates Biomimetic Calcium Phosphate Crystallization from Disordered Mineral Layers Covering Apatite Crystallites. Sci. Rep. 2020, 10, 15722. [Google Scholar] [CrossRef]
- Rodriguez, D.E.; Thula-Mata, T.; Toro, E.J.; Yeh, Y.-W.; Holt, C.; Holliday, L.S.; Gower, L.B. Multifunctional Role of Osteopontin in Directing Intrafibrillar Mineralization of Collagen and Activation of Osteoclasts. Acta Biomater. 2014, 10, 494–507. [Google Scholar] [CrossRef]
- Boyce, A.M.; Collins, M.T. Fibrous Dysplasia/McCune-Albright Syndrome: A Rare, Mosaic Disease of Gα s Activation. Endocr. Rev. 2020, 41, 345–370. [Google Scholar] [CrossRef]
- Si, J.; Wang, C.; Zhang, D.; Wang, B.; Zhou, Y. Osteopontin in Bone Metabolism and Bone Diseases. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e919159. [Google Scholar] [CrossRef]
- Riminucci, M.; Collins, M.T.; Fedarko, N.S.; Cherman, N.; Corsi, A.; White, K.E.; Waguespack, S.; Gupta, A.; Hannon, T.; Econs, M.J.; et al. FGF-23 in Fibrous Dysplasia of Bone and Its Relationship to Renal Phosphate Wasting. J. Clin. Investig. 2003, 112, 683–692. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Bhattacharyya, K.; Maitra, A. Possible Mechanisms of Interaction between Statins and Vitamin D. QJM 2012, 105, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Udagawa, N.; Takahashi, N. Action of RANKL and OPG for Osteoclastogenesis. Crit. Rev. Eukaryot. Gene Expr. 2009, 19, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Kawai-Kowase, K.; Matsui, H.; Sunaga, H.; Utsugi, T.; Iso, T.; Arai, M.; Tomono, S.; Kurabayashi, M. Fibroblast Growth Factor 23 Inhibits Osteoblastic Gene Expression and Induces Osteoprotegerin in Vascular Smooth Muscle Cells. Atherosclerosis 2016, 253, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.K.; Yadav, P.S.; Elliott, G.; Hu, D.Z.; Xu, R.; Yang, Y. Induced Gnas R201H Expression from the Endogenous Gnas Locus Causes Fibrous Dysplasia by up-Regulating Wnt/β-Catenin Signaling. Proc. Natl. Acad. Sci. USA 2018, 115, E418–E427. [Google Scholar] [CrossRef] [PubMed]
- Meier, M.E.; Hagelstein-Rotman, M.; Streefland, T.C.M.; Winter, E.M.; Bravenboer, N.; Appelman-Dijkstra, N.M. Clinical Value of RANKL, OPG, IL-6 and Sclerostin as Biomarkers for Fibrous Dysplasia/McCune-Albright Syndrome. Bone 2023, 171, 116744. [Google Scholar] [CrossRef]
- Lewiecki, E.M. Role of Sclerostin in Bone and Cartilage and Its Potential as a Therapeutic Target in Bone Diseases. Ther. Adv. Musculoskelet. Dis. 2014, 6, 48–57. [Google Scholar] [CrossRef]
- Greco, M.A.; Steiner, G.C. Ultrastructure of Fibrous Dysplasia of Bone: A Study of Its Fibrous, Osseous, and Cartilaginous Components. Ultrastruct. Pathol. 1986, 10, 55–66. [Google Scholar] [CrossRef]
- Piersanti, S.; Remoli, C.; Saggio, I.; Funari, A.; Michienzi, S.; Sacchetti, B.; Robey, P.G.; Riminucci, M.; Bianco, P. Transfer, Analysis and Reversion of the Fibrous Dysplasia Cellular Phenotype in Human Skeletal Progenitors. J. Bone Miner. Res. 2009, 25, 1103–1149. [Google Scholar] [CrossRef]
- Weinstein, L.S.; Collins, M.T. Gsα, Pseudohypoparathyroidism, Fibrous Dysplasia, and McCune–Albright Syndrome. In Genetics of Bone Biology and Skeletal Disease; Elsevier: Amsterdam, The Netherlands, 2018; pp. 637–653. [Google Scholar] [CrossRef]
- Yu, J.; Cao, J.; Li, H.; Liu, P.; Xu, S.; Zhou, R.; Yao, Z.; Guo, X. Bone Marrow Fibrosis with Fibrocytic and Immunoregulatory Responses Induced by β-Catenin Activation in Osteoprogenitors. Bone 2016, 84, 38–46. [Google Scholar] [CrossRef]
- Abraham, D.J.; Eckes, B.; Rajkumar, V.; Krieg, T. New Developments in Fibroblast and Myofibroblast Biology: Implications for Fibrosis and Scleroderma. Curr. Rheumatol. Rep. 2007, 9, 136–143. [Google Scholar] [CrossRef]
- Bartl, R.; Bartl, C. Bone Disorders; Springer International Publishing: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- Xu, J.; Kisseleva, T. Bone Marrow-Derived Fibrocytes Contribute to Liver Fibrosis. Exp. Biol. Med. 2015, 240, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Strieter, R.M.; Keeley, E.C.; Hughes, M.A.; Burdick, M.D.; Mehrad, B. The Role of Circulating Mesenchymal Progenitor Cells (Fibrocytes) in the Pathogenesis of Pulmonary Fibrosis. J. Leukoc. Biol. 2009, 86, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Galan, A.; Cowper, S.E.; Bucala, R. Nephrogenic Systemic Fibrosis (Nephrogenic Fibrosing Dermopathy). Curr. Opin. Rheumatol. 2006, 18, 614–617. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-O.; You, C.H.; Son, M.-Y.; Kim, Y.-D.; Jeon, H.; Chang, J.-S.; Cho, Y.S. Pro-Fibrotic Effects of PFKFB4-Mediated Glycolytic Reprogramming in Fibrous Dysplasia. Biomaterials 2016, 107, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.-M. Inflammatory Mediators and Renal Fibrosis. In Renal Fibrosis: Mechanisms and Therapies; Liu, B.-C., Lan, H.-Y., Lv, L.-L., Eds.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; Volume 1165, pp. 381–406. [Google Scholar] [CrossRef]
- Rao, B.; Malathi, N.; Narashiman, S.; Rajan, S.T. Evaluation of Myofibroblasts by Expression of Alpha Smooth Muscle Actin: A Marker in Fibrosis, Dysplasia and Carcinoma. J. Clin. Diagn. Res. JCDR 2014, 8, ZC14-17. [Google Scholar] [CrossRef]
- Krenning, G.; Zeisberg, E.M.; Kalluri, R. The Origin of Fibroblasts and Mechanism of Cardiac Fibrosis. J. Cell. Physiol. 2010, 225, 631–637. [Google Scholar] [CrossRef]
- Gabbiani, G. The Cellular Derivation and the Life Span of the Myofibroblast. Pathol. Res. Pract. 1996, 192, 708–711. [Google Scholar] [CrossRef]
- Kashima, T.G.; Nishiyama, T.; Shimazu, K.; Shimazaki, M.; Kii, I.; Grigoriadis, A.E.; Fukayama, M.; Kudo, A. Periostin, a Novel Marker of Intramembranous Ossification, Is Expressed in Fibrous Dysplasia and in c-Fos–Overexpressing Bone Lesions. Hum. Pathol. 2009, 40, 226–237. [Google Scholar] [CrossRef]
- Maeda, K.; Kobayashi, Y.; Koide, M.; Uehara, S.; Okamoto, M.; Ishihara, A.; Kayama, T.; Saito, M.; Marumo, K. The Regulation of Bone Metabolism and Disorders by Wnt Signaling. Int. J. Mol. Sci. 2019, 20, 5525. [Google Scholar] [CrossRef]
- Zhou, S.-H.; Yang, W.-J.; Liu, S.-W.; Li, J.; Zhang, C.-Y.; Zhu, Y.; Zhang, C.-P. Gene Expression Profiling of Craniofacial Fibrous Dysplasia Reveals ADAMTS2 Overexpression as a Potential Marker. Int. J. Clin. Exp. Pathol. 2014, 7, 8532–8541. [Google Scholar]
- Myneni, V.; Mezey, E. Regulation of Bone Remodeling by Vitamin K2. Oral Dis. 2017, 23, 1021–1028. [Google Scholar] [CrossRef]
- Kurra, S.; Reddy, S.; Gunupati, S.; Srikanth, K.; Reddy, S. Fibrous Dysplasia and Central Giant Cell Granuloma: A Report of Hybrid Lesion with Its Review and Hypotheticated Pathogenesis. J. Clin. Diagn. Res. JCDR 2013, 7, 954–958. [Google Scholar] [CrossRef]
- Stanton, R.P.; Hobson, G.M.; Montgomery, B.E.; Moses, P.A.; Smith-Kirwin, S.M.; Funanage, V.L. Glucocorticoids Decrease Interleukin-6 Levels and Induce Mineralization of Cultured Osteogenic Cells from Children with Fibrous Dysplasia. J. Bone Miner. Res. 1999, 14, 1104–1114. [Google Scholar] [CrossRef]
- Weinstein, L.S. Gsα Mutations in Fibrous Dysplasia and McCune-Albright Syndrome. J. Bone Miner. Res. 2006, 21, P120–P124. [Google Scholar] [CrossRef] [PubMed]
- Singer, F.R. Fibrous Dysplasia of Bone: The Bone Lesion Unmasked. Am. J. Pathol. 1997, 151, 1511–1515. [Google Scholar] [PubMed]
- Riminucci, M.; Kuznetsov, S.A.; Cherman, N.; Corsi, A.; Bianco, P.; Robey, P.G. Osteoclastogenesis in Fibrous Dysplasia of Bone: In Situ and in Vitro Analysis of IL-6 Expression. Bone 2003, 33, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Eller-Vainicher, C.; Rossi, D.S.; Guglielmi, G.; Beltramini, G.A.; Cairoli, E.; Russillo, A.; Mantovani, G.; Spada, A.; Chiodini, I. Prompt Clinical and Biochemical Response to Denosumab in a Young Adult Patient with Craniofacial Fibrous Dysplasia. Clin. Cases Miner. Bone Metab. Off. J. Ital. Soc. Osteoporos. Miner. Metab. Skelet. Dis. 2016, 13, 253–256. [Google Scholar] [CrossRef]
- Sun, Y.; Li, J.; Xie, X.; Gu, F.; Sui, Z.; Zhang, K.; Yu, T. Recent Advances in Osteoclast Biological Behavior. Front. Cell Dev. Biol. 2021, 9, 788680. [Google Scholar] [CrossRef]
- Collins, M.T.; De Castro, L.F.; Boyce, A.M. Denosumab for Fibrous Dysplasia: Promising, but Questions Remain. J. Clin. Endocrinol. Metab. 2020, 105, e4179–e4180. [Google Scholar] [CrossRef]
- de Castro, L.F.; Ovejero, D.; Boyce, A.M. Diagnosis of Endocrine Disease: Mosaic Disorders of FGF23 Excess: Fibrous Dysplasia/McCune-Albright Syndrome and Cutaneous Skeletal Hypophosphatemia Syndrome. Eur. J. Endocrinol. 2020, 182, R83–R99. [Google Scholar] [CrossRef]
- Sun, Y.; Li, J.; Xie, X.; Gu, F.; Sui, Z.; Zhang, K.; Yu, T. Macrophage-Osteoclast Associations: Origin, Polarization, and Subgroups. Front. Immunol. 2021, 12, 778078. [Google Scholar] [CrossRef] [PubMed]
- Kitaura, H.; Marahleh, A.; Ohori, F.; Noguchi, T.; Shen, W.-R.; Qi, J.; Nara, Y.; Pramusita, A.; Kinjo, R.; Mizoguchi, I. Osteocyte-Related Cytokines Regulate Osteoclast Formation and Bone Resorption. Int. J. Mol. Sci. 2020, 21, 5169. [Google Scholar] [CrossRef]
- Feng, X.; McDonald, J.M. Disorders of Bone Remodeling. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 121–145. [Google Scholar] [CrossRef] [PubMed]
- Tucker-Bartley, A.; Selen, D.J.; Golden, E.; Van Gool, R.; Ebb, D.; Mannstadt, M.; Upadhyay, J. Pharmacological Interventions Targeting Pain in Fibrous Dysplasia/McCune–Albright Syndrome. Int. J. Mol. Sci. 2023, 24, 2550. [Google Scholar] [CrossRef] [PubMed]
- Chapurlat, R.D.; Gensburger, D.; Jimenez-Andrade, J.M.; Ghilardi, J.R.; Kelly, M.; Mantyh, P. Pathophysiology and Medical Treatment of Pain in Fibrous Dysplasia of Bone. Orphanet J. Rare Dis. 2012, 7 (Suppl. S1), S3. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.H.; Brillante, B.; Collins, M.T. Pain in Fibrous Dysplasia of Bone: Age-Related Changes and the Anatomical Distribution of Skeletal Lesions. Osteoporos. Int. 2008, 19, 57–63. [Google Scholar] [CrossRef]
- Majoor, B.C.J.; Traunmueller, E.; Maurer-Ertl, W.; Appelman-Dijkstra, N.M.; Fink, A.; Liegl, B.; Hamdy, N.A.T.; Sander Dijkstra, P.D.; Leithner, A. Pain in Fibrous Dysplasia: Relationship with Anatomical and Clinical Features. Acta Orthop. 2019, 90, 401–405. [Google Scholar] [CrossRef]
- Meier, M.E.; Hagelstein-Rotman, M.; Van De Ven, A.C.; Van Der Geest, I.C.M.; Donker, O.; Pichardo, S.E.C.; Hissink Muller, P.C.E.; Van Der Meeren, S.W.; Dorleijn, D.M.J.; Winter, E.M.; et al. A Multidisciplinary Care Pathway Improves Quality of Life and Reduces Pain in Patients with Fibrous Dysplasia/McCune-Albright Syndrome: A Multicenter Prospective Observational Study. Orphanet J. Rare Dis. 2022, 17, 439. [Google Scholar] [CrossRef]
- Chao, K.; Katznelson, L. Use of High-Dose Oral Bisphosphonate Therapy for Symptomatic Fibrous Dysplasia of the Skull: Case Report. J. Neurosurg. 2008, 109, 889–892. [Google Scholar] [CrossRef]
- Mäkitie, A.A.; Törnwall, J.; Mäkitie, O. Bisphosphonate Treatment in Craniofacial Fibrous Dysplasia—A Case Report and Review of the Literature. Clin. Rheumatol. 2008, 27, 809–812. [Google Scholar] [CrossRef]
- Boyce, A.M.; Chong, W.H.; Yao, J.; Gafni, R.I.; Kelly, M.H.; Chamberlain, C.E.; Bassim, C.; Cherman, N.; Ellsworth, M.; Kasa-Vubu, J.Z.; et al. Denosumab Treatment for Fibrous Dysplasia. J. Bone Miner. Res. 2012, 27, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Majoor, B.C.J.; Papapoulos, S.E.; Dijkstra, P.D.S.; Fiocco, M.; Hamdy, N.A.T.; Appelman-Dijkstra, N.M. Denosumab in Patients with Fibrous Dysplasia Previously Treated with Bisphosphonates. J. Clin. Endocrinol. Metab. 2019, 104, 6069–6078. [Google Scholar] [CrossRef]
- Benarroch, E.E. Ion Channels in Nociceptors: Recent Developments. Neurology 2015, 84, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.-S. Molecular Mechanism of Inflammatory Pain. World J. Anesthesiol. 2014, 3, 71. [Google Scholar] [CrossRef]
- Nagae, M.; Hiraga, T.; Wakabayashi, H.; Wang, L.; Iwata, K.; Yoneda, T. Osteoclasts Play a Part in Pain Due to the Inflammation Adjacent to Bone. Bone 2006, 39, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Middlemiss, T.; Laird, B.J.A.; Fallon, M.T. Mechanisms of Cancer-Induced Bone Pain. Clin. Oncol. 2011, 23, 387–392. [Google Scholar] [CrossRef]
- Mantyh, P.W. Bone Cancer Pain: From Mechanism to Therapy. Curr. Opin. Support. Palliat. Care 2014, 8, 83–90. [Google Scholar] [CrossRef]
- Bloom, A.P.; Jimenez-Andrade, J.M.; Taylor, R.N.; Castañeda-Corral, G.; Kaczmarska, M.J.; Freeman, K.T.; Coughlin, K.A.; Ghilardi, J.R.; Kuskowski, M.A.; Mantyh, P.W. Breast Cancer-Induced Bone Remodeling, Skeletal Pain, and Sprouting of Sensory Nerve Fibers. J. Pain 2011, 12, 698–711. [Google Scholar] [CrossRef]
- Gordon-Williams, R.M.; Dickenson, A.H. Central Neuronal Mechanisms in Cancer-Induced Bone Pain. Curr. Opin. Support. Palliat. Care 2007, 1, 6–10. [Google Scholar] [CrossRef]
- Spencer, T.L.; Watts, L.; Soni, A.; Pinedo-Villanueva, R.; Heegaard, A.-M.; Boyce, A.M.; Javaid, M.K. Neuropathic-like Pain in Fibrous Dysplasia/McCune-Albright Syndrome. J. Clin. Endocrinol. Metab. 2022, 107, e2258–e2266. [Google Scholar] [CrossRef]
- Mercadante, S. Malignant Bone Pain: Pathophysiology and Treatment. Pain 1997, 69, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, H.; Rauch, F.; Zeitlin, L.; Munns, C.; Travers, R.; Glorieux, F.H. Effect of Pamidronate Treatment in Children with Polyostotic Fibrous Dysplasia of Bone. J. Clin. Endocrinol. Metab. 2003, 88, 4569–4575. [Google Scholar] [CrossRef]
- Hatcher, L.; Henry, N.; Hooten, W.M.; D’Souza, R.S. Intravenous Bisphosphonate Therapy as a Rescue Analgesic in Refractory Fibrous Dysplasia. Pain Med. 2022, 23, 2085–2089. [Google Scholar] [CrossRef] [PubMed]
- Trojani, M.C.; Gensburger, D.; Bagouet, F.; Cortet, B.; Couture, G.; Marcelli, C.; Mehsen Cetre, N.; Breuil, V.; Chapurlat, R. Denosumab Use in Bone Fibrous Dysplasia Refractory to Bisphosphonate: A Retrospective Multicentric Study. Bone 2023, 174, 116819. [Google Scholar] [CrossRef]
- Chapurlat, R.; Legrand, M.A. Bisphosphonates for the Treatment of Fibrous Dysplasia of Bone. Bone 2021, 143, 115784. [Google Scholar] [CrossRef] [PubMed]
- Boyce, A.M.; Kelly, M.H.; Brillante, B.A.; Kushner, H.; Wientroub, S.; Riminucci, M.; Bianco, P.; Robey, P.G.; Collins, M.T. A Randomized, Double Blind, Placebo-Controlled Trial of Alendronate Treatment for Fibrous Dysplasia of Bone. J. Clin. Endocrinol. Metab. 2014, 99, 4133–4140. [Google Scholar] [CrossRef] [PubMed]
- Chapurlat, R.D.; Hugueny, P.; Delmas, P.D.; Meunier, P.J. Treatment of Fibrous Dysplasia of Bone with Intravenous Pamidronate: Long-Term Effectiveness and Evaluation of Predictors of Response to Treatment. Bone 2004, 35, 235–242. [Google Scholar] [CrossRef]
- Tripathy, S.K.; Swaroop, S.; Velagada, S.; Priyadarshini, D.; Das, R.R.; Satpathy, A.K.; Agrawal, K. Response to Zoledronic Acid Infusion in Children with Fibrous Dysplasia. Front. Pediatr. 2020, 8, 582316. [Google Scholar] [CrossRef]
- Javaid, M.K.; Boyce, A.; Appelman-Dijkstra, N.; Ong, J.; Defabianis, P.; Offiah, A.; Arundel, P.; Shaw, N.; Pos, V.D.; Underhil, A.; et al. Best Practice Management Guidelines for Fibrous Dysplasia/McCune-Albright Syndrome: A Consensus Statement from the FD/MAS International Consortium. Orphanet J. Rare Dis. 2019, 14, 139. [Google Scholar] [CrossRef]
- Raborn, L.N.; Burke, A.B.; Ebb, D.H.; Collins, M.T.; Kaban, L.B.; Boyce, A.M. Denosumab for Craniofacial Fibrous Dysplasia: Duration of Efficacy and Post-Treatment Effects. Osteoporos. Int. 2021, 32, 1889–1893. [Google Scholar] [CrossRef]
- Palmisano, B.; Spica, E.; Remoli, C.; Labella, R.; Di Filippo, A.; Donsante, S.; Bini, F.; Raimondo, D.; Marinozzi, F.; Boyde, A.; et al. RANKL Inhibition in Fibrous Dysplasia of Bone: A Preclinical Study in a Mouse Model of the Human Disease. J. Bone Miner. Res. 2019, 34, 2171–2182. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yin, Y.; Wang, Z.; Xie, L.; Deng, P.; Wang, D.; Ji, N.; Zhao, H.; Han, X.; Chen, Q.; et al. RANKL Inhibition Halts Lesion Progression and Promotes Bone Remineralization in Mice with Fibrous Dysplasia. Bone 2022, 156, 116301. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Tang, X.; Zhang, J.; Li, M.; Xu, N.; Qi, W.; Tan, J.; Lou, X.; Yu, Z.; Sun, J.; et al. Interleukin-6-Knockdown of Chimeric Antigen Receptor-Modified T Cells Significantly Reduces IL-6 Release from Monocytes. Exp. Hematol. Oncol. 2020, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. Immunotherapeutic Implications of IL-6 Blockade for Cytokine Storm. Immunotherapy 2016, 8, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Chapurlat, R.; Gensburger, D.; Trolliet, C.; Rouanet, S.; Mehsen-Cetre, N.; Orcel, P. Inhibition of IL-6 in the Treatment of Fibrous Dysplasia of Bone: The Randomized Double-Blind Placebo-Controlled TOCIDYS Trial. Bone 2022, 157, 116343. [Google Scholar] [CrossRef]
- Sehgal, A.; Irvine, K.M.; Hume, D.A. Functions of Macrophage Colony-Stimulating Factor (CSF1) in Development, Homeostasis, and Tissue Repair. Semin. Immunol. 2021, 54, 101509. [Google Scholar] [CrossRef]
- Bush, S.J.; McCulloch, M.E.B.; Lisowski, Z.M.; Muriuki, C.; Clark, E.L.; Young, R.; Pridans, C.; Prendergast, J.G.D.; Summers, K.M.; Hume, D.A. Species-Specificity of Transcriptional Regulation and the Response to Lipopolysaccharide in Mammalian Macrophages. Front. Cell Dev. Biol. 2020, 8, 661. [Google Scholar] [CrossRef]
- Keshvari, S.; Caruso, M.; Teakle, N.; Batoon, L.; Sehgal, A.; Patkar, O.L.; Ferrari-Cestari, M.; Snell, C.E.; Chen, C.; Stevenson, A.; et al. CSF1R-Dependent Macrophages Control Postnatal Somatic Growth and Organ Maturation. PLOS Genet. 2021, 17, e1009605. [Google Scholar] [CrossRef]
- MacDonald, K.P.A.; Palmer, J.S.; Cronau, S.; Seppanen, E.; Olver, S.; Raffelt, N.C.; Kuns, R.; Pettit, A.R.; Clouston, A.; Wainwright, B.; et al. An Antibody against the Colony-Stimulating Factor 1 Receptor Depletes the Resident Subset of Monocytes and Tissue- and Tumor-Associated Macrophages but Does Not Inhibit Inflammation. Blood 2010, 116, 3955–3963. [Google Scholar] [CrossRef]
- Sauter, K.A.; Pridans, C.; Sehgal, A.; Tsai, Y.T.; Bradford, B.M.; Raza, S.; Moffat, L.; Gow, D.J.; Beard, P.M.; Mabbott, N.A.; et al. Pleiotropic Effects of Extended Blockade of CSF1R Signaling in Adult Mice. J. Leukoc. Biol. 2014, 96, 265–274. [Google Scholar] [CrossRef]
- Mantyh, P.W.; Koltzenburg, M.; Mendell, L.M.; Tive, L.; Shelton, D.L.; Warner, D.S. Antagonism of Nerve Growth Factor-TrkA Signaling and the Relief of Pain. Anesthesiology 2011, 115, 189–204. [Google Scholar] [CrossRef]
- Matsuda, H.; Kannan, Y.; Ushio, H.; Kiso, Y.; Kanemoto, T.; Suzuki, H.; Kitamura, Y. Nerve Growth Factor Induces Development of Connective Tissue-Type Mast Cells in Vitro from Murine Bone Marrow Cells. J. Exp. Med. 1991, 174, 7–14. [Google Scholar] [CrossRef]
- Skaper, S.D. Nerve Growth Factor. Mol. Neurobiol. 2001, 24, 183–200. [Google Scholar] [CrossRef]
- Rotman, M.; Hamdy, N.A.T.; Appelman-Dijkstra, N.M. Clinical and Translational Pharmacological Aspects of the Management of Fibrous Dysplasia of Bone. Br. J. Clin. Pharmacol. 2019, 85, 1169–1179. [Google Scholar] [CrossRef]
- Shelton, D.L.; Zeller, J.; Ho, W.-H.; Pons, J.; Rosenthal, A. Nerve Growth Factor Mediates Hyperalgesia and Cachexia in Auto-Immune Arthritis. Pain 2005, 116, 8–16. [Google Scholar] [CrossRef]
- Bishop, T.; Hewson, D.W.; Yip, P.K.; Fahey, M.S.; Dawbarn, D.; Young, A.R.; McMahon, S.B. Characterisation of Ultraviolet-B-Induced Inflammation as a Model of Hyperalgesia in the Rat. Pain 2007, 131, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Sevcik, M.A.; Ghilardi, J.R.; Peters, C.M.; Lindsay, T.H.; Halvorson, K.G.; Jonas, B.M.; Kubota, K.; Kuskowski, M.A.; Boustany, L.; Shelton, D.L.; et al. Anti-NGF Therapy Profoundly Reduces Bone Cancer Pain and the Accompanying Increase in Markers of Peripheral and Central Sensitization. Pain 2005, 115, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Lane, N.E.; Corr, M. Anti-NGF Treatments for Pain—Two Steps Forward, One Step Back? Nat. Rev. Rheumatol. 2017, 13, 76–78. [Google Scholar] [CrossRef]
- Park, C.W.; Kim, B.J.; Lee, Y.W.; Won, C.; Park, C.O.; Chung, B.Y.; Lee, D.H.; Jung, K.; Nam, H.-J.; Choi, G.; et al. Asivatrep, a TRPV1 Antagonist, for the Topical Treatment of Atopic Dermatitis: Phase 3, Randomized, Vehicle-Controlled Study (CAPTAIN-AD). J. Allergy Clin. Immunol. 2022, 149, 1340–1347.e4. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, C.; Chiang, C.; Xiao, T.; Chen, Y.; Zhao, Y.; Zheng, D. The Impact of TRPV1 on Cancer Pathogenesis and Therapy: A Systematic Review. Int. J. Biol. Sci. 2021, 17, 2034–2049. [Google Scholar] [CrossRef] [PubMed]
- Späth, E.; Darling, S.F. Synthese Des Capsaicins. Berichte Dtsch. Chem. Ges. B Ser. 1930, 63, 737–743. [Google Scholar] [CrossRef]
- Cordell, G.A.; Araujo, O.E. Capsaicin: Identification, Nomenclature, and Pharmacotherapy. Ann. Pharmacother. 1993, 27, 330–336. [Google Scholar] [CrossRef]
- Koivisto, A.-P.; Belvisi, M.G.; Gaudet, R.; Szallasi, A. Advances in TRP Channel Drug Discovery: From Target Validation to Clinical Studies. Nat. Rev. Drug Discov. 2022, 21, 41–59. [Google Scholar] [CrossRef] [PubMed]
- Jurczak, A.; Delay, L.; Barbier, J.; Simon, N.; Krock, E.; Sandor, K.; Agalave, N.M.; Rudjito, R.; Wigerblad, G.; Rogóż, K.; et al. Antibody-Induced Pain-like Behavior and Bone Erosion: Links to Subclinical Inflammation, Osteoclast Activity, and Acid-Sensing Ion Channel 3-Dependent Sensitization. Pain 2022, 163, 1542–1559. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Chen, F. Acid-Sensing Ion Channel-1a in Articular Chondrocytes and Synovial Fibroblasts: A Novel Therapeutic Target for Rheumatoid Arthritis. Front. Immunol. 2021, 11, 580936. [Google Scholar] [CrossRef]
- Chung, W.-C.; Kermode, J.C. Suramin Disrupts Receptor-G Protein Coupling by Blocking Association of G Protein α and Βγ Subunits. J. Pharmacol. Exp. Ther. 2005, 313, 191–198. [Google Scholar] [CrossRef]
- Freissmuth, M.; Waldhoer, M.; Bofill-Cardona, E.; Nanoff, C. G Protein Antagonists. Trends Pharmacol. Sci. 1999, 20, 237–245. [Google Scholar] [CrossRef]
- Hohenegger, M.; Waldhoer, M.; Beindl, W.; Böing, B.; Kreimeyer, A.; Nickel, P.; Nanoff, C.; Freissmuth, M. Gsα-Selective G Protein Antagonists. Proc. Natl. Acad. Sci. USA 1998, 95, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Beindl, W.; Mitterauer, T.; Hohenegger, M.; Ijzerman, A.P.; Nanoff, C.; Freissmuth, M. Inhibition of Receptor/G Protein Coupling by Suramin Analogues. Mol. Pharmacol. 1996, 50, 415–423. [Google Scholar] [PubMed]
- Kawabata, H.; Ono, Y.; Tamamura, N.; Oyama, K.; Ueda, J.; Sato, H.; Takahashi, K.; Taniue, K.; Okada, T.; Fujibayashi, S.; et al. Mutant GNAS Limits Tumor Aggressiveness in Established Pancreatic Cancer via Antagonizing the KRAS-Pathway. J. Gastroenterol. 2022, 57, 208–220. [Google Scholar] [CrossRef]
- Ding, H.; Zhang, X.; Su, Y.; Jia, C.; Dai, C. GNAS Promotes Inflammation-Related Hepatocellular Carcinoma Progression by Promoting STAT3 Activation. Cell. Mol. Biol. Lett. 2020, 25, 8. [Google Scholar] [CrossRef]
- Lung, H.; Hsiao, E.C.; Wentworth, K.L. Advances in Models of Fibrous Dysplasia/McCune-Albright Syndrome. Front. Endocrinol. 2020, 10, 925. [Google Scholar] [CrossRef]
- Saggio, I. Perils and Promises of Therapeutic Approaches for the Stem Cell Disease Fibrous Dysplasia. Stem Cells Transl. Med. 2019, 8, 110–111. [Google Scholar] [CrossRef] [PubMed]
- Kantor, A.; McClements, M.; MacLaren, R. CRISPR-Cas9 DNA Base-Editing and Prime-Editing. Int. J. Mol. Sci. 2020, 21, 6240. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, Y.; Tamai, K.; Ito, H. Oral Alendronate Treatment for Polyostotic Fibrous Dysplasia: A Case Report. J. Orthop. Sci. 2004, 9, 521–525. [Google Scholar] [CrossRef]
- Jayaraman, M.; Karikumar, K.; Verma, A.; Modi, K.D. Alendronate Therapy in Polyostotic Fibrous Dysplasia Presenting with Pathologic Fracture. Am. J. Orthop. Belle Mead NJ 2011, 40, E48-50. [Google Scholar] [PubMed]
- Liens, D.; Delmas, P.D.; Meunier, P. Long-Term Effects of Intravenous Pamidronate in Fibrous Dysplasia of Bone. Lancet 1994, 343, 953–954. [Google Scholar] [CrossRef] [PubMed]
- Lala, R.; Matarazzo, P.; Bertelloni, S.; Buzi, F.; Rigon, F.; De Sanctis, C. Pamidronate Treatment of Bone Fibrous Dysplasia in Nine Children with McCune-Albright Syndrome. Acta Paediatr. 2000, 89, 188–193. [Google Scholar] [CrossRef]
- Matarazzo, P.; Lala, R.; Masi, G.; Andreo, M.; Altare, F.; de Sanctis, C. Pamidronate Treatment in Bone Fibrous Dysplasia in Children and Adolescents with McCune-Albright Syndrome. J. Pediatr. Endocrinol. Metab. JPEM 2002, 15 (Suppl. S3), 929–937. [Google Scholar]
- Wang, Y.; Wang, O.; Jiang, Y.; Li, M.; Xia, W.; Meng, X.; Xing, X. Efficacy and Safety of Bisphosphonate Therapy in Mccune-Albright Syndrome–Related Polyostotic Fibrous Dysplasia: A Single-Center Experience. Endocr. Pract. 2019, 25, 23–30. [Google Scholar] [CrossRef]
- Valadares, L.P.; Ferreira, B.S.D.A.; Cunha, B.M.D.; Moreira, L.A.; Batista, F.G.A.; Hottz, C.D.F.; Magalhães, G.G.R. Effects of Zoledronic Acid Therapy in Fibrous Dysplasia of Bone: A Single-Center Experience. Arch. Endocrinol. Metab. 2022, 66, 247–255. [Google Scholar] [CrossRef]
- Meier, M.E.; Van Der Bruggen, W.; Van De Sande, M.A.J.; Appelman-Dijkstra, N.M. Regression of Fibrous Dysplasia in Response to Denosumab Therapy: A Report of Two Cases. Bone Rep. 2021, 14, 101058. [Google Scholar] [CrossRef] [PubMed]
- De Castro, L.F.; Michel, Z.; Pan, K.; Taylor, J.; Szymczuk, V.; Paravastu, S.; Saboury, B.; Papadakis, G.Z.; Li, X.; Milligan, K.; et al. Safety and Efficacy of Denosumab for Fibrous Dysplasia of Bone. N. Engl. J. Med. 2023, 388, 766–768. [Google Scholar] [CrossRef] [PubMed]
- de Boysson, H.; Johnson, A.; Hablani, N.; Hajlaoui, W.; Auzary, C.; Geffray, L. Tocilizumab in the Treatment of a Polyostotic Variant of Fibrous Dysplasia of Bone. Rheumatol. Oxf. Engl. 2015, 54, 1747–1749. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Wang, S.; Guo, R.; Liu, D. CSF1R Inhibitors Are Emerging Immunotherapeutic Drugs for Cancer Treatment. Eur. J. Med. Chem. 2023, 245, 114884. [Google Scholar] [CrossRef] [PubMed]
- Fallon, M.; Sopata, M.; Dragon, E.; Brown, M.T.; Viktrup, L.; West, C.R.; Bao, W.; Agyemang, A. A Randomized Placebo-Controlled Trial of the Anti-Nerve Growth Factor Antibody Tanezumab in Subjects with Cancer Pain Due to Bone Metastasis. Oncologist 2023, XX, 1–11. [Google Scholar] [CrossRef]
- Manitpisitkul, P.; Flores, C.M.; Moyer, J.A.; Romano, G.; Shalayda, K.; Tatikola, K.; Hutchison, J.S.; Mayorga, A.J. A Multiple-Dose Double-Blind Randomized Study to Evaluate the Safety, Pharmacokinetics, Pharmacodynamics and Analgesic Efficacy of the TRPV1 Antagonist JNJ-39439335 (Mavatrep). Scand. J. Pain 2018, 18, 151–164. [Google Scholar] [CrossRef]
- Lv, M.; Li, X.; Huang, Y.; Wang, N.; Zhu, X.; Sun, J. Inhibition of Fibrous Dysplasia via Blocking Gsα with Suramin Sodium Loaded with an Alendronate-Conjugated Polymeric Drug Delivery System. Biomater. Sci. 2016, 4, 1113–1122. [Google Scholar] [CrossRef]
- Watanabe, K.; Nakamura, T.; Onodera, S.; Saito, A.; Shibahara, T.; Azuma, T. A Novel GNAS-Mutated Human Induced Pluripotent Stem Cell Model for Understanding GNAS-Mutated Tumors. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2020, 42, 1010428320962588. [Google Scholar] [CrossRef]


| DNA Nucleotide Modification | Predicted Protein Modification | Reference Sequences |
|---|---|---|
| c.601C>T | p.Arg201Cys (R201C) | NM_000516.7 NP_000507.1 |
| c.601C>G | p.Arg201Gly (R201G) | |
| c.601C>A | p.Arg201Ser (R201S) | |
| c.602G>A | p.Arg201His (R201H) | |
| c.602G>C | p.Arg201Pro (R201P) | |
| c.602G>T | p.Arg201Leu (R201L) | |
| c.679C>A | p.Gln227Lys (Q227K) | |
| c.680A>T | p.Gln227Leu (Q227L) | |
| c.680A>G | p.Gln227Arg (Q227R) | |
| c.681G>T | p.Gln227His (Q227H) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.-Y.; Shim, J.-H.; Heo, C.-Y. A Rare Skeletal Disorder, Fibrous Dysplasia: A Review of Its Pathogenesis and Therapeutic Prospects. Int. J. Mol. Sci. 2023, 24, 15591. https://doi.org/10.3390/ijms242115591
Kim H-Y, Shim J-H, Heo C-Y. A Rare Skeletal Disorder, Fibrous Dysplasia: A Review of Its Pathogenesis and Therapeutic Prospects. International Journal of Molecular Sciences. 2023; 24(21):15591. https://doi.org/10.3390/ijms242115591
Chicago/Turabian StyleKim, Ha-Young, Jung-Hee Shim, and Chan-Yeong Heo. 2023. "A Rare Skeletal Disorder, Fibrous Dysplasia: A Review of Its Pathogenesis and Therapeutic Prospects" International Journal of Molecular Sciences 24, no. 21: 15591. https://doi.org/10.3390/ijms242115591
APA StyleKim, H. -Y., Shim, J. -H., & Heo, C. -Y. (2023). A Rare Skeletal Disorder, Fibrous Dysplasia: A Review of Its Pathogenesis and Therapeutic Prospects. International Journal of Molecular Sciences, 24(21), 15591. https://doi.org/10.3390/ijms242115591