
Arrhythmia-Associated Calmodulin E105A Mutation Alters the Binding Affinity of CaM to a Ryanodine Receptor 2 CaM-Binding Pocket

 , and
, and
Abstract
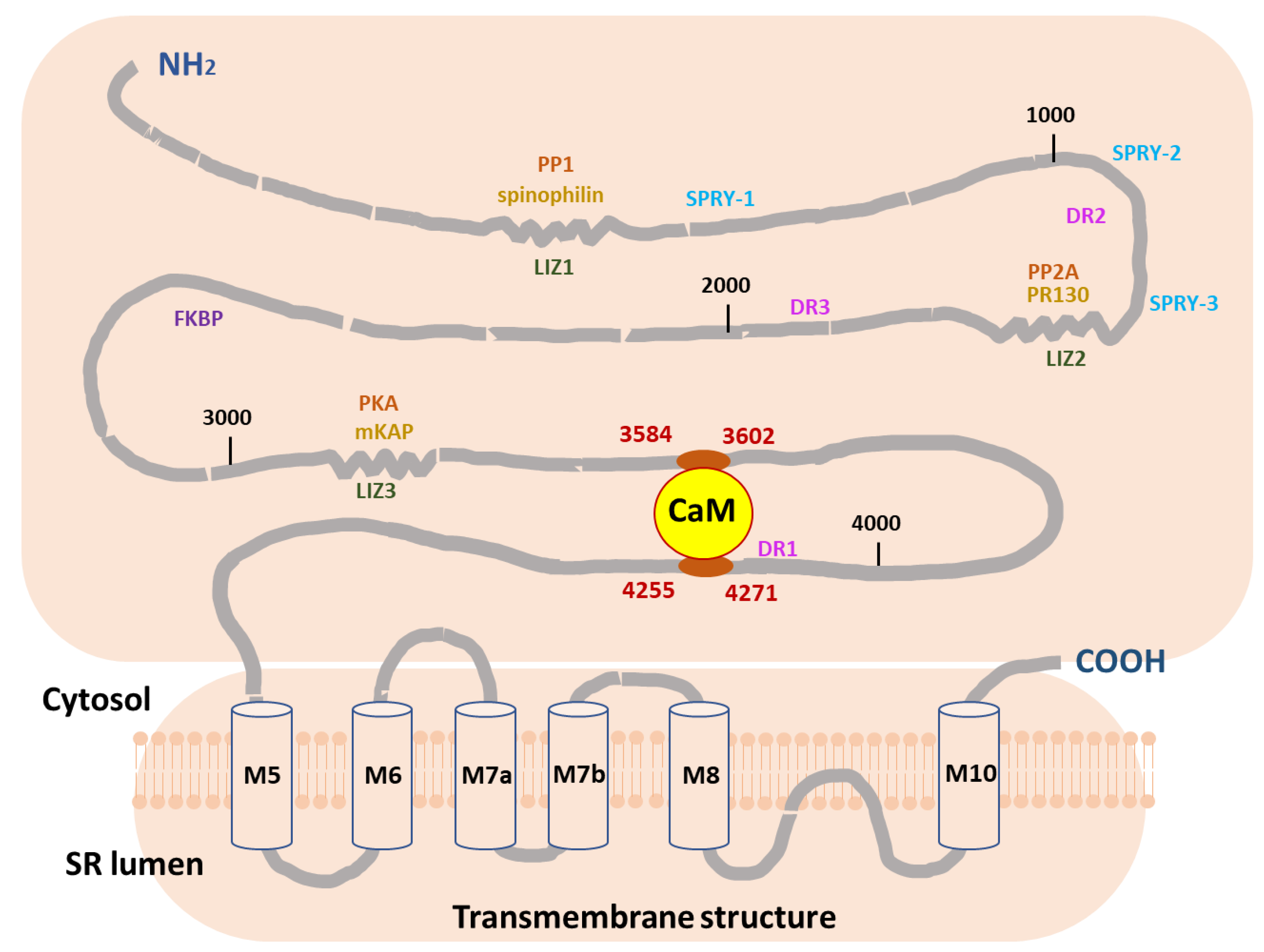
:1. Introduction
2. Results
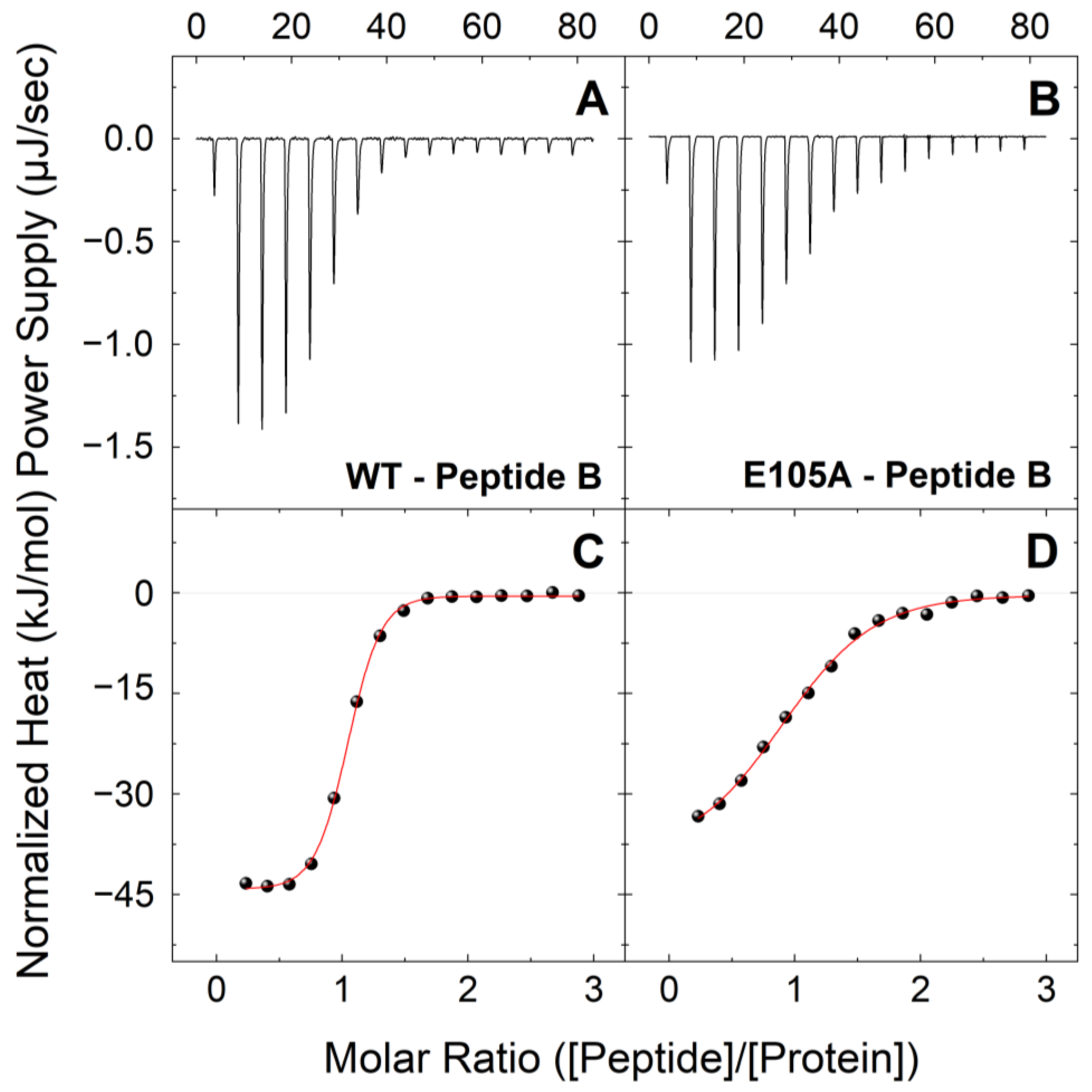
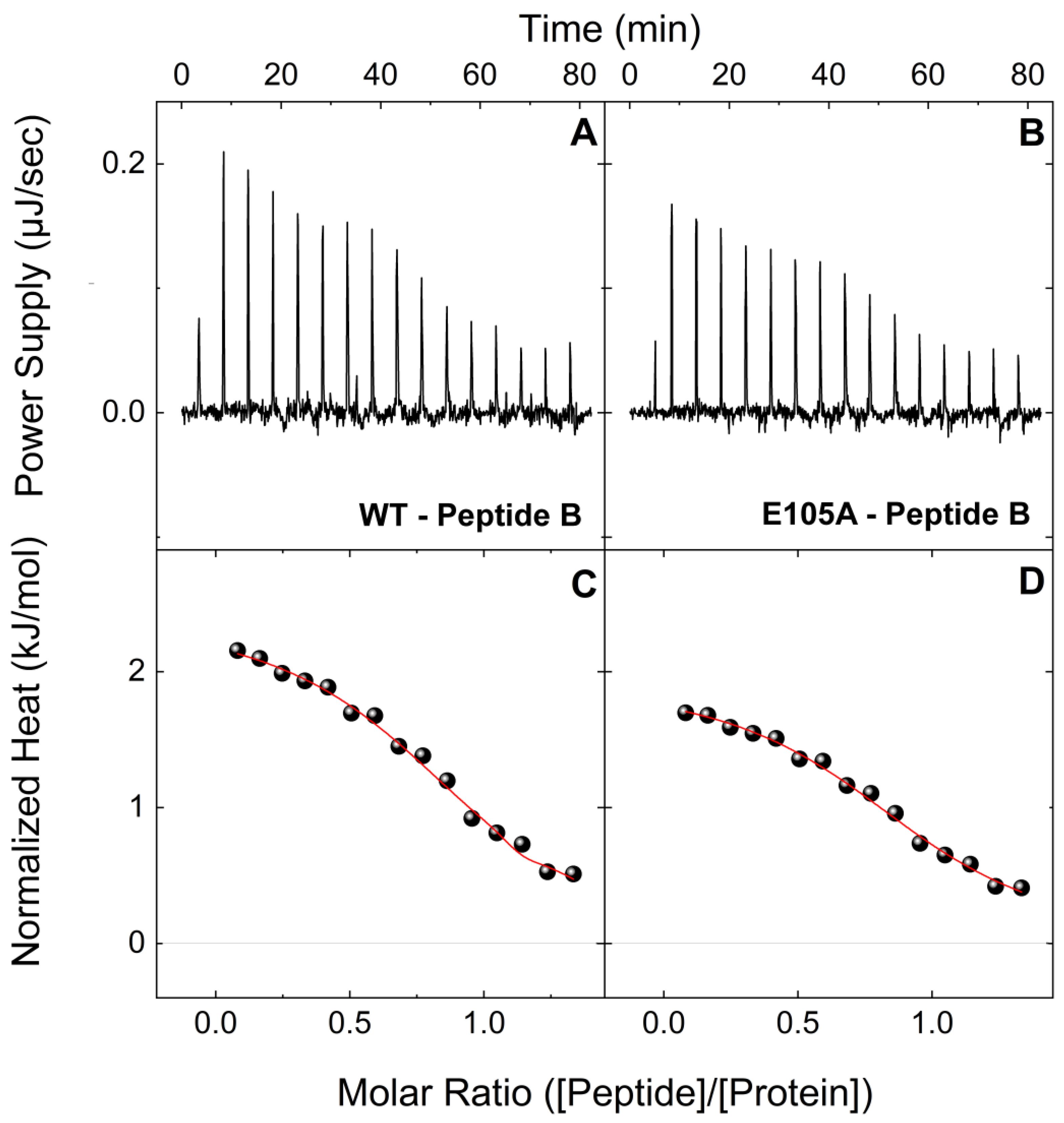
2.1. E105A Mutation Alters the Affinity of CaM for Peptide B (RyR2 3581-3607aa) in the Presence and Absence of Ca2+
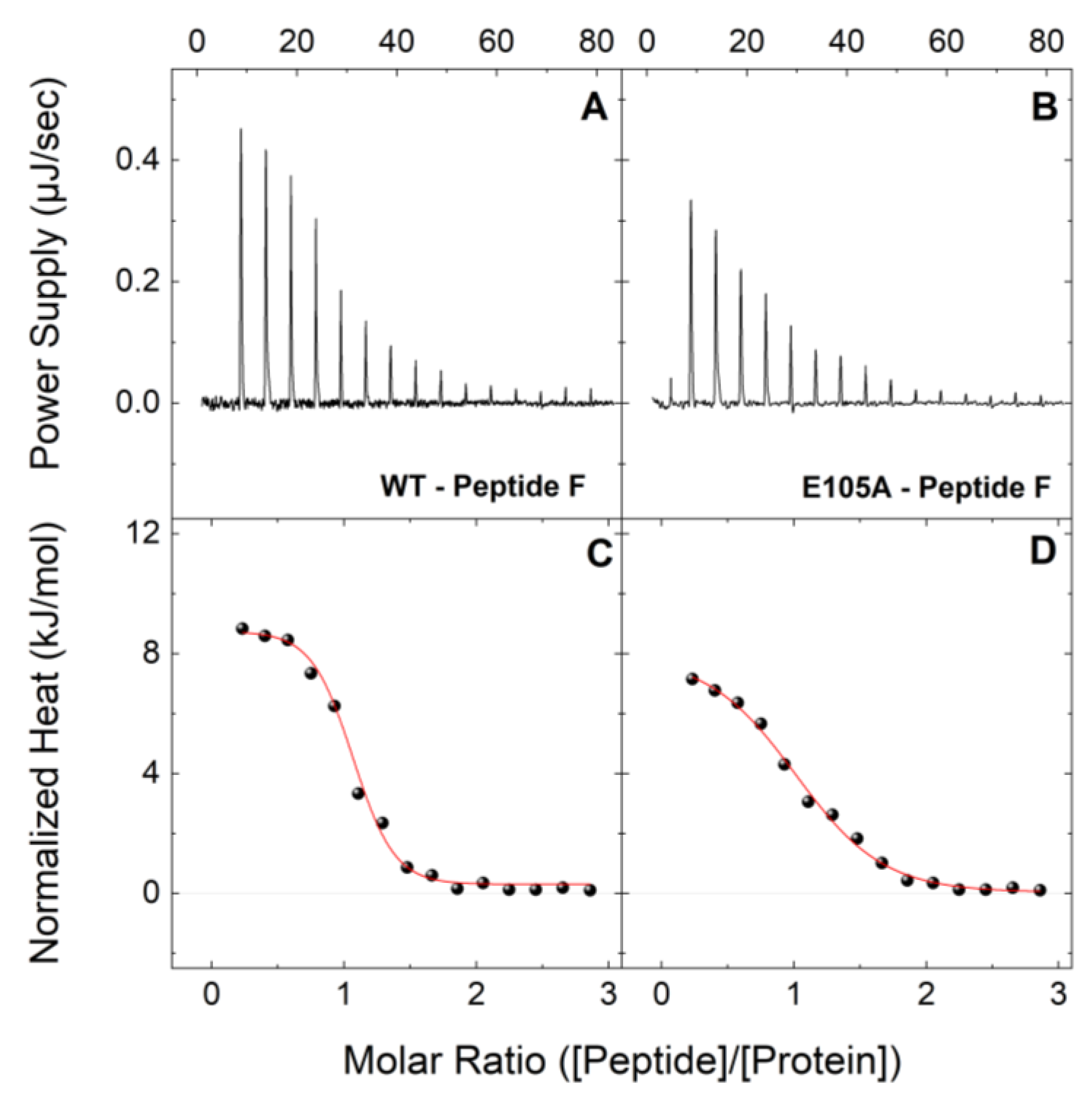
2.2. E105A Mutation Alters the Affinity of CaM for Peptide F (RyR2 4255-4271aa) in the Presence and Absence of Ca2+
3. Discussion
4. Materials and Methods
4.1. Plasmid Construction
4.2. Protein Expression and Purification
4.3. Isothermal Titration Calorimetry
4.4. Molecular Modeling
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Yuchi, Z.; Van Petegem, F. Ryanodine receptors under the magnifying lens: Insights and limitations of cryo-electron microscopy and X-ray crystallography studies. Cell Calcium 2016, 59, 209–227. [Google Scholar] [CrossRef]
- Peng, W.; Shen, H.; Wu, J.; Guo, W.; Pan, X.; Wang, R.; Chen, S.R.W.; Yan, N. Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science 2016, 354, aah5324. [Google Scholar] [CrossRef]
- Bers, D.M. Sarcoplasmic reticulum Ca release in intact ventricular myocytes. Front. Biosci. 2002, 7, 1697–1711. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.; Gong, D.; Ren, K.; Zhou, G.; Huang, G.; Lei, J.; Zhou, Q.; Yan, N. Molecular Basis for Allosteric Regulation of the Type 2 Ryanodine Receptor Channel Gating by Key Modulators. Proc. Natl. Acad. Sci. USA 2019, 116, 25575–25582. [Google Scholar] [CrossRef] [PubMed]
- George, C.H.; Jundi, H.; Thomas, N.L.; Fry, D.L.; Lai, F.A. Ryanodine Receptors and Ventricular Arrhythmias: Emerging Trends in Mutations, Mechanisms and Therapies. J. Mol. Cell Cardiol. 2007, 42, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, P.J.; Brown, K.M.; Piippo, K.; Swan, H.; Devaney, J.M.; Brahmbhatt, B.; Donarum, E.A.; Marino, M.; Tiso, N.; Viitasalo, M.; et al. Mutations of the Cardiac Ryanodine Receptor (RyR2) Gene in Familial Polymorphic Ventricular Tachycardia. Circulation 2001, 103, 485–490. [Google Scholar] [CrossRef]
- Medeiros-Domingo, A.; Bhuiyan, Z.A.; Tester, D.J.; Hofman, N.; Bikker, H.; van Tintelen, J.P.; Mannens, M.M.; Wilde, A.A.; Ackerman, M.J. The Ryr2-Encoded Ryanodine Receptor/Calcium Release Channel in Patients Diagnosed Previously with Either Catecholaminergic Polymorphic Ventricular Tachycardia or Genotype Negative, Exercise-Induced Long Qt Syndrome: A Comprehensive Open Reading Frame Mutational Analysis. J. Am. Coll. Cardiol. 2009, 54, 2065–2074. [Google Scholar] [PubMed]
- Priori, S.G.; Napolitano, C.; Memmi, M.; Colombi, B.; Drago, F.; Gasparini, M.; DeSimone, L.; Coltorti, F.; Bloise, R.; Keegan, R.; et al. Clinical and Molecular Characterization of Patients with Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2002, 106, 69–74. [Google Scholar] [CrossRef]
- Priori, S.G.; Chen, S.R. Inherited Dysfunction of Sarcoplasmic Reticulum Ca2+ Handling and Arrhythmogenesis. Circ. Res. 2011, 108, 871–883. [Google Scholar] [CrossRef]
- Rousseau, E.; Ladine, J.; Liu, Q.-Y.; Meissner, G. Activation of the Ca2+ release channel of skeletal muscle sarcoplasmic reticulum by caffeine and related compounds. Arch. Biochem. Biophys. 1988, 267, 75–86. [Google Scholar] [CrossRef]
- Meissner, G.; Henderson, J.S. Rapid Calcium Release from Cardiac Sarcoplasmic Reticulum Vesicles Is Dependent on Ca2+ and Is Modulated by Mg2+, Adenine Nucleotide, and Calmodulin. J. Biol. Chem. 1987, 262, 3065–3073. [Google Scholar] [CrossRef]
- Smith, G.L.; O’Neill, S.C. A comparison of the effects of ATP and tetracaine on spontaneous Ca2+ release from rat permeabilised cardiac myocytes. J. Physiol. 2001, 534, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Reiken, S.; Hisamatsu, Y.; Jayaraman, T.; Burkhoff, D.; Rosemblit, N.; Marks, A.R. PKA Phosphorylation Dissociates FKBP12.6 from the Calcium Release Channel (Ryanodine Receptor): Defective Regulation in Failing Hearts. Cell 2000, 101, 365–376. [Google Scholar] [CrossRef]
- Seidler, T.; Loughrey, C.M.; Zibrova, D.; Kettlewell, S.; Teucher, N.; Kogler, H.; Hasenfuss, G.; Smith, G.L. Overexpression of Fk-506 Binding Protein 12.0 Modulates Excitation Contraction Coupling in Adult Rabbit Ventricular Cardiomyocytes. Circ. Res. 2007, 101, 1020–1029. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Takahashi, N.; Xu, L.; Smithies, O.; Meissner, G. Early cardiac hypertrophy in mice with impaired calmodulin regulation of cardiac muscle Ca2+ release channel. J. Clin. Investig. 2007, 117, 1344–1353. [Google Scholar] [CrossRef]
- Arnáiz-Cot, J.J.; Damon, B.J.; Zhang, X.H.; Cleemann, L.; Yamaguchi, N.; Meissner, G.; Morad, M. Cardiac calcium signalling pathologies associated with defective calmodulin regulation of type 2 ryanodine receptor. J. Physiol. 2013, 591, 4287–4299. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yano, M.; Uchinoumi, H.; Hino, A.; Suetomi, T.; Ono, M.; Tateishi, H.; Oda, T.; Okuda, S.; Doi, M.; et al. Defective calmodulin binding to the cardiac ryanodine receptor plays a key role in CPVT-associated channel dysfunction. Biochem. Biophys. Res. Commun. 2010, 394, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Balshaw, D.M.; Xu, L.; Yamaguchi, N.; Pasek, D.A.; Meissner, G. Calmodulin Binding and Inhibition of Cardiac Muscle Calcium Release Channel (Ryanodine Receptor). J. Biol. Chem. 2001, 276, 20144–20153. [Google Scholar] [CrossRef] [PubMed]
- Fruen, B.R.; Bardy, J.M.; Byrem, T.M.; Strasburg, G.M.; Louis, C.F. Differential Ca2+ Sensitivity of Skeletal and Cardiac Muscle Ryanodine Receptors in the Presence of Calmodulin. Am. J. Physiol. Cell Physiol. 2000, 279, C724–C733. [Google Scholar] [CrossRef]
- Tadross, M.R.; Dick, I.E.; Yue, D.T. Mechanism of Local and Global Ca2+ Sensing by Calmodulin in Complex with a Ca2+ Channel. Cell 2008, 133, 1228–1240. [Google Scholar] [CrossRef]
- Zhang, M.; Abrams, C.; Wang, L.; Gizzi, A.; He, L.; Lin, R.; Chen, Y.; Loll, P.J.; Pascal, J.M.; Zhang, J.-F. Structural Basis for Calmodulin as a Dynamic Calcium Sensor. Structure 2012, 20, 911–923. [Google Scholar] [CrossRef]
- Fischer, R.; Koller, M.; Flura, M.; Mathews, S.; Strehler-Page, M.A.; Krebs, J.; Penniston, J.T.; Carafoli, E.; Strehler, E.E. Multiple divergent mRNAs code for a single human calmodulin. J. Biol. Chem. 1988, 263, 17055–17062. [Google Scholar] [CrossRef] [PubMed]
- Nyegaard, M.; Overgaard, M.T.; Søndergaard, M.T.; Vranas, M.; Behr, E.R.; Hildebrandt, L.L.; Lund, J.; Hedley, P.L.; Camm, A.J.; Wettrell, G.; et al. Mutations in Calmodulin Cause Ventricular Tachycardia and Sudden Cardiac Death. Am. J. Hum. Genet. 2012, 91, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Crotti, L.; Johnson, C.N.; Graf, E.; De Ferrari, G.M.; Cuneo, B.F.; Ovadia, M.; Papagiannis, J.; Feldkamp, M.D.; Rathi, S.G.; Kunic, J.D.; et al. Calmodulin Mutations Associated with Recurrent Cardiac Arrest in Infants. Circulation 2013, 127, 1009–1017. [Google Scholar] [CrossRef]
- Marsman, R.F.; Barc, J.; Beekman, L.; Alders, M.; Dooijes, D.; van den Wijngaard, A.; Ratbi, I.; Sefiani, A.; Bhuiyan, Z.A.; Wilde, A.A.; et al. A Mutation in Calm1 Encoding Calmodulin in Familial Idiopathic Ventricular Fibrillation in Childhood and Adolescence. J. Am. Coll. Cardiol. 2014, 63, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Makita, N.; Yagihara, N.; Crotti, L.; Johnson, C.N.; Beckmann, B.-M.; Roh, M.S.; Shigemizu, D.; Lichtner, P.; Ishikawa, T.; Aiba, T.; et al. Novel Calmodulin Mutations Associated with Congenital Arrhythmia Susceptibility. Circ. Cardiovasc. Genet. 2014, 7, 466–474. [Google Scholar] [CrossRef]
- Takahashi, K.; Ishikawa, T.; Makita, N.; Takefuta, K.; Nabeshima, T.; Nakayashiro, M. A novel de novo calmodulin mutation in a 6-year-old boy who experienced an aborted cardiac arrest. Hear. Case Rep. 2017, 3, 69–72. [Google Scholar] [CrossRef]
- Hussey, J.W.; Limpitikul, W.B.; Dick, I.E. Calmodulin Mutations in Human Disease. Channels 2023, 17, 2165278. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Hurtado, N.; Boczek, N.J.; Kryshtal, D.O.; Johnson, C.N.; Sun, J.; Nitu, F.R.; Cornea, R.L.; Chazin, W.J.; Calvert, M.L.; Tester, D.J.; et al. Novel CPVT-Associated Calmodulin Mutation in CALM3 (CALM3-A103V) Activates Arrhythmogenic Ca Waves and Sparks. Circ. Arrhythmia Electrophysiol. 2016, 9, e004161. [Google Scholar] [CrossRef]
- Hwang, H.S.; Nitu, F.R.; Yang, Y.; Walweel, K.; Pereira, L.; Johnson, C.N.; Faggioni, M.; Chazin, W.J.; Laver, D.; George, A.L.; et al. Divergent Regulation of Ryanodine Receptor 2 Calcium Release Channels by Arrhythmogenic Human Calmodulin Missense Mutants. Circ. Res. 2014, 114, 1114–1124. [Google Scholar] [CrossRef]
- Limpitikul, W.B.; Dick, I.E.; Joshi-Mukherjee, R.; Overgaard, M.T.; George, A.L., Jr.; Yue, D.T. Calmodulin Mutations Associated with Long Qt Syndrome Prevent Inactivation of Cardiac L-Type Ca2+ Currents and Promote Proarrhythmic Behavior in Ventricular Myocytes. J. Mol. Cell Cardiol. 2014, 74, 115–124. [Google Scholar] [CrossRef]
- Jensen, H.H.; Brohus, M.; Nyegaard, M.; Overgaard, M.T. Human Calmodulin Mutations. Front. Mol. Neurosci. 2018, 11, 396. [Google Scholar] [CrossRef] [PubMed]
- Søndergaard, M.T.; Liu, Y.; Brohus, M.; Guo, W.; Nani, A.; Carvajal, C.; Fill, M.; Overgaard, M.T.; Chen, S.R.W. Diminished inhibition and facilitated activation of RyR2-mediated Ca2+ release is a common defect of arrhythmogenic calmodulin mutations. FEBS J. 2019, 286, 4554–4578. [Google Scholar] [CrossRef] [PubMed]
- Sondergaard, M.T.; Liu, Y.; Guo, W.; Wei, J.; Wang, R.; Brohus, M.; Overgaard, M.T.; Chen, S.R.W. Role of Cardiac Ryanodine Receptor Calmodulin-Binding Domains in Mediating the Action of Arrhythmogenic Calmodulin N-Domain Mutation N54i. FEBS J. 2020, 287, 2256–2280. [Google Scholar] [CrossRef]
- Nomikos, M.; Thanassoulas, A.; Beck, K.; Vassilakopoulou, V.; Hu, H.; Calver, B.L.; Theodoridou, M.; Kashir, J.; Blayney, L.; Livaniou, E.; et al. Altered RyR2 regulation by the calmodulin F90L mutation associated with idiopathic ventricular fibrillation and early sudden cardiac death. FEBS Lett. 2014, 588, 2898–2902. [Google Scholar] [CrossRef] [PubMed]
- Vassilakopoulou, V.; Calver, B.L.; Thanassoulas, A.; Beck, K.; Hu, H.; Buntwal, L.; Smith, A.; Theodoridou, M.; Kashir, J.; Blayney, L.; et al. Distinctive malfunctions of calmodulin mutations associated with heart RyR2-mediated arrhythmic disease. Biochim. Biophys. Acta (BBA) Gen. Subj. 2015, 1850, 2168–2176. [Google Scholar] [CrossRef]
- Da’as, S.I.; Thanassoulas, A.; Calver, B.L.; Beck, K.; Salem, R.; Saleh, A.; Kontogianni, I.; Al-Maraghi, A.; Nasrallah, G.K.; Safieh-Garabedian, B.; et al. Arrhythmogenic Calmodulin E105a Mutation Alters Cardiac Ryr2 Regulation Leading to Cardiac Dysfunction in Zebrafish. Ann. N. Y. Acad. Sci. 2019, 1448, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Thanassoulas, A.; Vassilakopoulou, V.; Calver, B.L.; Buntwal, L.; Smith, A.; Lai, C.; Kontogianni, I.; Livaniou, E.; Nounesis, G.; Lai, F.A.; et al. Life-Threatening Arrhythmogenic Cam Mutations Disrupt Cam Binding to a Distinct Ryr2 Cam-Binding Pocket. Biochim. Biophys. Acta Gen. Subj. 2023, 1867, 130313. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Xu, L.; Pasek, D.A.; Evans, K.E.; Meissner, G. Molecular Basis of Calmodulin Binding to Cardiac Muscle Ca2+ Release Channel (Ryanodine Receptor). J. Biol. Chem. 2003, 278, 23480–23486. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Chakraborty, A.; Huang, T.-Q.; Xu, L.; Gomez, A.C.; Pasek, D.A.; Meissner, G. Cardiac hypertrophy associated with impaired regulation of cardiac ryanodine receptor by calmodulin and S100A1. Am. J. Physiol. Circ. Physiol. 2013, 305, H86–H94. [Google Scholar] [CrossRef]
- Sondergaard, M.T.; Tian, X.; Liu, Y.; Wang, R.; Chazin, W.J.; Chen, S.R.; Overgaard, M.T. Arrhythmogenic Calmodulin Mutations Affect the Activation and Termination of Cardiac Ryanodine Receptor-Mediated Ca2+ Release. J. Biol. Chem. 2015, 290, 26151–26162. [Google Scholar] [CrossRef]
- Brohus, M.; Søndergaard, M.T.; Chen, S.R.W.; van Petegem, F.; Overgaard, M.T. Ca2+-dependent calmodulin binding to cardiac ryanodine receptor (RyR2) calmodulin-binding domains. Biochem. J. 2019, 476, 193–209. [Google Scholar] [CrossRef]
- Tsai, W.C.; Chen, P.S.; Rubart, M. Calmodulinopathy in inherited arrhythmia syndromes. Tzu Chi Med. J. 2021, 33, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Nyegaard, M.; Overgaard, M.T. The International Calmodulinopathy Registry: Recording the diverse phenotypic spectrum of un-CALM hearts. Eur. Heart J. 2019, 40, 2976–2978. [Google Scholar] [CrossRef]
- Etheridge, S.P.; Niu, M.C. Calmodulinopathies: Throwing back the veil on the newest life-threatening genetic arrhythmia syndrome. Curr. Opin. Cardiol. 2021, 36, 61–66. [Google Scholar] [CrossRef]
- Crotti, L.; Spazzolini, C.; Tester, D.J.; Ghidoni, A.; Baruteau, A.-E.; Beckmann, B.-M.; Behr, E.R.; Bennett, J.S.; Bezzina, C.R.; A Bhuiyan, Z.; et al. Calmodulin mutations and life-threatening cardiac arrhythmias: Insights from the International Calmodulinopathy Registry. Eur. Heart J. 2019, 40, 2964–2975. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Brohus, M.; Holt, C.; Overgaard, M.T.; Wimmer, R.; Van Petegem, F. Arrhythmia mutations in calmodulin can disrupt cooperativity of Ca2+ binding and cause misfolding. J. Physiol. 2020, 598, 1169–1186. [Google Scholar] [CrossRef] [PubMed]
- Holt, C.; Hamborg, L.; Lau, K.; Brohus, M.; Sorensen, A.B.; Larsen, K.T.; Sommer, C.; Van Petegem, F.; Overgaard, M.T.; Wimmer, R. The Arrhythmogenic N53i Variant Subtly Changes the Structure and Dynamics in the Calmodulin N-Terminal Domain, Altering Its Interaction with the Cardiac Ryanodine Receptor. J. Biol. Chem. 2020, 295, 7620–7634. [Google Scholar] [CrossRef]
- Wang, K.; Holt, C.; Lu, J.; Brohus, M.; Larsen, K.T.; Overgaard, M.T.; Wimmer, R.; Van Petegem, F. Arrhythmia mutations in calmodulin cause conformational changes that affect interactions with the cardiac voltage-gated calcium channel. Proc. Natl. Acad. Sci. USA 2018, 115, E10556–E10565. [Google Scholar] [CrossRef]
- Søndergaard, M.T.; Liu, Y.; Larsen, K.T.; Nani, A.; Tian, X.; Holt, C.; Wang, R.; Wimmer, R.; Van Petegem, F.; Fill, M.; et al. The Arrhythmogenic Calmodulin p.Phe142Leu Mutation Impairs C-domain Ca2+ Binding but Not Calmodulin-dependent Inhibition of the Cardiac Ryanodine Receptor. J. Biol. Chem. 2017, 292, 1385–1395. [Google Scholar] [CrossRef]
- Sondergaard, M.T.; Sorensen, A.B.; Skov, L.L.; Kjaer-Sorensen, K.; Bauer, M.C.; Nyegaard, M.; Linse, S.; Oxvig, C.; Overgaard, M.T. Calmodulin Mutations Causing Catecholaminergic Poly-morphic Ventricular Tachycardia Confer Opposing Functional and Biophysical Molecular Changes. FEBS J. 2015, 282, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyaya, R.; Meador, W.E.; Means, A.R.; Quiocho, F.A. Calmodulin Structure Refined at 1.7 Å Resolution. J. Mol. Biol. 1992, 228, 1177–1192. [Google Scholar] [CrossRef]
- Yano, M.; Yamamoto, T.; Ikeda, Y.; Matsuzaki, M. Mechanisms of Disease: Ryanodine receptor defects in heart failure and fatal arrhythmia. Nat. Clin. Pr. Cardiovasc. Med. 2006, 3, 43–52. [Google Scholar] [CrossRef]
- Lau, K.; Chan, M.M.Y.; Van Petegem, F. Lobe-Specific Calmodulin Binding to Different Ryanodine Receptor Isoforms. Biochemistry 2014, 53, 932–946. [Google Scholar] [CrossRef]
- Huang, X.; Liu, Y.; Wang, R.; Zhong, X.; Liu, Y.; Koop, A.; Chen, S.R.; Wagenknecht, T.; Liu, Z. Two Potential Calmodulin-Binding Sequences in the Ryanodine Receptor Contribute to a Mobile, Intra-Subunit Calmodulin-Binding Domain. J. Cell Sci. 2013, 126, 4527–4535. [Google Scholar] [CrossRef]
- Yu, Q.; Anderson, D.E.; Kaur, R.; Fisher, A.J.; Ames, J.B. The Crystal Structure of Calmodulin Bound to the Cardiac Ryanodine Receptor (Ryr2) at Residues Phe4246-Val4271 Reveals a Fifth Calcium Binding Site. Biochemistry 2021, 60, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Venetucci, L.; Denegri, M.; Napolitano, C.; Priori, S.G. Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat. Rev. Cardiol. 2012, 9, 561–575. [Google Scholar] [CrossRef]
- Lahat, H.; Pras, E.; Eldar, M. Ryr2 and Casq2 Mutations in Patients Suffering from Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2003, 107, e29. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Titration | Dissociation Constant (Kd) (μM) | Stoichiometry [N] | Binding Enthalpy (ΔrH) (kJ/mol) | Entropic Term (−T∙ΔrS) (kJ/mol) | Gibbs Free Energy Change (ΔrG) (kJ/mol) | |
|---|---|---|---|---|---|---|
| Peptide B | Holo-CaMWT | 0.41 ± 0.05 | 0.95 ± 0.01 | −44.5 ± 1.8 | 8.1 ± 1.8 | −36.5 ± 0.3 |
| Holo-CaME105A | 2.29 ± 0.24 | 0.93 ± 0.01 | −34.2 ± 1.5 | 2.0 ± 1.5 | −32.2 ± 0.3 | |
| Peptide B | Apo-CaMWT | 4.54 ± 0.51 | 0.91 ± 0.01 | 2.3 ± 0.1 | −32.9 ± 0.3 | −30.5 ± 0.3 |
| Apo-CaME105A | 7.04 ± 1.02 | 0.87 ± 0.01 | 1.9 ± 0.6 | −31.3 ± 0.4 | −29.4 ± 0.4 | |
| Titration | Dissociation Constant (Kd) (μM) | Stoichiometry (N) | Binding Enthalpy (ΔrH) (kJ/mol) | Entropic Term (−T∙ΔrS) (kJ/mol) | Gibbs Free Energy Change (ΔrG) (kJ/mol) | |
|---|---|---|---|---|---|---|
| Peptide F | Holo-CaMWT | 0.63 ± 0.07 | 0.97 ± 0.01 | 9.1 ± 0.5 | −44.5 ± 0.7 | −35.4 ± 0.3 |
| Holo-CaME105A | 2.58 ± 0.28 | 0.94 ± 0.01 | 7.8 ± 0.4 | −39.7 ± 0.5 | −31.9 ± 0.3 | |
| Peptide F | Apo-CaMWT | 11.05 ± 1.36 | 0.93 ± 0.01 | 6.6 ± 0.5 | −34.9 ± 0.6 | −28.3 ± 0.3 |
| Apo-CaME105A | 24.48 ± 3.29 | 0.89 ± 0.01 | 5.2 ± 0.5 | −31.6 ± 0.6 | −26.3 ± 0.3 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thanassoulas, A.; Theodoridou, M.; Barrak, L.; Riguene, E.; Alyaarabi, T.; Elrayess, M.A.; Lai, F.A.; Nomikos, M. Arrhythmia-Associated Calmodulin E105A Mutation Alters the Binding Affinity of CaM to a Ryanodine Receptor 2 CaM-Binding Pocket. Int. J. Mol. Sci. 2023, 24, 15630. https://doi.org/10.3390/ijms242115630
Thanassoulas A, Theodoridou M, Barrak L, Riguene E, Alyaarabi T, Elrayess MA, Lai FA, Nomikos M. Arrhythmia-Associated Calmodulin E105A Mutation Alters the Binding Affinity of CaM to a Ryanodine Receptor 2 CaM-Binding Pocket. International Journal of Molecular Sciences. 2023; 24(21):15630. https://doi.org/10.3390/ijms242115630
Chicago/Turabian StyleThanassoulas, Angelos, Maria Theodoridou, Laila Barrak, Emna Riguene, Tamader Alyaarabi, Mohamed A. Elrayess, F. Anthony Lai, and Michail Nomikos. 2023. "Arrhythmia-Associated Calmodulin E105A Mutation Alters the Binding Affinity of CaM to a Ryanodine Receptor 2 CaM-Binding Pocket" International Journal of Molecular Sciences 24, no. 21: 15630. https://doi.org/10.3390/ijms242115630
APA StyleThanassoulas, A., Theodoridou, M., Barrak, L., Riguene, E., Alyaarabi, T., Elrayess, M. A., Lai, F. A., & Nomikos, M. (2023). Arrhythmia-Associated Calmodulin E105A Mutation Alters the Binding Affinity of CaM to a Ryanodine Receptor 2 CaM-Binding Pocket. International Journal of Molecular Sciences, 24(21), 15630. https://doi.org/10.3390/ijms242115630