1. Introduction
Multipotent mesenchymal stem/stromal cells (MSCs) are found in almost all connective tissues of adult organisms [
1]. Their ability for self-renewal and differentiation, as well as the secretion of cytokines and signaling hormones in particular, with angiogenic and anti-apoptotic effects, makes them promising tools for biomedical applications [
2].
The use of monolayer cultures, including MSCs, has become widespread as a model that makes it easy to manipulate the state of these cells by adding various agents to the culture medium and studying the evoked cellular responses. A monolayer can be considered a two-dimensional (or 2D) system in which each cell has the same access to nutrients and signaling molecules, which ensures uniform behavior of the entire cell population during the experiment. However, the cells in tissues, depending on their position in the three-dimensional (3D) structure characterized by natural gradients of oxygen, signaling molecules, nutrients, and metabolic products, have unequal access to these vital substances, which affects the intracellular processes in individual cells. Obviously, a 3D configuration is much more physiological, and 3D culture systems are actively introduced into experimental practice in order to mimic natural conditions. It should be emphasized that studies of this kind are mainly devoted to spheroids obtained from tumor cells, while the behavior of MSC-derived spheroids is analyzed to a significantly lesser extent. Suffice it to say that more than 21 thousand publications on tumor spheroids and less than 400 on those obtained from MSCs are cited in PubMed up to now. However, recent studies show (see [
3] for a review) that 3D MSCs demonstrate increased stemness, regenerative potential, and other benefits compared to those cultured in 2D. Thus, understanding these differences is extremely important for both fundamental research and practical applications such as regenerative and reparative medicine.
One of the key factors affecting cellular metabolism is the level of available oxygen, since O
2 is a basic substrate for mitochondrial ATP production and numerous intracellular redox reactions also taking place both in the cytosol and in membrane compartments such as lysosomes. It is important to note that the metabolic environment of the niche in which MSCs usually grow is characterized by low oxygen tension [
4], therefore cells cultured under such conditions exhibited increased proliferation, migration, and angiogenesis, and decreased senescence and apoptosis [
5,
6,
7]. It has been shown [
8] that cell populations derived from MSCs demonstrate higher proliferative activity when cultivated at low oxygen tension (2 to 5% O
2, corresponding to 15 to 38 mmHg), compared to normal oxygen tension (21% O
2, corresponding to 160 mmHg, according to [
9]). In general, the cells in tissue layers more distant from vessels normally exist under conditions of moderate, or physiological, hypoxia. There are also reports on inherent hypoxia in some tumors [
10], and distinguishing of physiological levels of hypoxia from those typical for pathologies can be of great value in identifying certain diseases.
Another very significant factor in the normal activity of cells of all types is the level of acidification of the endolysosomal apparatus, provided by the action of the vesicular proton pump V
0/V
1 [
11,
12]. It is important to note that MSC senescence and various pathologies are characterized by changes in both endolysosome acidification [
13,
14] and ROS production in lysosomes [
15]; however, understanding of links between these two factors is still obscure. Obviously, knowledge of the actual levels and local microgradients of O
2 and pH at the cellular level and their precise control in vitro and in vivo in complex 2D and 3D cell models is currently an urgent task in biology and medicine. Consequently, appropriate tools are needed to solve it, and the development and characterization of various biosensors is a hot spot of modern biomedical research.
In this context, the sensors allowing simultaneous determination of several parameters, for example, pH level and oxygen pressure, in the same intracellular structures are of particular interest. Phosphorescent cyclometallated Ir(III) complexes are a convenient basis for creating highly sensitive biolabels and sensors that visualize the O
2 level in tissues [
16,
17,
18]. It is also worth noting that cyclometallated Ir(III) complexes usually remain stable under exposure to photoexcitation that makes them suitable candidates for prolonged biological experiments. The probe response in the imaging and sensing measurements with the phosphorescent Ir(III) complexes can be quantified in two modes, namely either by using a ratiometric approach [
19] or by phosphorescence lifetime imaging (PLIM) [
20]. However, the latter has some evident advantages due to the independence of the probe signal from its concentration and optical properties of the sample under study, thus giving high-resolution images at cellular level and providing accurate information about the microenvironment along with real-time changes in O
2 levels. As for the estimation of media acidity, numerous fluorescent organic probes are commercially available, among which fluorescein is widely used because its intensity decreases with increasing acidity of the microenvironment [
21].
Despite the fact that dual pH/O
2 sensors based on ratiometric sensing mode are well-known [
22,
23,
24], the development of their lifetime-based analogs, which are more promising due to higher measurements reliability, appeared to be more challenging [
25]. Recently, we have developed the dual luminescence pH/O
2 lifetime sensor (Ir-HSA-FITC) based on the covalent conjugation of fluorescein in the form of fluorescein isothiocyanate (FITC, fluorescent pH sensor) and an orthometalated iridium complex (phosphorescent O
2 sensor) to human serum albumin (HSA) [
26]. The emission and excitation spectra of the dual sensor represent a linear combination of the corresponding spectroscopic patterns of individual chromophores (fluorescein and iridium complex), while overlapping excitation spectra allow pumping of both emitters at 405 nm. The location of the sensor emission components (fluorescein fluorescence centered at 520 nm and structured phosphorescence of Ir band at 580–720 nm) provides independent acquisition of the fluorescence and phosphorescence decays in the corresponding spectral windows. Moreover, phosphorescence signal is also separated from that of fluorescence in “time domain” mode of lifetime measurements that additionally increase sensor selectivity to the target parameters. The dual sensor features mentioned above make possible application of the advanced lifetime microscopy technique, namely the combined FLIM/PLIM mode of the data acquisition, which allows simultaneous spatially co-localized recording of the fluorescence and phosphorescence lifetimes in one pulse sequence [
26].
Linking these two chromophores to HSA increases the overall size of the sensor to the value of ca. 8 nm (i.e., to the size of HSA) that, in turn, makes endocytosis the only route for its entry into the cells [
27,
28]. Theoretically, in this case, the dual sensor will be localized preferentially in endosomes and lysosomes that allow avoiding a high fluorescence background of cytoplasmically located sensor, and makes possible detection of pH and O
2 levels and their external condition-generated changes in the same endolysosomal structures. In the case of intracellular measurements in combined FLIM/PLIM mode, the independence of the fluorophore and phosphor lifetime from the probe concentration becomes quite beneficial, since the number of sensor molecules can vary greatly in individual endosomes and lysosomes and can differ at different stages of endocytosis.
However, practical implementation of any sensor needs detailed analysis of possible limitations of its application and optimization of experimental protocols for both cultured cell models and tissues of living organisms to avoid misinterpretations of obtained data. The supposed limitations of the dual sensor may be related to two main points. First, the cells possess various endogenous physiologically relevant molecules (such as FAD [
29], or NAD(P)H [
30]) responsible for so called autofluorescence that can also be recorded by FLIM technique, and its level can compromise measurements of exogenously added fluorophores. Second, endolysosomal system organization is dependent of the cell type which can be different in the number, size and localization of endolysosomes, as well as concentration of fluorophore inside them.
In this article, we have analyzed in detail the possibility of application of the internalizable Ir-HSA-FITC dual sensor for the simultaneous detection of the acidification level and oxygen concentration in human endometrial MSC (enMSC) endolysosomes in a monolayer and in spheroids as the models of 2D- and 3D-cell growth. Endolysosomes in enMSCs are smaller and their number is higher than in the cells like CHO and A549 and are characterized by higher level of autofluorescence in green region of spectrum. Using monolayer enMSCs and enMSC spheroids models, we have performed the experiments aimed at elucidating cellular uptake, intracellular distribution, and excretion of Ir-HSA-FITC probe and possibility to monitor the level of pH and O2 consumption by using several approaches such as confocal microscopy, flow cytometry, and a combined FLIM/PLIM technique. We have found that high autofluorescence level compromise FLIM data, but accumulation of FITC component is perfectly recorded by cytofluorometry. We believe that autofluorescense level must be taken into account as important parameter that can influence interpretation of acquired data. At the same time, PLIM experiments make possible monitoring of fast variations in O2 level both in monolayer cells and in spheroids at a spheroid depth greater than that allowed by confocal microscopy. Our findings are critical to understanding what dual sensor design improvements are needed to achieve a more versatile product.
3. Discussion
MSCs are currently considered promising candidates for cell therapy and regenerative medicine, and one of the main areas of research is the development of tools that allow monitoring of such key parameters of cellular homeostasis as oxygen consumption and lysosomal acidification. These two processes may be interrelated, since MSC senescence is accompanied by a decrease in lysosome acidification which, in turn, leads to an increase in reactive oxygen species production and a number of other consequences [
14]. Dysfunction in both acidification and oxygen consumption is also observed in neoplastic processes and neurodegenerative diseases [
10,
42].
It was established that MSCs function in tissues under conditions of hypoxia, but the exact hypoxia level can determine cell survival or death. The control of the oxygen level and its changes, as well as the level of acidification of lysosomes in tissue cells, is currently an extremely urgent task, which has stimulated numerous studies devoted to the development and verification of various biosensors aimed at the measurement of oxygen concentration and pH in biological systems. Phosphorescence of complexes based on iridium, ruthenium and platinum, which is quenched by oxygen, makes these compounds excellent oxygen sensors [
20,
43,
44]. Various compounds based on fluorescein, cyanine and other fluorophores, as well as nanoparticles, are used as pH sensors [
45,
46].
In this work, we used a dual pH/O
2 sensor, Ir-HSA-FITC, in which the pH-sensitive FITC is combined with the O
2-sensitive iridium complex through conjugation of both chromophores to HSA [
26]. The presence of a large protein component determines the uptake of this complex only by endocytosis and, in principle, makes it possible to simultaneously determine the level of pH and oxygen in vesicles of the endolysosomal system of the cell which, in itself, is an interesting scientific problem. Although there is an evidence of a clathrin-dependent receptor-mediated pathway for albumin internalization [
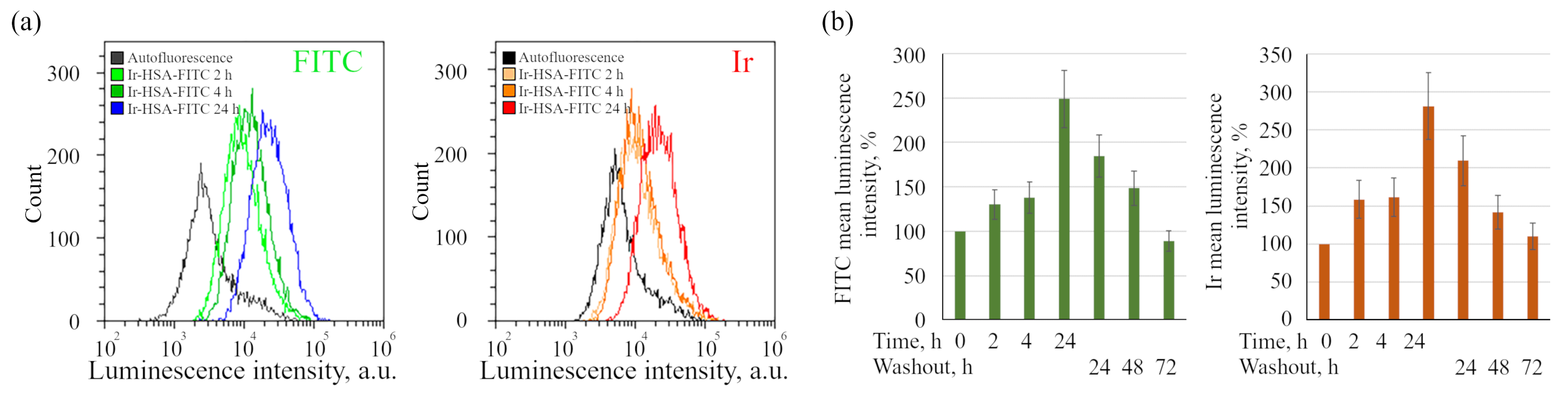
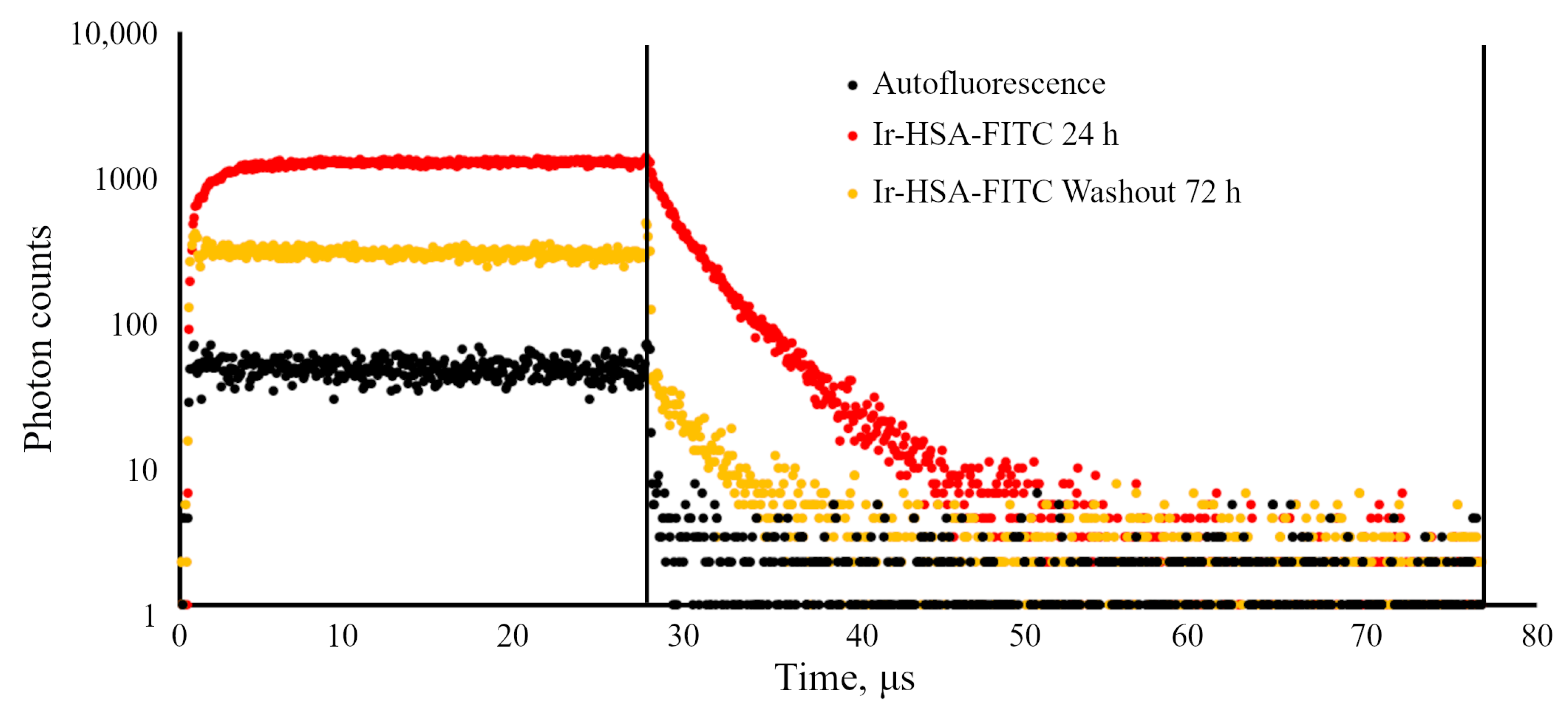
47], the dynamics of sensor accumulation in cells, determined using the flow cytometry method (
Figure 3), suggests that the sensor behaves like a fluid-phase marker, which is characterized by slow entry through all endocytic portals followed by accumulation in late endosomes and lysosomes. Indeed, it should be taken into account that the internalization of albumin through binding to proteins of the scavenger receptors family is typically restricted to the cells of the immune system [
28,
31].
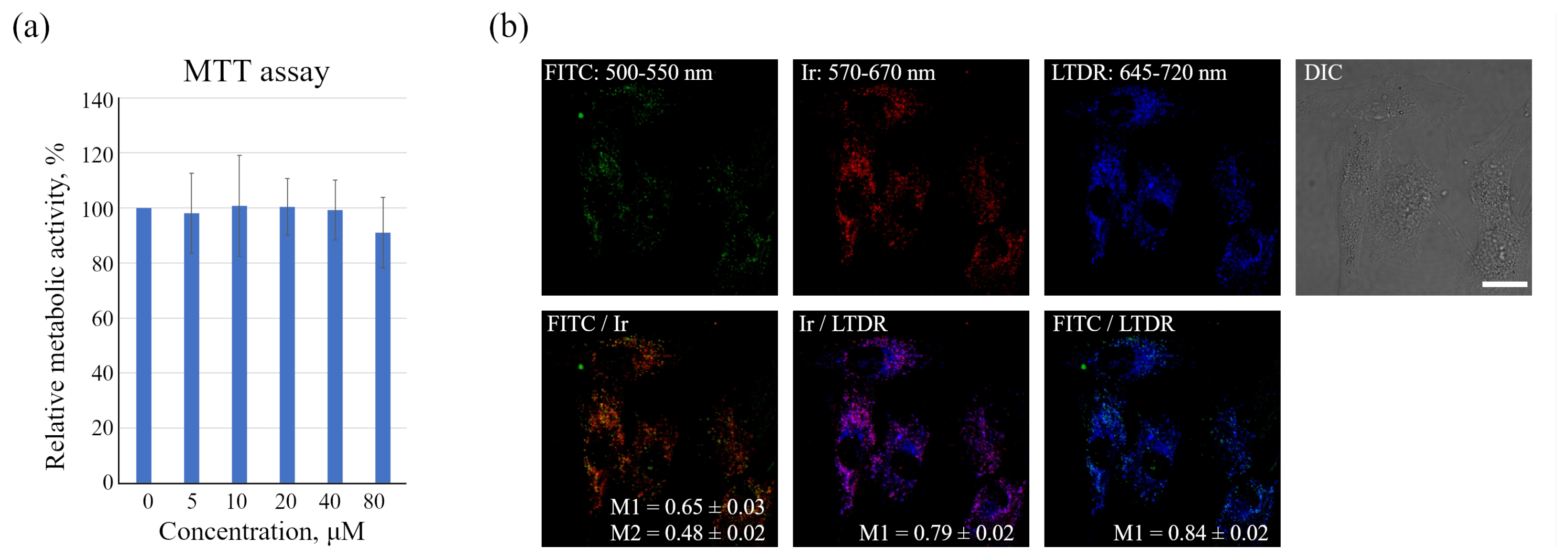
The data obtained, however, show that in monolayer enMSCs, the FITC signal is not clearly detected by confocal microscopy and makes single vesicle analysis impossible in this type of cell. In our opinion, there may be several reasons for this observation: firstly, enMSCs have a very high level of autofluorescence in the range of FITC signal emission, and secondly, the endolysosomal apparatus of enMSCs is represented by a very large number of small vesicles distributed evenly throughout the cytoplasm of highly spread out cells. This indicates that, in particular, in enMSCs fusions of endosomes lead to an insignificant concentration of cargo, in comparison with the cells of HeLa or A549 type in which a strong concentration of cargo occurs in the early stages of endocytosis. The sensor internalized by enMSCs enters endosomes through all endocytic portals, but the low concentration in early endosomes with a pH of 6.8–6.0 makes it impossible to detect the signal by confocal microscopy. Within 24 h, when the sensor reaches lysosomes and concentrates there, the pH level typical for these compartments (5.5–4.5) leads to partial quenching of FITC fluorescence that was confirmed by the data (
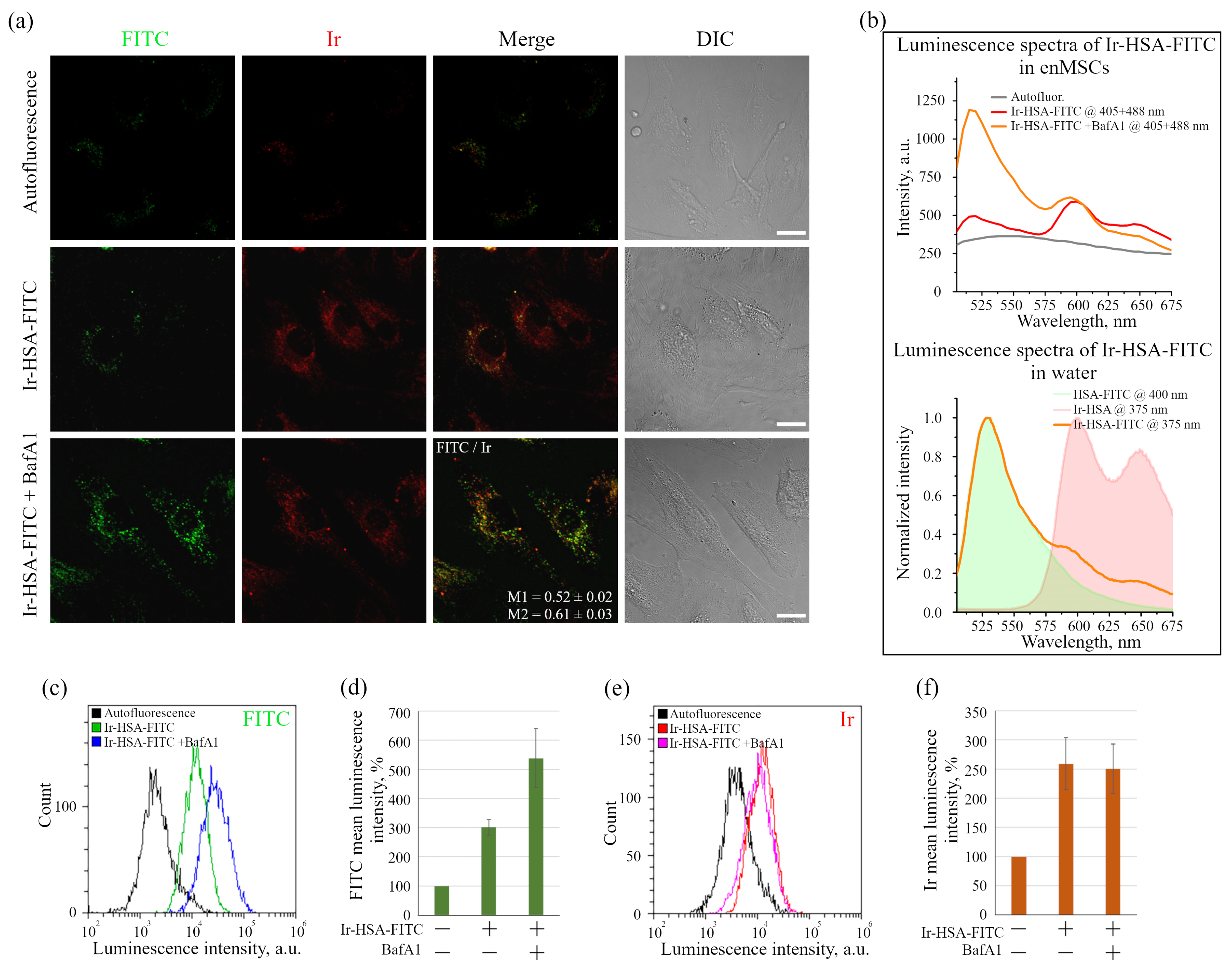
Figure 1b) on the co-localization of sensor chromophores (M1 = 0.65 ± 0.03 and M2 = 0.48 ± 0.02). It is interesting to note that in the presence of BafA1, which abolishes acidification of endosomes and thus leads to the increase in the fluorescence intensity of quenched FITC molecules, a much larger number of bright vesicles with the corresponding FITC signals was detected. However, even in this case, the co-localization of FITC and Ir signals is incomplete with Manders’ coefficients comparable to the above-mentioned values (compare
Figure 1b and
Figure 2a). We assume that this effect is associated with the degradation of HSA in lysosomes that could result in separation of the FITC and Ir chromophores to give independent localization of the pH and oxygen sensors. Since lysosomes are able to exchange contents during multiple fusions and fissions of endolysosomes, the FITC:Ir ratio, which was initially close to 1:1, can be significantly violated.
However, both chromophores remain in lysosomes. Firstly, the most expected cleavage products containing emitters are their conjugates with short peptides or single amino acids, mainly lysine, since this amino acid is the most abundant source of primary amino groups, the target conjugation functions for both FITC and Ir complex [
48]. We suppose that the complete detachment of emitters from peptides or amino acids is unlikely due to their much lower susceptibility to cleavage by various lysosomal hydrolytic enzymes. Linking of the emitters to one or several amino acid residues makes them rather hydrophilic and, therefore, can suppress their lysosomal escape. Second, FITC is known as hydrophilic and thus as a membrane non-permeable compound. As a result, we have found rather high co-localization of both emitters with LysoTracker Deep Red (LTDR), with Manders’ co-localization coefficients (M1) of 0.84 ± 0.02 and 0.79 ± 0.02 for FITC and Ir complex, correspondingly (
Figure 1b), even at 24 h incubation. Additionally, we have not observed the emission of Ir complex inside the cytoplasm, which also corroborates our conclusion regarding the primary localization of both emitters inside lysosomes.
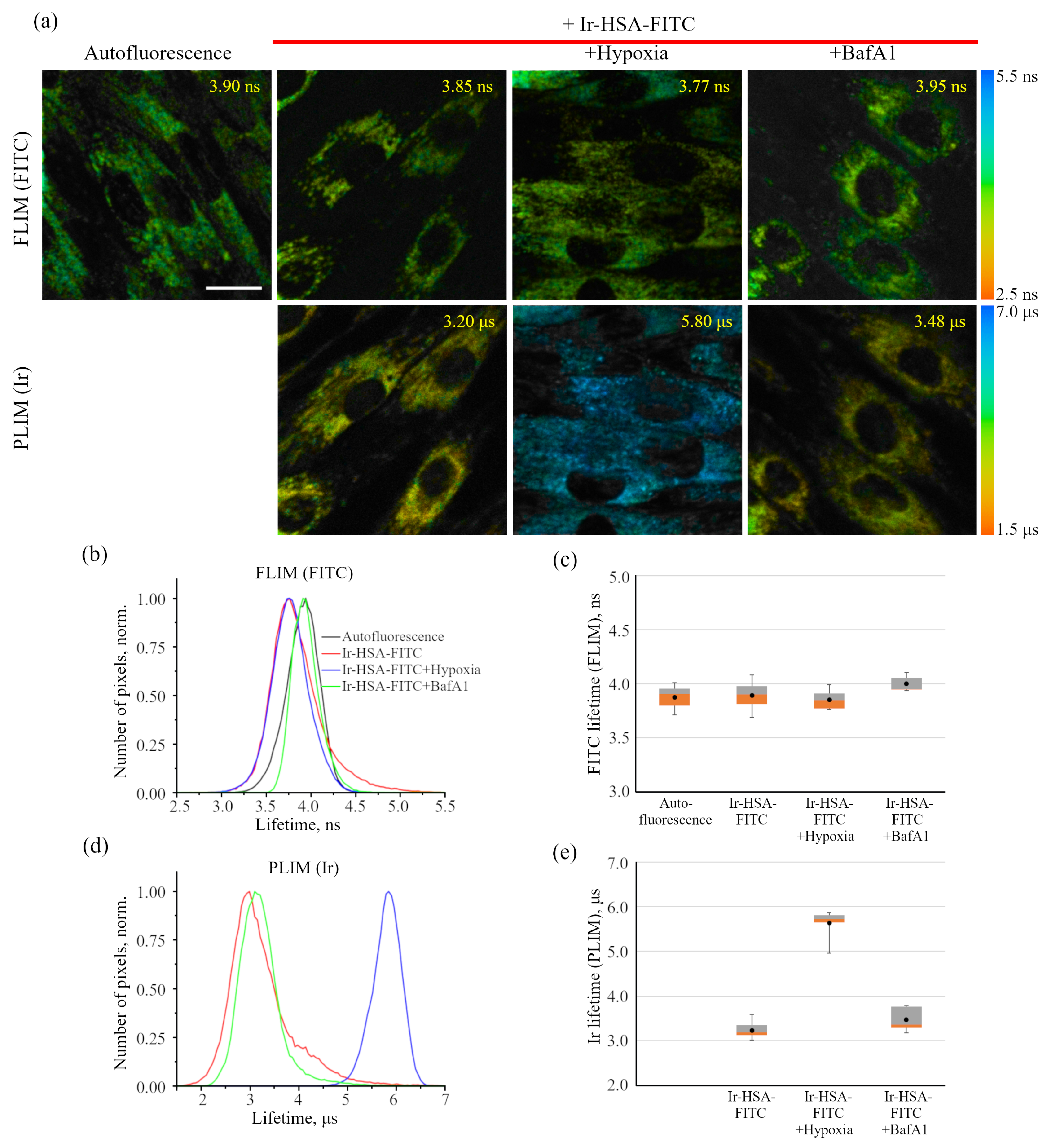
As was stated above, it was not possible to use Ir-HSA-FITC as a FLIM pH sensor in enMSCs due to high autofluorescence typical for these cells (
Figure 4a and
Figure 8). Nevertheless, this conjugate contains two independent emitters and, thus, it could be used as a ratiometric probe like was recently demonstrated for another pH-sensor based on albumin [
49]. Unfortunately, this possibility also seems unreliable because of two main reasons mentioned above: (i) the Ir-HSA-FITC probe can be (at least partially) cleaved in lysosomes, and (ii) the Ir:FITC ratio can be significantly deviate from the starting 1:1 value due to numerous fusions and fissions of endolysosomal vesicles. To avoid this drawback in future, further research should be aimed at the development of constructions stable towards lysosomal degradation.
It is important to stress that flow cytometry approach seems to be more sensitive than confocal microscopy, but can result in misinterpretations due to the fact that monolayer cells in this case must be transferred into suspension before analysis, which can lead to rapid changes in both endolysosomal acidification and, what is more realistic, in the redox status of cells. We have found here that Ir chromophore lifetime reacts rather fast onto the changes in oxygen availability, even when the emitter is localized in lysosomes. Nevertheless, this approach seems to be the most adequate for determining the dynamics of changes in the amount of internalized sensors per cell by fluorescence intensity estimations.
On the other hand, there could be another way to quantitatively detect the FITC signal in individual endosomes by using as a carrier the protein compounds, which are able to enter the cells itself or through interaction with some intermediate via highly efficient receptor-mediated endocytosis. Streptavidin, capable of binding biotinylated ligands or antibodies to efficiently internalized cytokine receptors, and similar high affinity systems, can be imagined as a more promising alternative for HSA. Such carriers are rapidly concentrated in endosomes as early as 5–15 min after internalization due to active homotypic fusions [
50,
51], while the pH in such endosomes does not yet fall below 6.0, which would give a stronger FITC emission and higher signal-to-noise ratio.
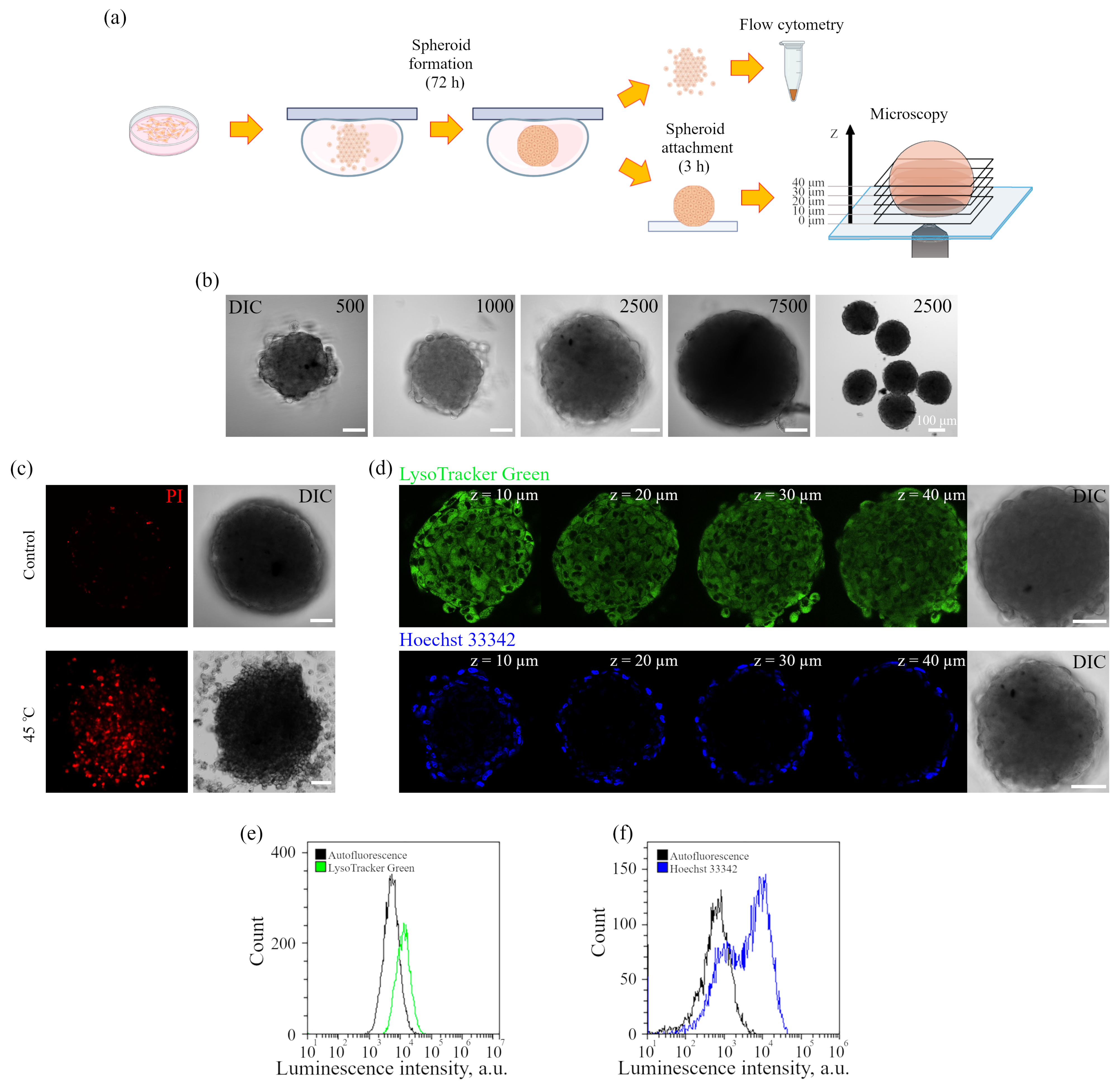
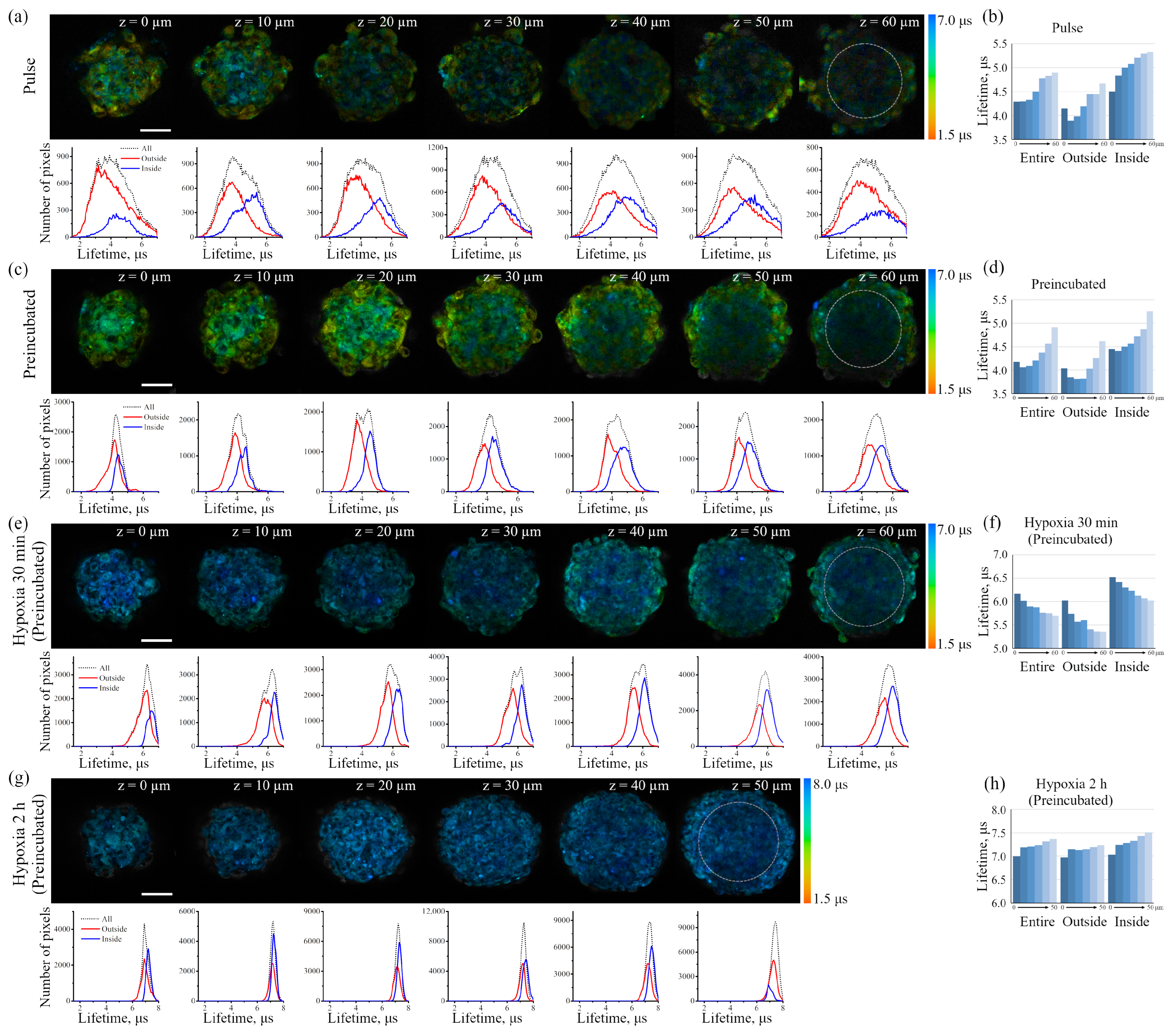
The iridium component of the sensor has demonstrated good sensitivity to hypoxia in both cell monolayers and spheroids loaded with the sensor according to both protocols (see above). In contrast to FITC, the Ir luminescence intensity, which is not very strong by itself, remains easily detectable and strongly different from background fluorescence, giving substantial variations in its lifetime (from 3–4 μs to 6–7 μs) upon changes from normoxia to hypoxia, respectively. Such a wide range of changes makes it possible to detect the effects of hypoxia with a high degree of reliability. However, given the peculiarities of the endolysosomal apparatus of enMSCs, segmentation of PLIM images with a large number of small vesicles is a significant obstacle for single vesicle analysis, so we averaged the data per cell. The obtained spheroids contain about 2500 cells each, and display an average diameter of about 200 μm (
Figure 6), which made it possible to avoid cell death in the internal space, which is believed to be characteristic of spheroids with a diameter of more than 500 μm [
37,
38,
39]. At the same time, such spheroids demonstrate cell heterogeneity and metabolic gradients of oxygen, glucose and ATP, which appear in spheroids only when they are larger than 150 μm. The obtained data show that the spheroids do not have a necrotic nucleus and all the cells in them are alive (
Figure 6c and
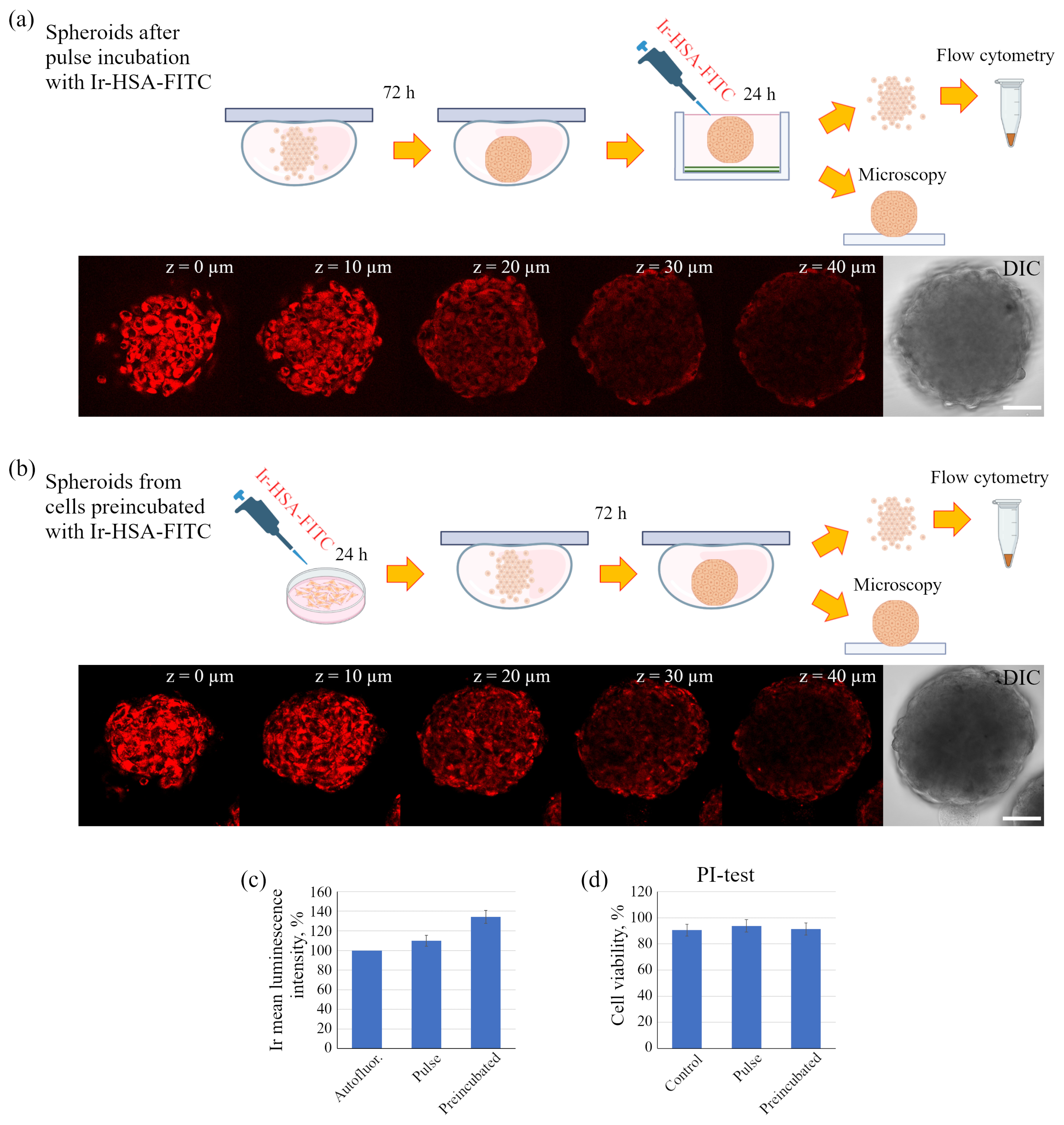
Figure 7d), and substances such as PI or LysoTracker Green freely penetrate the inner layers and are fairly evenly distributed among all cells. Since the sensor has a rather large size due to albumin, one would expect difficulties in its penetration into the inner layers of spheroid cells in the pulse staining protocol. A comparison of the distribution of the Ir signal in spheroids, all cells of which were pre-loaded by the Ir-HSA-FITC and hence contained the same amount of the sensor, with those that were incubated with the sensor after their formation, showed that the sensor penetrates the cells of all layers; however, in the second case, the PLIM signal was more heterogeneous, with wide lifetime distribution peaks. Nevertheless, the median values coincided with those in the preincubated cells (see the histograms under the images of the corresponding cells in
Figure 9a,c). It is important to take into account that the pulse staining approach is much more compatible with various practical applications, e.g., for the analysis of the oxygen level in tissues, where it could give reliable results.
However, for in vitro experiments, it is preferable to use a protocol with preincubation. It should also be emphasized that the fluorescent signal of cells in the inner layers of the spheroid is practically not detected at depths of more than 40 μm due to light absorption/scattering, while the PLIM signal is reliably recorded up to 60 μm due to higher sensitivity of the corresponding detectors. It also should be taken into account that, in contrast to luminescence intensity, PLIM data are more reliable due to lifetime independence of the sensor concentration. The advantage of phosphorescence lifetime visualization was directly demonstrated in a detailed comparison of the two approaches performed on spheroids from the Colon26 mouse colon carcinoma cell line using a platinum-based O
2 sensor [
52].
Data on the accessibility of the inner layers of the spheroid to small molecules support the idea that a small molecule such as oxygen should also be distributed evenly in the depth of the spheroids; however, O
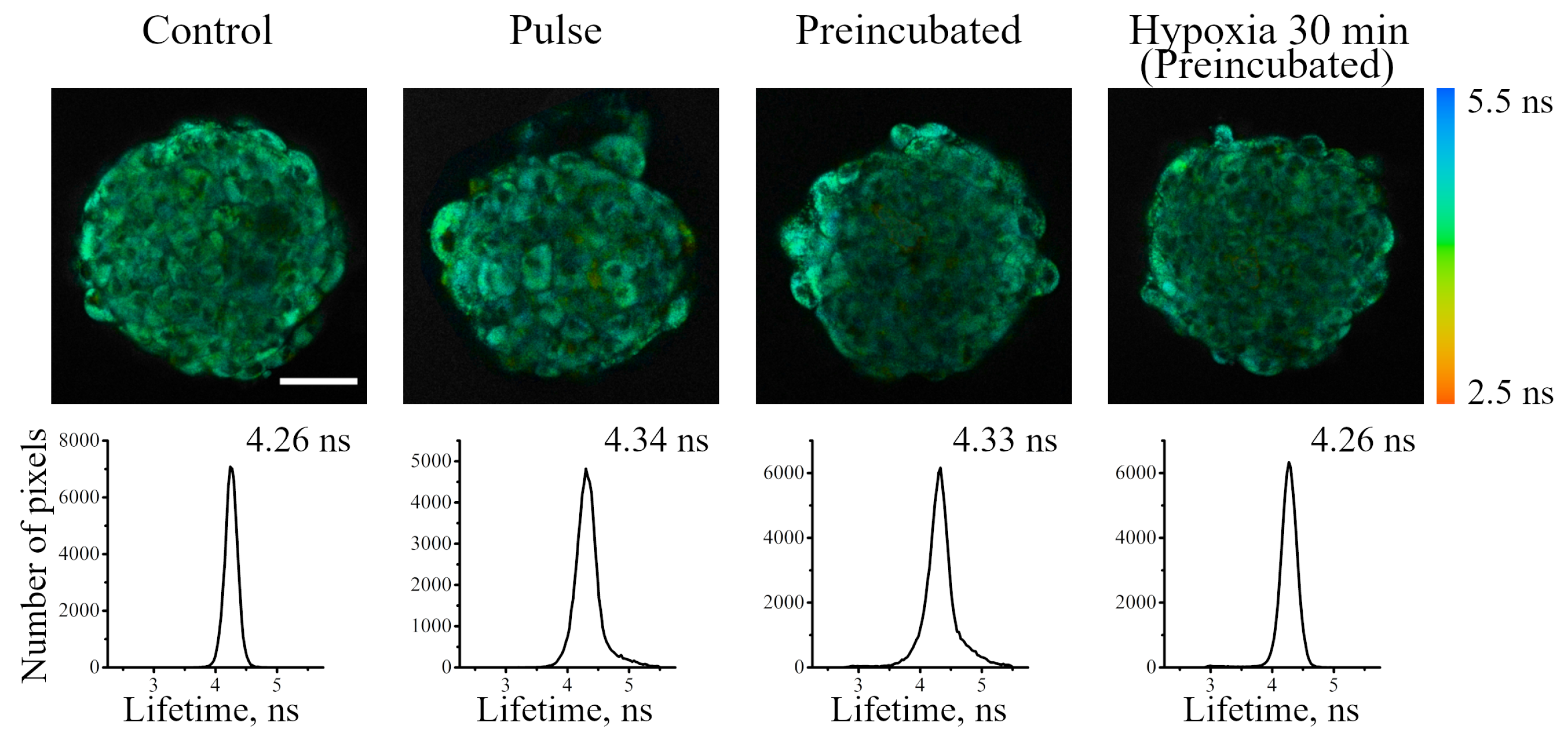
2 is not simply accumulated in the cells, but is involved in the metabolism, which results in its consumption. Indeed, PLIM analysis has demonstrated lifetime gradient (from about 4.5 to 5.3 μs) in spheroids, indicating a decrease in oxygen availability in the inner layers (
Figure 9a–d). This phenomenon can be interpreted as the appearance of a stationary state that emerges as the result of interplay of two processes: (i) O
2 diffusion into the spheroid, and (ii) irreversible oxygen consumption inside cells. This interplay leads to lower oxygen accessibility and, hence, its lower stationary concentration, in inner parts of spheroids. The gradients of sensor lifetimes in enMSC spheroids were similar to those obtained in spheroids of other origin using both Ir- and Pt-based oxygen sensors [
53,
54,
55]. To understand how quickly the sensor in cell lysosomes reacts to hypoxia, we replaced the air with nitrogen in a special chamber, and kept the spheroid under such conditions for 30 min before PLIM recording (
Figure 9e,f). In this experiment, we observed an “inverted” lifetime gradient, indicating moderate increase in lifetimes in the outer layer of cells (ranging from 6.0 to 5.3 μs), and lifetime increase to higher values (ranging from 6.5 to 6.0 μs) in the inner layers of the spheroids.
Comparing normoxia with such a short-term (30 min) hypoxia, it must be stressed that the cells in the inner layers maintain “mild hypoxia”, which is characterized not by the complete inaccessibility of oxygen, but only by its reduced level. Keeping the cells under conditions of complete hypoxia for 2 h showed that, eventually, all the cells of the spheroids lost oxygen, and this conditional level of “absolute hypoxia” corresponds to the sensor phosphorescence lifetime of about 7.5 μs measured throughout the entire spheroid (
Figure 9g,h). This value is in good agreement with the Ir lifetimes measured for the same sensor in deeply deoxygenated phosphate-buffered saline at pH 7.4 [
26].
The obtained results indicate that the sensor can be successfully used to assess the dynamics of changes in the oxygen level in cell lysosomes under different external stimuli, and it, in turn, reflects the oxygen availability in the cytoplasm.
It is known that cells in tissues are subject to changes in oxygen concentration and, consequently, in the level of hypoxia. For example, cyclic fluctuations in oxygen concentration have been demonstrated in tumors in vivo due to changes in erythrocyte flow, as well as in vascular remodeling, and thermoregulation [
56]. It is very important to have experimental models that (i) adequately mimic in vivo tissue oxygen fluctuations and (ii) allow simultaneous control of O
2 levels. The spheroids loaded with PLIM oxygen sensors appear to be an experimental model that bridges the gap between standard experimental protocols and clinical settings.
4. Materials and Methods
4.1. Cell Culture
Human endometrial mesenchymal stem/stromal cells (enMSCs) were isolated from a desquamated endometrium of menstrual blood from healthy donors [
57] and demonstrated properties typical for the mesenchymal stem cell cultures. enMSCs were cultured in DMEM/F12 medium (GIBCO, Waltham, MA, USA) supplemented with 10% fetal bovine serum (GIBCO, Waltham, MA, USA), 1% L-glutamine, and 1% penicillin-streptomycin (GIBCO, Waltham, MA, USA), in an atmosphere of 5% CO
2 at 37 °C. The cells from the third to ninth passages were reseeded twice a week at the split ratio 1:3. These cells are characterized by high rate of cell proliferation (doubling time 22–23 h).
For experiments, the cells were harvested by trypsinization and plated on Petri dishes with glass coverslips (Nunc, Thermo Scientific, Rochester, NY, USA) or on 35 mm imaging μ-dishes with a polymer coverslip bottom (Ibidi GmbH, Gräfelfing, Germany) or 12-well plates (Nunc, Thermo Scientific, Rochester, NY, USA) in DMEM/F12 medium, supplemented with 10% fetal bovine serum, or were used to form spheroids. The experiments were performed at 60–70% confluence 48 h after seeding.
The experiments were approved by the Ethics Committee of the Almazov National Medical Research Centre (Saint-Petersburg, Russia) and performed in accordance with the institutional guidelines. All cell donors signed an informed consent for voluntary participation.
4.2. Spheroid Formation
Spheroids (3D-cultures) were formed from monolayer enMSCs (2D cultures) using the hanging drop technique. A total of 2500 cells in suspension per 35 μL of DMEM/F-12 medium containing 10% fetal bovine serum were placed in drops on the cover of 5 cm Petri dishes (Nunc, Thermo Scientific, Rochester, NY, USA) and inverted. During the next 72 h, cells in hanging drops spontaneously formed spheroids.
For confocal microscopy, already formed spheroids were placed in dishes with a polymer coverslip bottom with full growth medium and were left for 3–4 h to ensure attachment and immobility of spheroids.
For the flow cytometry assay, the single-cell suspension from spheroids was obtained. For this purpose, spheroids with culture medium were transferred to a 15 mL conical tube and centrifuged for 3 min at 1500 rpm. After centrifugation, the supernatant was removed; 10 mL of phosphate-buffered saline solution was added, and the cells were centrifuged again. Then, the supernatant was removed, and spheroids were incubated with 0.05% trypsin/EDTA solution for 5 min at 37 °C. After the incubation was completed, the cell suspension was gently pipetted 2–3 times to achieve spheroid dissociation. Finally, a three-fold volume of the culture media was added for trypsin inactivation.
4.3. Incubation with Ir-HSA-FITC Sensor
The Ir-HSA-FITC sensor based on human serum albumin (HSA) conjugated with a pH-sensitive fluorophore (FITC) and an O
2-sensitive phosphor represented by cyclometalated iridium complex
used in this study were synthesized and characterized according to the methods described in our previous publication [
26].
Ir-HSA-FITC was dissolved in the water at concentration of 250 μm and added to the enMSC monolayers in a final concentration of 20 μm. After incubation with the probe for indicated time, cells were washed with fresh media and analyzed.
The sensor was introduced into spheroids according two protocols: in the first one, already formed spheroids were transferred to dishes coated with a nonadhesive substrate (2-hydroxyethyl methacrylate, Sigma-Aldrich, St. Louis, MO, USA) and cultured in full growth medium with Ir-HSA-FITC for the next 24 h (pulse); in the second one, 2D monolayer enMSCs were preincubated with Ir-HSA-FITC for 24 h followed by the formation of spheroids in the absence of the sensor (preincubated).
4.4. Intracellular Compartment Identification
For vital staining of lysosomes and late endosomes in enMSCs or spheroids, LysoTracker Deep Red or LysoTracker Green DND-26 (Invitrogen, Eugene, OR, USA) at a concentration of 50 nM was used. LysoTracker was added into the culture medium for 20 min prior to confocal imaging. For vital staining of nuclei in spheroids, Hoechst 33342 (Invitrogen, Eugene, OR, USA) was used at a concentration of 1.6 μm for 20 min before analysis.
4.5. Cell Treatments
Bafilomycin A1 (BafA1, Sigma-Aldrich, St. Louis, MO, USA), a specific inhibitor of vacuolar type H+-ATPase, at a concentration of 100 nM was added for 30 min to the cells, preliminarily incubated with Ir-HSA-FITC (20 μm) for 24 h.
Experiments were carried out under a normal oxygenated atmosphere, containing 5% CO2, 37 °C (normoxia). In case of hypoxia, air was completely substituted with nitrogen while the CO2 level remained unchanged; the cells or spheroids were incubated under such conditions for 20–30 min and for 2 h.
4.6. Evaluation of Cell Metabolic Activity by MTT Test
enMSCs were seeded in 96-well plates at 104 cells per well and allowed to grow for 24 h. After that, Ir-HSA-FITC were added to cells for 24 h at concentrations of 0–80 μm. At the end of the incubation, Ir-HSA-FITC was washed out, the medium was exchanged for fresh DMEM/F12 without phenol red (100 μL/well), and then, 10 μL/well of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide reagent (MTT, 5 mg/mL, Invitrogen, Eugene, OR, USA) was added. After 4 h of additional incubation, the media were removed, and 50 μL/well of DMSO was added to solve the formed formazan crystals produced by the metabolic active cells, and a Thermo Scientific Multiskan FC microplate format photometer (Thermo Fisher Scientific, Waltham, MA, USA) determining the optical absorption at a wavelength of 570 nm was used to measure the cell viability.
4.7. Analysis of Cell-Associated Fluorescence by Flow Cytometry
Flow cytometry analysis was carried out by CytoFLEX cytometer (Beckman Coulter, Brea, CA, USA) at a maximum sample feed rate (1 μL/s) for 100 s. The data obtained were analyzed using the CytExpert 2.0.0.152 software (Beckman Coulter, Brea, CA, USA) and are presented as diagrams and histograms.
Laser excitation wavelengths at 405 (for Ir) and 488 (for FITC) nm were used to estimate the amount of Ir-HSA-FITC associated with cells in monolayer culture and in spheroids. Luminescence was recorded using combination of 610/20 and 660/10 BP filters for Ir and 525/40 BP filter for FITC.
Hoechst 33342 was excited at 405 nm and registered with 450/45 BP filter; LysoTracker Green was excited at 488 nm and registered with 525/40 BP filter; PI was excited at 488 nm and registered with 690/50 BP filter. Autofluorescence of cells was analyzed under the same conditions for each case.
4.8. Estimation of Cell Survival with PI Test
Living cells have membranes that are still intact and exclude propidium iodide (PI, Sigma, St. Louis, MO, USA) that easily penetrates the damaged, permeable membranes of non-viable cells. PI at a concentration of 50 μg/ml was added for 1–2 min to the suspensions of control cells and the cell suspension obtained after disintegration of spheroids incubated with Ir-HSA-FITC. Then, the fluorescence of cells was analyzed by flow cytometry. The results were expressed in the proportion of live cells (not stained with PI) relative to the total number of cells in each sample.
Spheroids were incubated with PI (40 μg/mL, 20 min at 37 °C) and analyzed using a confocal microscope. In this case, a positive control visualization of the dead cells in spheroids was achieved using PI staining performed 4 h after the stress insult by heat shock (spheroids were incubated at 45 °C for 30 min in the water bath).
4.9. Confocal Microscopy
Cell imaging was performed using an Olympus FV3000 laser scanning confocal microscope (Olympus, Tokyo, Japan). The samples were observed with a 40/1.42× oil immersion objective, obtaining images of pixels. Images were captured in one, two or three spectral channels in sequential scan mode, with only one laser operating at a time to avoid spectral overlap, and in a channel of differential interference contrast in transmitted light (DIC). Z-series optical sections for monolayer cells were taken at 0.5 μm steps from bottom to top (14–16 sections) and for spheroids at 10 μm steps from bottom to top (5–7 sections). When indicated, the luminescence of Ir-HSA-FITC was studied in spectral scanning mode (Mode/xy) in the range of 505–675 nm with a step of 5 nm.
The dual emission of Ir-HSA-FITC was excited separately with 405 and 488 nm lasers. The emission of iridium chromophore was recorded in the 570–670 nm range and FITC fluorescence signal was recorded in the 500–550 nm range. In the spectral scanning mode, the emission of Ir-HSA-FITC was excited simultaneously with 405 and 488 nm lasers.
LysoTracker Green was excited at 488 nm and recorded in the 500–550 nm range. LysoTracker Deep Red fluorescence was excited at 640 nm and recorded at 650–750 nm. Hoechst 33342 fluorescence was excited at 405 nm and recorded in the range of 430–480 nm. PI fluorescence was excited at 488 nm and recorded in the range of 600–680 nm.
In each experiment, 5–10 fields containing 20–60 cells totally were imaged for each experimental point. Images were processed and analyzed using Fiji software 1.52v (National Institutes of Health, Bethesda, MD, USA). The most representative single sections from a Z-series of typical cells were selected for demonstration. For quantitative analysis, raw images were used.
4.10. Combined FLIM/PLIM Experiments
Combined fluorescence and phosphorescence lifetime imaging microscopy (FLIM/PLIM) of enMSCs or spheroids was carried out using a time-correlated single photon counting (TCSPC) DCS-120 module (Becker & Hickl GmbH, Berlin, Germany) integrated into the Nikon Eclipse Ti2 (Nikon Corporation, Tokyo, Japan) confocal instrument. All measurements were performed in humidified Stage Top Incubator Tokai HIT (Tokyo, Japan) at 37 °C and 5% CO2. Required CO2, air and N2 percentage was maintained by Tokai HIT Digital gas mixer GM-8000.
Emission was excited with a picosecond laser at 405 nm, light from the sample was divided on a beam splitter, and fluorescence and phosphorescence were recorded in two channels. FLIM was recorded in the channel with 435 nm long pass filter and 535/30 nm band pass filter and pinhole of 0.25–0.5. PLIM was recorded using 575 nm long pass filter and 630/75 nm band pass filter and pinhole of 0.25–0.5. The following settings were used: frame time 21.74 s, pixel dwell time 81.60 μs, total acquisition time 173.9 s, and image size 512 × 512 pixels. Water immersion 40× objective with zoom 5.33 for cells and 1.43 for spheroids. Fluorescence and phosphorescence lifetime distributions were calculated using SPCImage 8.1 software (Becker & Hickl GmbH, Berlin, Germany). Fluorescence decays were fitted monoexponentially with an average goodness of the fit 0.9 ≤ ≤ 1.3. The phosphorescence decay curves were fitted in monoexponential decay mode with an average goodness of the fit 0.8 ≤ ≤ 1.2. The average number of photons per curve were not less than 5000 at binning 4 (monolayer cells) or 7 (spheroids). The data are presented as distribution of fluorescence or phosphorescence lifetime collected from the scanning field, as indicated in figure legends.
4.11. Statistical and Co-Localization Analyses
The images were processed and analyzed using Fiji software 1.52v (National Institutes of Health, Bethesda, MY, USA). The quantitative co-localization analysis was performed using ImageJ JACoP Plugin to determine Manders’ co-localization coefficients (M), which are defined using thresholds as the sum of intensities of co-localized pixels from one channel divided by their integrated density. Thresholds were set by a visually estimated value for each channel. The results are represented as mean ± standard error of the mean (SEM).
Statistical data processing was performed using Microsoft Office Excel 2021 (Microsoft Corporation, Albuquerque, NM, USA). The graphs were built using the Origin 8.5 software (OriginLab, Northampton, MA, USA), the bar charts (mean ± SEM) and box plots using Microsoft Office Excel 2021. All results were obtained from at least three independent experiments.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








