
In Utero Cell Treatment of Hemophilia A Mice via Human Amniotic Fluid Mesenchymal Stromal Cell Engraftment

 ,
,  ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
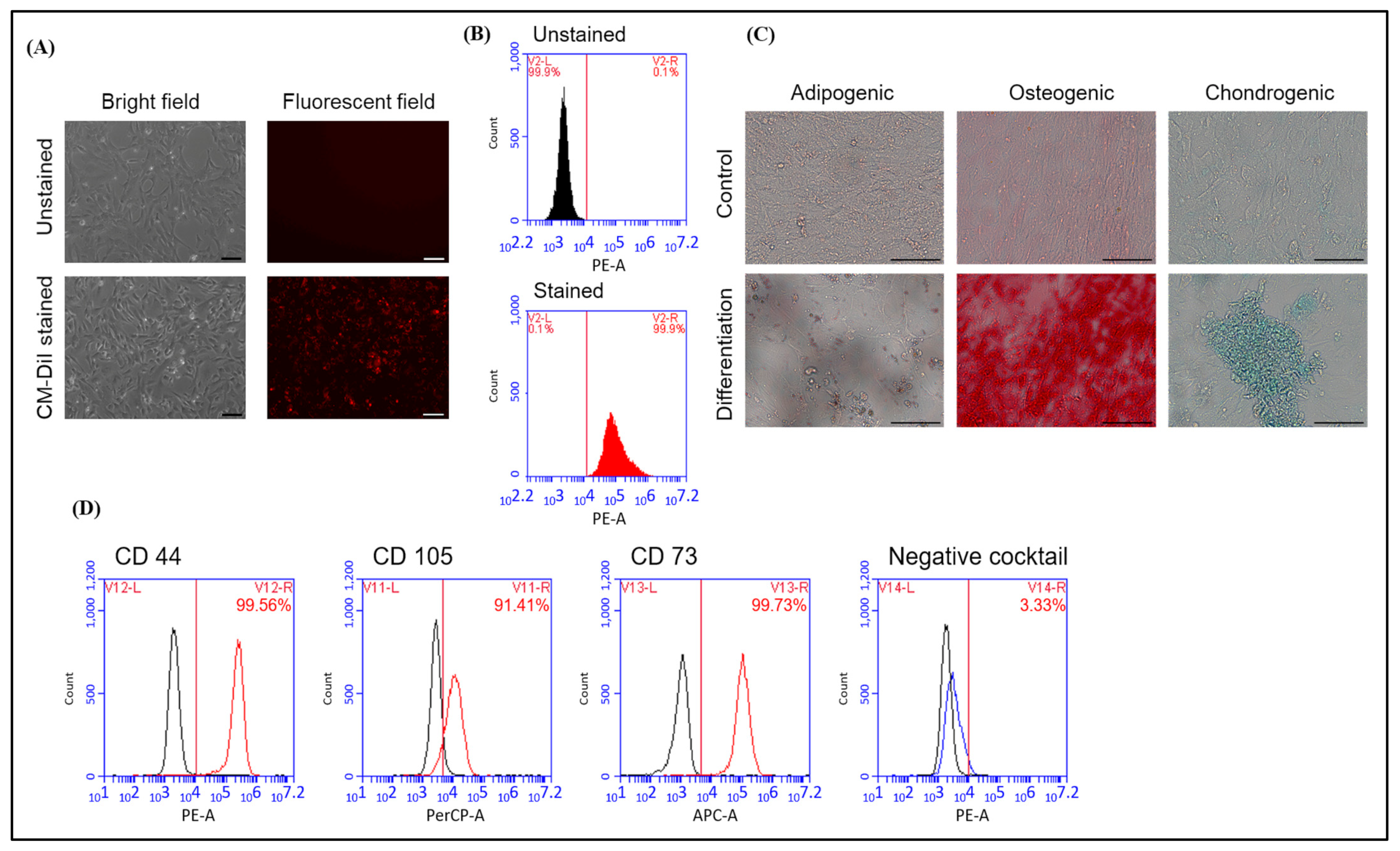
2.1. Characterization of Human AFMSCs
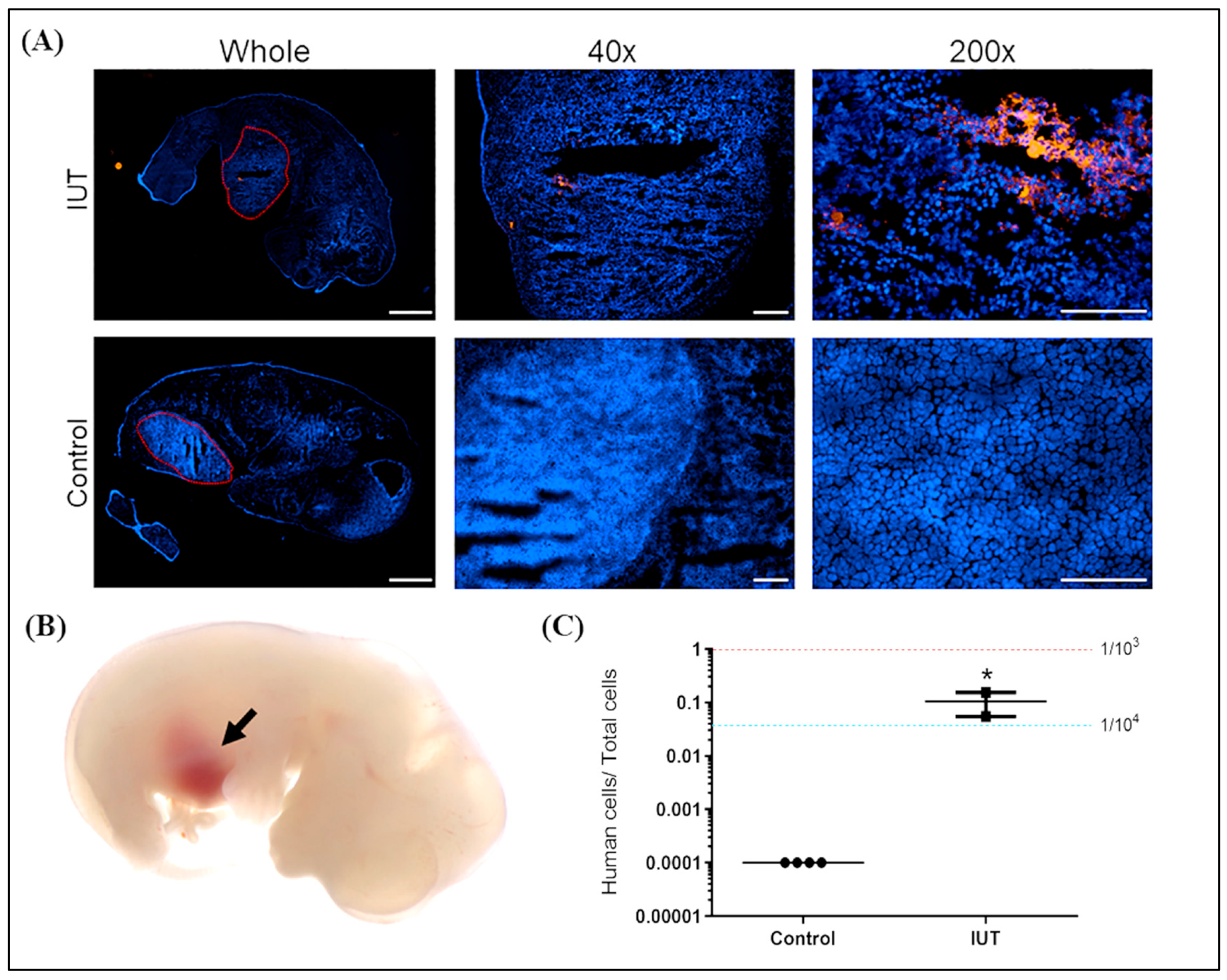
2.2. Detection of hAFMSCs in the Livers of IUT Fetuses
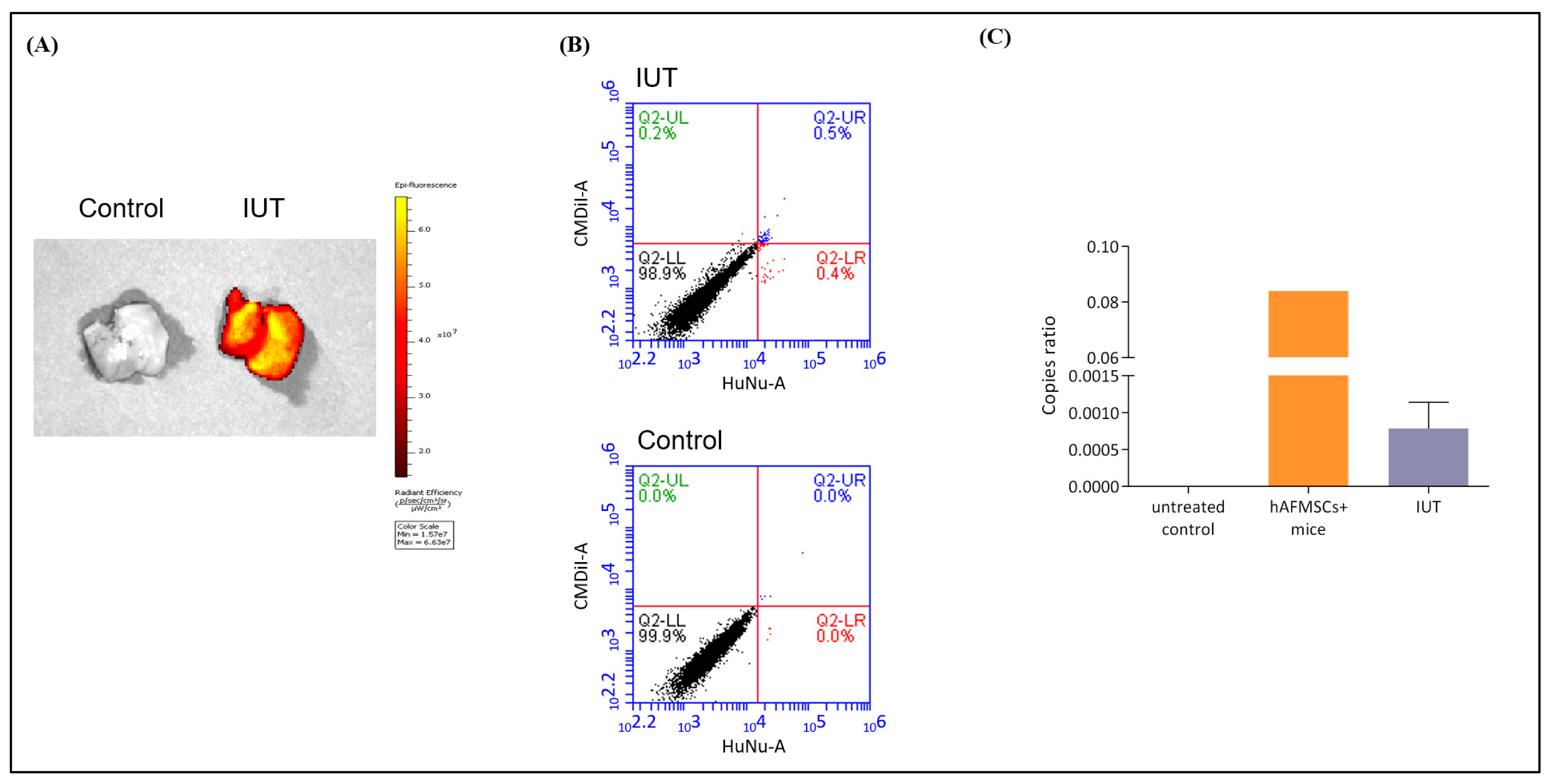
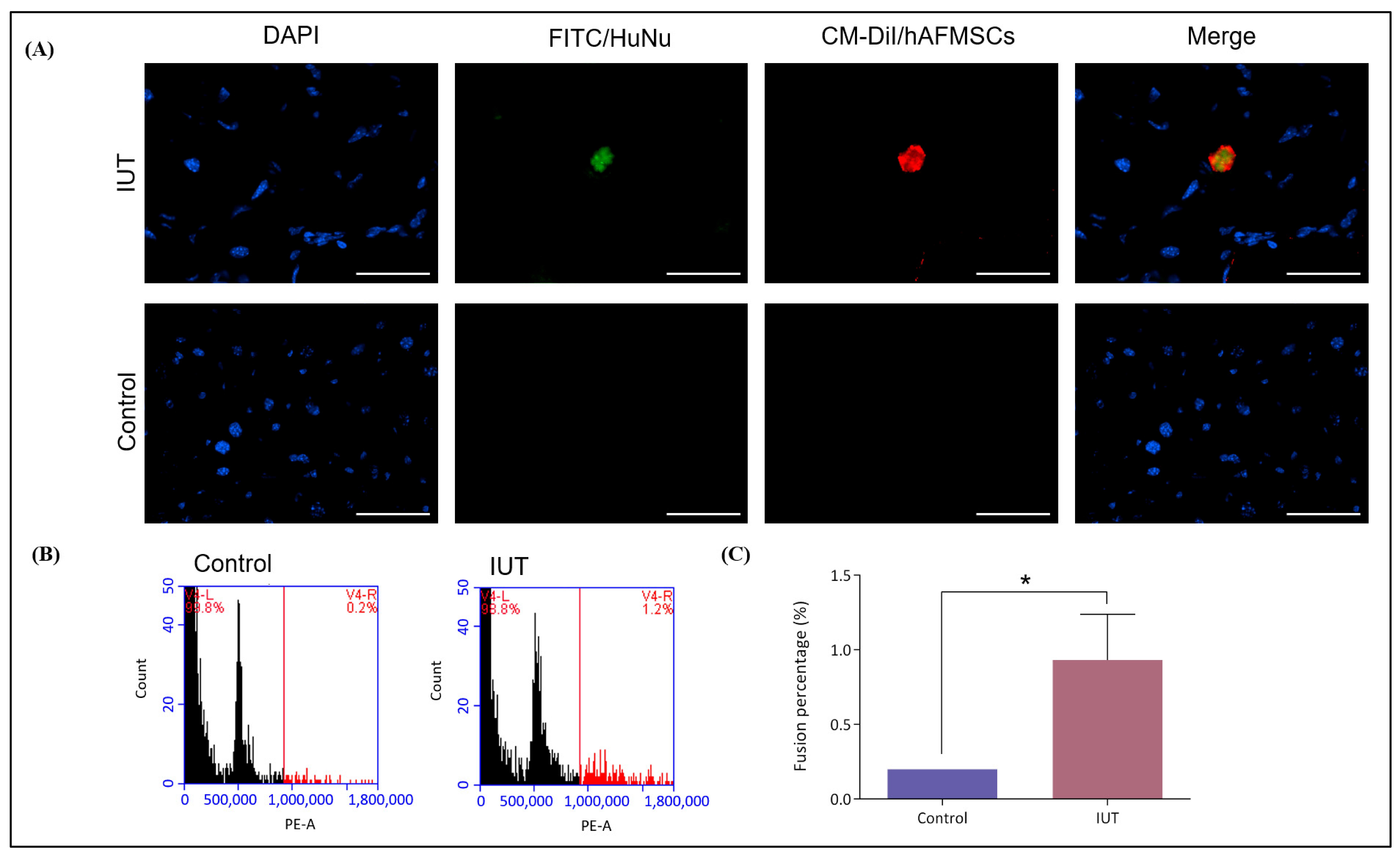
2.3. Detection of Human Cells in the Livers of Recipient Mice
2.4. Examination of the Human–Mousen Cell Fusion Phenomenon in Recipient Mice
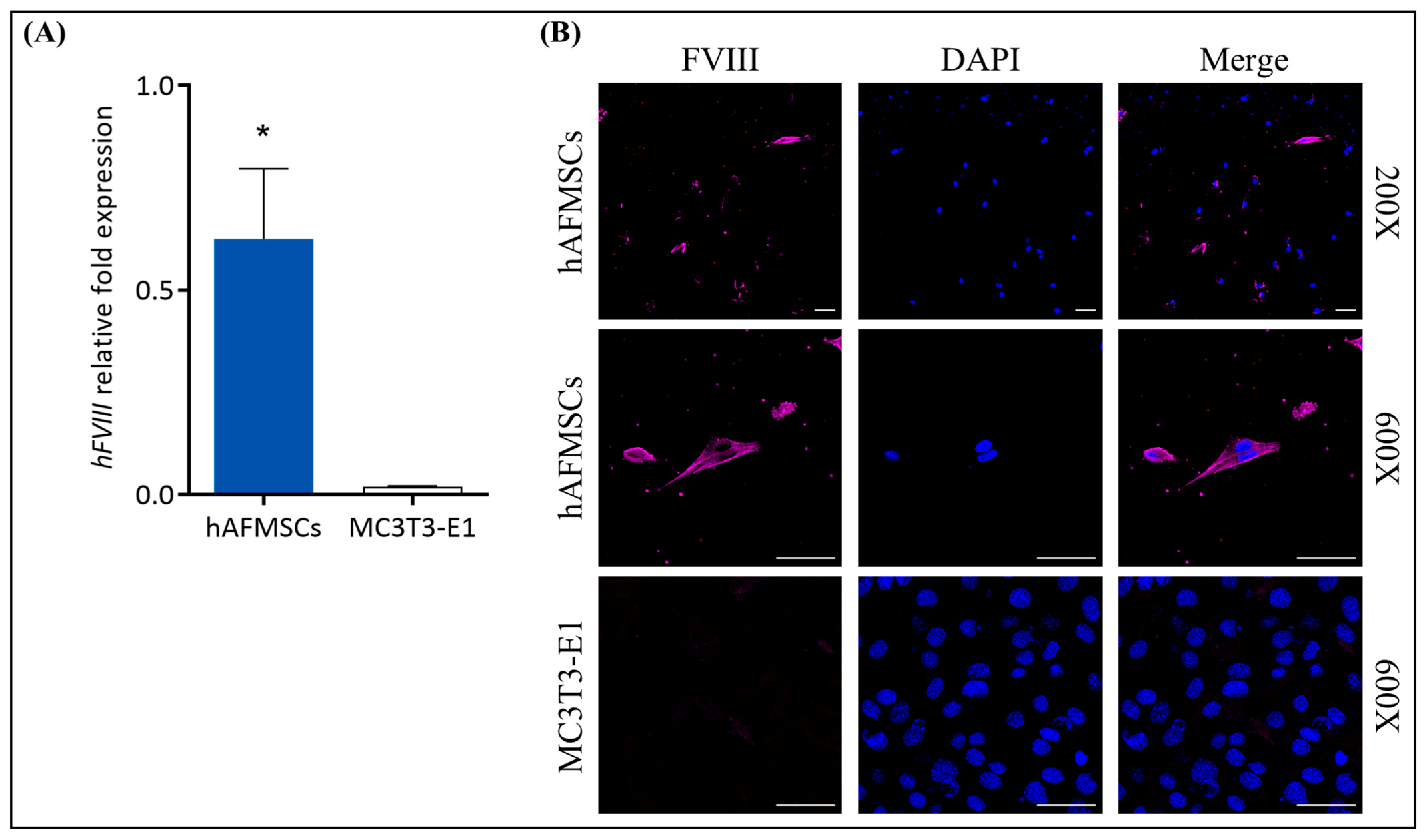
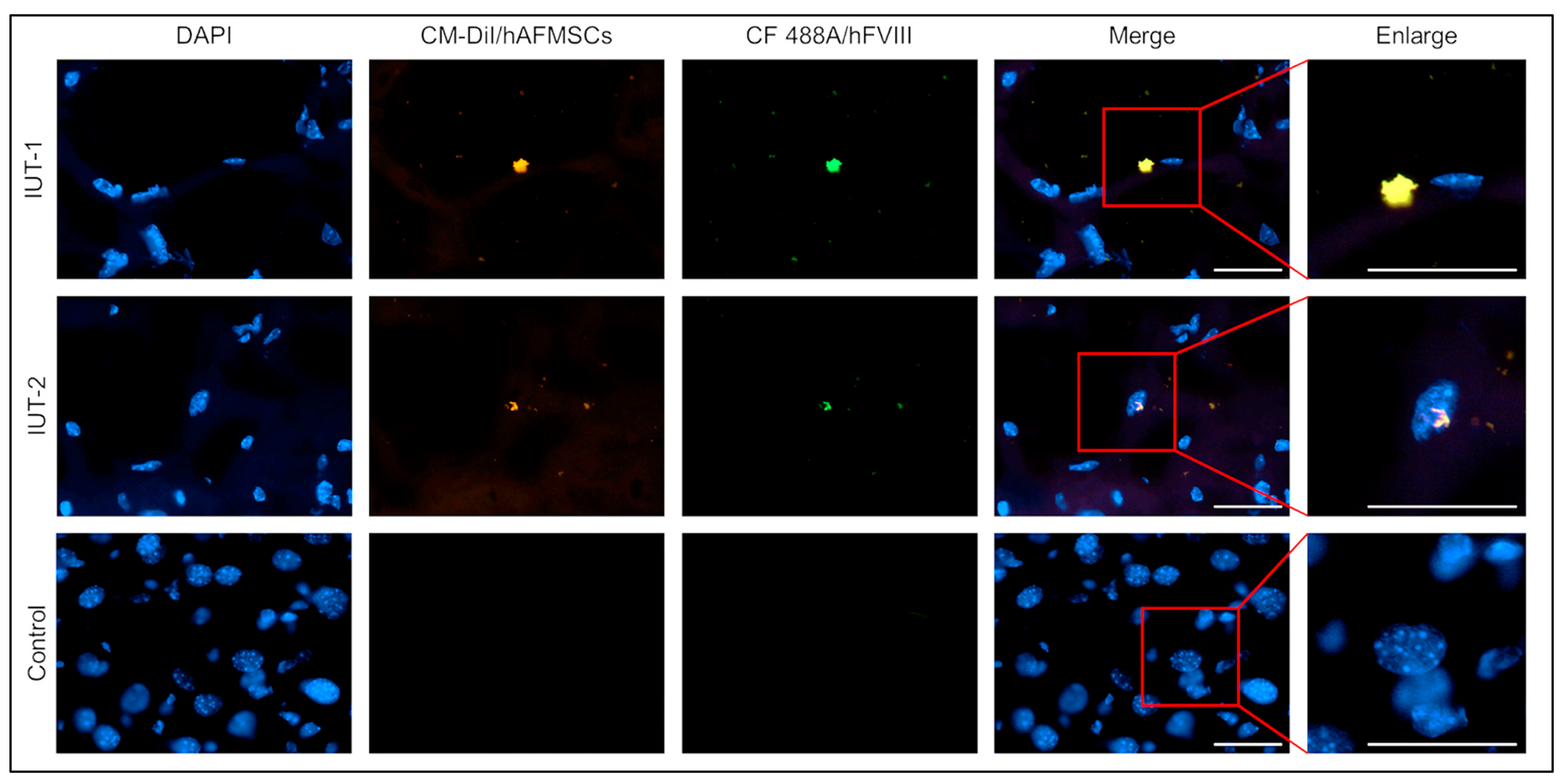
2.5. Observation of Human FVIII Protein in the Livers of Recipient Mice
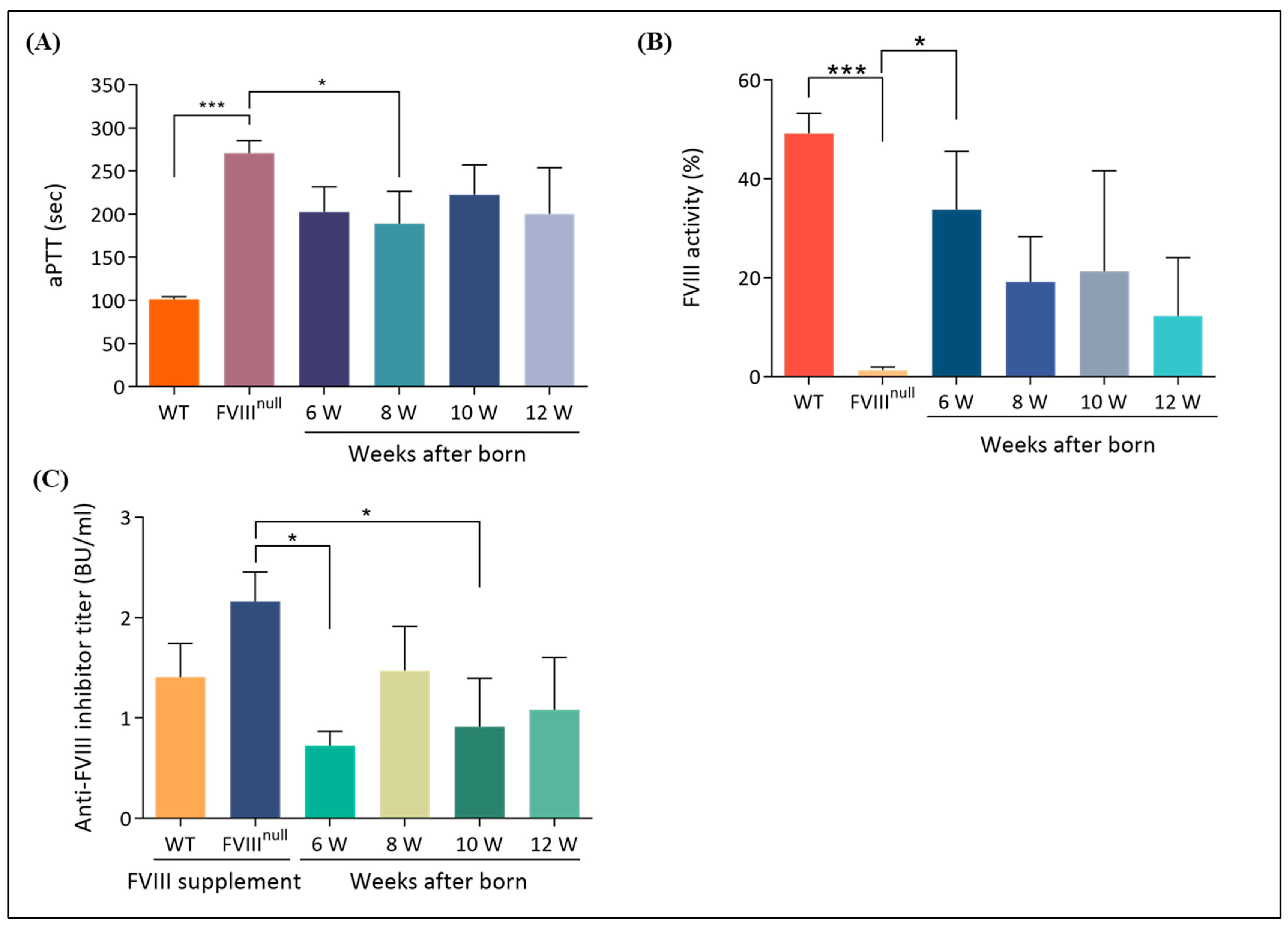
2.6. Coagulation Problems in Hemophilia A Mice Were Corrected via hAFMSCs In Utero Therapy
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Culture of Human Amniotic Fluid Mesenchymal Stromal Cells
4.3. Characterized Human MSC CD Markers of hAFMSCs
4.4. In Vitro Differentiation Test of hAFMSCs
4.5. Detection of FVIII mRNA Expression in hAFMSCs via qRT-PCR
4.6. Immunofluorescence Staining
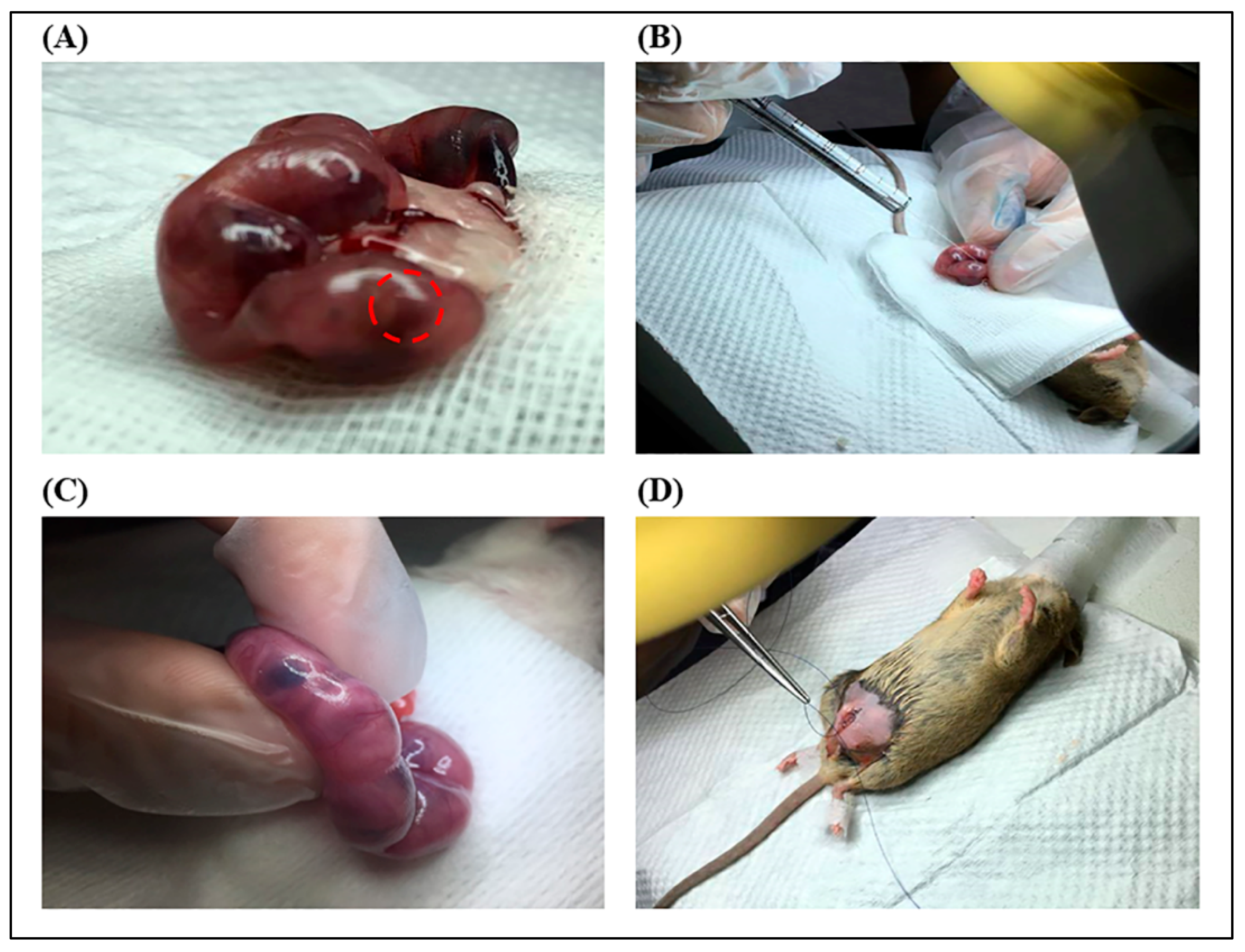
4.7. In Utero Transplantation (IUT) Procedure
4.8. Flow Cytometry Detection of Human Cells in Mice Livers
4.9. DNA Extraction and PCR
4.10. Droplet Digital PCR (ddPCR)
4.11. In Vivo Imaging System (IVIS)
4.12. Histological Examination
4.13. Cell Fusion Chromosome Test
4.14. The activated Partial Thromboplastin Time (aPTT) Assay
4.15. The FVIII Activity Assay
4.16. The FVIII Inhibitor Antibody Assay
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Srivastava, A.; Santagostino, E.; Dougall, A.; Kitchen, S.; Sutherland, M.; Pipe, S.W.; Carcao, M.; Mahlangu, J.; Ragni, M.V.; Windyga, J.; et al. WFH Guidelines for the Management of Hemophilia, 3rd Edition. Haemophilia 2020, 26, 1–158. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Brewer, A.K.; Mauser-Bunschoten, E.P.; Key, N.S.; Kitchen, S.; Llinas, A.; Ludlam, C.A.; Mahlangu, J.N.; Mulder, K.; Poon, M.C.; et al. Guidelines for the Management of Hemophilia. Haemophilia 2013, 19, e1–e47. [Google Scholar] [CrossRef]
- Yen, C.C.; Liu, Y.W.; Chang, G.R.L.; Lan, Y.W.; Kao, Y.T.; Cheng, S.N.; Chen, W.; Chen, C.M. Therapeutic effects of kefir peptides on hemophilia-induced osteoporosis in mice with deficient coagulation factor VIII. Front. Cell Dev. Biol. 2022, 10, 794198. [Google Scholar] [CrossRef] [PubMed]
- Paris, L.Q. Foundations of hemophilia and epidemiology. Blood Coagul. Fibrinolysis 2023, 34, S35–S36. [Google Scholar] [CrossRef]
- D’Angiolella, L.S.; Cortesi, P.A.; Rocino, A.; Coppola, A.; Hassan, H.J.; Giampaolo, A.; Solimeno, L.P.; Lafranconi, A.; Micale, M.; Mangano, S.; et al. The socioeconomic burden of patients affected by hemophilia with inhibitors. Eur. J. Haematol. 2018, 101, 435–456. [Google Scholar] [CrossRef]
- Hermans, C.; Makris, M. Disruptive technology and hemophilia care: The multiple impacts of emicizumab. Res. Pract. Thromb. Haemost. 2021, 5, e12508. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.T.; Chen, Y.T.; Fan, H.C.; Tsai, T.C.; Cheng, S.N.; Lai, P.S.; Chen, J.K.; Chen, C.M. Novel coagulation factor VIII gene therapy in a mouse model of hemophilia A by lipid-coated Fe3O4 nanoparticles. Biomedicines 2021, 9, 1116. [Google Scholar] [CrossRef] [PubMed]
- Astermark, J. FVIII inhibitors: Pathogenesis and avoidance. Blood 2015, 125, 2045–2051. [Google Scholar] [CrossRef]
- Ananyeva, N.M.; Lacroix-Desmazes, S.; Hauser, C.A.; Shima, M.; Ovanesov, M.V.; Khrenov, A.V.; Saenko, E.L. Inhibitors in hemophilia A: Mechanisms of inhibition, management and perspectives. Blood Coagul. Fibrinolysis 2004, 15, 109–124. [Google Scholar] [CrossRef]
- Perin, L.; Sedrakyan, S.; Da Sacco, S.; De Filippo, R. Characterization of human amniotic fluid stem cells and their pluripotential capability. In Methods in Cell Biology; Stem Cell Culture; Academic Press: Cambridge, MA, USA, 2008; Volume 86, pp. 85–99. [Google Scholar]
- Phermthai, T.; Odglun, Y.; Julavijitphong, S.; Titapant, V.; Chuenwattana, P.; Vantanasiri, C.; Pattanapanyasat, K. A novel method to derive amniotic fluid stem cells for therapeutic purposes. BMC Cell Biol. 2010, 11, 79. [Google Scholar] [CrossRef]
- Siegel, N.; Rosner, M.; Hanneder, M.; Valli, A.; Hengstschläger, M. Stem cells in amniotic fluid as new tools to study human genetic diseases. Stem Cell Rev. 2007, 3, 256–264. [Google Scholar] [CrossRef]
- Siegel, N.; Rosner, M.; Hanneder, M.; Freilinger, A.; Hengstschläger, M. Human amniotic fluid stem cells: A new perspective. Amino Acids 2008, 35, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.C.; Pitacco, P.; Nulty, J.; Cunniffe, G.M.; Kelly, D.J. 3D printed microchannel networks to direct vascularization during endochondral bone repair. Biomaterials 2018, 162, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Ibraheim, H.; Giacomini, C.; Kassam, Z.; Dazzi, F.; Powell, N. Advances in mesenchymal stromal cell therapy in the management of Crohn’s disease. Expert. Rev. Gastroenterol. Hepatol. 2018, 12, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.Y.; Liu, S.A.; Sheu, M.L.; Chen, F.C.; Chen, C.J.; Su, H.L.; Pan, H.C. Feasibility of human amniotic fluid derived stem cells in alleviation of neuropathic pain in chronic constrictive injury nerve model. PLoS ONE 2016, 11, e0159482. [Google Scholar] [CrossRef]
- Götherström, C.; Westgren, M.; Shaw, S.W.S.; Åström, E.; Biswas, A.; Byers, P.H.; Mattar, C.N.Z.; Graham, G.E.; Taslimi, J.; Ewald, U.; et al. Pre- and postnatal transplantation of fetal mesenchymal stem cells in osteogenesis imperfecta: A two-center experience. Stem Cells Transl. Med. 2014, 3, 255–264. [Google Scholar] [CrossRef]
- Sharma, A.; George, L.; Erskin, K. Osteogenesis imperfecta in pregnancy: Two case reports and review of literature. Obs. Gynecol. Surv. 2001, 56, 563–566. [Google Scholar] [CrossRef]
- Peranteau, W.H.; Flake, A.W. The future of in utero gene therapy. Mol. Diagn. Ther. 2020, 24, 135–142. [Google Scholar] [CrossRef]
- Clapp, D.W. Developmental regulation of the immune system. Semin. Perinatol. 2006, 30, 69–72. [Google Scholar] [CrossRef]
- Miles, B.S.; Anderson, P.; Agostino, A.; Golomb, M.R.; Achonu, C.; Blanchette, V.; Feldman, B.M.; McLimont, M.; Revel-Vilk, S.; Stain, A.; et al. Effect of intracranial bleeds on the neurocognitive, academic, behavioural and adaptive functioning of boys with haemophilia. Haemophilia 2012, 18, 229–234. [Google Scholar] [CrossRef]
- Stieltjes, N.; Calvez, T.; Demiguel, V.; Torchet, M.F.; Briquel, M.E.; Fressinaud, E.; Claeyssens, S.; Coatmelec, B.; Chambost, H.; Group, T.F.I.S. Intracranial haemorrhages in French haemophilia patients (1991–2001): Clinical presentation, management and prognosis factors for death. Haemophilia 2005, 11, 452–458. [Google Scholar] [CrossRef]
- Kulkarni, R.; Soucie, J.M.; Lusher, J.; Presley, R.; Shapiro, A.; Gill, J.; Manco-Johnson, M.; Koerper, M.; Mathew, P.; Abshire, T.; et al. Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: A report from the Centers for Disease Control and Prevention’s (CDC) universal data collection (UDC) project. Haemophilia 2009, 15, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Stem, C.; Rodman, C.; Ramamurthy, R.M.; George, S.; Meares, D.; Farland, A.; Atala, A.; Doering, C.B.; Spencer, H.T.; Porada, C.D.; et al. Investigating optimal autologous cellular platforms for prenatal or perinatal factor VIII delivery to treat hemophilia a. Front. Cell Dev. Biol. 2021, 9, 678117. [Google Scholar] [CrossRef]
- Velasquillo, C.; Madrazo-Ibarra, A.; Gutiérrez-Gómez, C.; Pérez-Dosal, M.R.; Melgarejo-Ramírez, Y.; Ibarra, C. Stem cells and tissue engineering: An alternative treatment for craniofacial congenital malformations and articular degenerative diseases. Plast. Aesthetic Res. 2020, 7, 31. [Google Scholar] [CrossRef]
- Ghoryani, M.; Shariati-Sarabi, Z.; Tavakkol-Afshari, J.; Ghasemi, A.; Poursamimi, J.; Mohammadi, M. Amelioration of clinical symptoms of patients with refractory rheumatoid arthritis following treatment with autologous bone marrow-derived mesenchymal stem cells: A successful clinical trial in Iran. Biomed. Pharmacother. 2019, 109, 1834–1840. [Google Scholar] [CrossRef]
- Kim, S.H.; Cho, J.H.; Lee, Y.H.; Lee, J.H.; Kim, S.S.; Kim, M.Y.; Lee, M.G.; Kang, W.Y.; Lee, K.S.; Ahn, Y.K.; et al. Improvement in left ventricular function with intracoronary mesenchymal stem cell therapy in a patient with anterior wall ST-segment elevation myocardial infarction. Cardiovasc. Drugs Ther. 2018, 32, 329–338. [Google Scholar] [CrossRef]
- Oh, K.W.; Noh, M.Y.; Kwon, M.S.; Kim, H.Y.; Oh, S.I.; Park, J.; Kim, H.J.; Ki, C.S.; Kim, S.H. Repeated intrathecal mesenchymal stem cells for amyotrophic lateral sclerosis. Ann. Neurol. 2018, 84, 361–373. [Google Scholar] [CrossRef]
- Lan, Y.W.; Yang, J.C.; Yen, C.C.; Huang, T.T.; Chen, Y.C.; Chen, H.L.; Chong, K.Y.; Chen, C.M. Predifferentiated amniotic fluid mesenchymal stem cells enhance lung alveolar epithelium regeneration and reverse elastase-induced pulmonary emphysema. Stem Cell Res. Ther. 2019, 10, 163. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Xie, M.; Zhou, M.; Liu, X.; Hu, Z.; Wu, L. Restoration of FVIII function and phenotypic rescue in hemophilia A mice by transplantation of MSCs derived from F8-modified iPSCs. Front. Cell Dev. Biol. 2021, 9, 630353. [Google Scholar] [CrossRef]
- Boelig, M.M.; Kim, A.G.; Stratigis, J.D.; McClain, L.E.; Li, H.; Flake, A.W.; Peranteau, W.H. The intravenous route of injection optimizes engraftment and survival in the murine model of in utero hematopoietic cell transplantation. Biol Blood Marrow Transpl. 2016, 22, 991–999. [Google Scholar] [CrossRef]
- Nijagal, A.; Le, T.; Wegorzewska, M.; Mackenzie, T.C. A mouse model of in utero transplantation. J. Vis. Exp. 2011, 47, 2303. [Google Scholar] [CrossRef]
- De Santis, M.; De Luca, C.; Mappa, I.; Cesari, E.; Quattrocchi, T.; Spagnuolo, T.; Visconti, D.; Noia, G.; Caruso, A. In-utero stem cell transplantation: Clinical use and therapeutic potential. Minerva Ginecol. 2011, 63, 387–398. [Google Scholar]
- Vrecenak, J.D.; Flake, A.W. In utero hematopoietic cell transplantation—Recent progress and the potential for clinical application. Cytotherapy 2013, 15, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Cantore, A.; Ranzani, M.; Bartholomae, C.C.; Volpin, M.; Valle, P.D.; Sanvito, F.; Sergi, L.S.; Gallina, P.; Benedicenti, F.; Bellinger, D.; et al. Liver-directed lentiviral gene therapy in a dog model of hemophilia B. Sci. Transl. Med. 2015, 7, 277ra28. [Google Scholar] [CrossRef] [PubMed]
- Alwahsh, S.M.; Rashidi, H.; Hay, D.C. Liver cell therapy: Is this the end of the beginning? Cell. Mol. Life Sci. 2018, 75, 1307–1324. [Google Scholar] [CrossRef] [PubMed]
- Ekblad-Nordberg, Å.; Walther-Jallow, L.; Westgren, M.; Götherström, C. Prenatal stem cell therapy for inherited diseases: Past, present, and future treatment strategies. Stem Cells Transl. Med. 2020, 9, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Lipshutz, G.S.; Gruber, C.A.; Cao, Y.; Hardy, J.; Contag, C.H.; Gaensler, K.M.L. In utero delivery of adeno-associated viral vectors: Intraperitoneal gene transfer produces long-term expression. Mol. Ther. 2001, 3, 284–292. [Google Scholar] [CrossRef]
- Waddington, S.N.; Buckley, S.M.K.; Nivsarkar, M.; Jezzard, S.; Schneider, H.; Dahse, T.; Kemball-Cook, G.; Miah, M.; Tucker, N.; Dallman, M.J.; et al. In utero gene transfer of human factor IX to fetal mice can induce postnatal tolerance of the exogenous clotting factor. Blood 2003, 101, 1359–1366. [Google Scholar] [CrossRef]
- Almeida-Porada, G.; Atala, A.; Porada, C.D. In utero stem cell transplantation and gene therapy: Rationale, history, and recent advances toward clinical application. Mol. Ther. Methods Clin. Dev. 2016, 3, 16020. [Google Scholar] [CrossRef]
- Mattar, C.N.; Nathwani, A.C.; Waddington, S.N.; Dighe, N.; Kaeppel, C.; Nowrouzi, A.; Mcintosh, J.; Johana, N.B.; Ogden, B.; Fisk, N.M.; et al. Stable human FIX expression after 0.9G intrauterine gene transfer of self-complementary adeno-associated viral vector 5 and 8 in macaques. Mol. Ther. 2011, 19, 1950–1960. [Google Scholar] [CrossRef]
- Vanover, M.; Wang, A.; Farmer, D. Potential clinical applications of placental stem cells for use in fetal therapy of birth defects. Placenta 2017, 59, 107–112. [Google Scholar] [CrossRef]
- Antonucci, I.; Stuppia, L.; Kaneko, Y.; Yu, S.; Tajiri, N.; Bae, E.C.; Chheda, S.H.; Weinbren, N.L.; Borlongan, C.V. Amniotic fluid as a rich source of mesenchymal stromal cells for transplantation therapy. Cell Transpl. 2011, 20, 789–796. [Google Scholar] [CrossRef]
- Rezaee, F.; Peppelenbosch, M.; Dashty, M. Donor chimera model for tolerance induction in transplantation. Hum. Immunol. 2013, 74, 550–556. [Google Scholar] [CrossRef]
- Peng, S.Y.; Chen, Y.H.; Chou, C.J.; Wang, Y.H.; Lee, H.M.; Cheng, W.T.K.; Shaw, S.W.S.; Wu, S.C. Cell fusion phenomena detected after in utero transplantation of Ds-red-harboring porcine amniotic fluid stem cells into EGFP transgenic mice. Prenat. Diagn. 2014, 34, 487–495. [Google Scholar] [CrossRef]
- Freeman, B.T.; Ogle, B.M. Viral-mediated fusion of mesenchymal stem cells with cells of the infarcted heart hinders healing via decreased vascularization and immune modulation. Sci. Rep. 2016, 6, 20283. [Google Scholar] [CrossRef]
- Sokal, E.M.; Lombard, C.; Mazza, G. Mesenchymal stem cell treatment for hemophilia: A review of current knowledge. J. Thromb. Haemost. 2015, 13, S161–S166. [Google Scholar] [CrossRef]
- Baranovskii, D.S.; Klabukov, I.D.; Arguchinskaya, N.V.; Yakimova, A.O.; Kisel, A.A.; Yatsenko, E.M.; Ivanov, S.A.; Shegay, P.V.; Kaprin, A.D. Adverse events, side effects and complications in mesenchymal stromal cell-based therapies. Stem Cell Investig. 2022, 9, 7. [Google Scholar] [CrossRef]
- von Bahr, L.; Sundberg, B.; Lönnies, L.; Sander, B.; Karbach, H.; Hägglund, H.; Ljungman, P.; Gustafsson, B.; Karlsson, H.; Le Blanc, K.; et al. Long-term complications, immunologic effects, and role of passage for outcome in mesenchymal stromal cell therapy. Biol Blood Marrow Transpl. 2012, 18, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Yu, K.R. Impact of mesenchymal stem cell senescence on inflammaging. BMB Rep. 2020, 53, 65. [Google Scholar] [CrossRef] [PubMed]
- Lyamina, S.; Baranovskii, D.; Kozhevnikova, E.; Ivanova, T.; Kalish, S.; Sadekov, T.; Klabukov, I.; Maev, I.; Govorun, V. Mesechymal stromal cells as a driver of inflammaging. Int. J. Mol. Sci. 2023, 24, 6372. [Google Scholar] [CrossRef] [PubMed]
- Cafuir, L.A.; Kempton, C.L. Current and emerging factor VIII replacement products for hemophilia A. Ther. Adv. Hematol. 2017, 8, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Porada, C.D.; Almeida-Porada, G. Mechanistic insights into factor VIII immune tolerance induction via prenatal cell therapy in hemophilia A. Curr. Stem Cell Rep. 2019, 5, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Gao, K.; Wang, C.; Pivetti, C.; Lankford, L.; Farmer, D.; Wang, A. In utero transplantation of placenta-derived mesenchymal stromal cells for potential fetal treatment of hemophilia A. Cell Transpl. 2018, 27, 130–139. [Google Scholar] [CrossRef]
- Mold, J.E.; McCune, J.M. Immunological tolerance during fetal development: From mouse to man. Adv. Immunol. 2012, 115, 73–111. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Lan, Y.W.; Huang, S.M.; Yen, C.C.; Chen, W.; Wu, W.J.; Staniczek, T.; Chong, K.Y.; Chen, C.M. Human amniotic fluid mesenchymal stem cells attenuate pancreatic cancer cell proliferation and tumor growth in an orthotopic xenograft mouse model. Stem Cell Res. Ther. 2022, 13, 235. [Google Scholar] [CrossRef]
- Wen, S.T.; Chen, W.; Chen, H.L.; Lai, C.W.; Yen, C.C.; Lee, K.H.; Wu, S.C.; Chen, C.M. Amniotic fluid stem cells from EGFP transgenic mice attenuate hyperoxia-induced acute lung injury. PLoS ONE 2013, 8, e75383. [Google Scholar] [CrossRef]
- Chuang, K.C.; Lai, Y.W.; Ko, C.H.; Yen, C.C.; Chen, H.L.; Lan, Y.W.; Chen, C.F.; Chen, W.; Chen, C.M. Therapeutic effects of kefir peptides on adjuvant-induced arthritis in rats through anti-inflammation and downregulation of matrix metalloproteinases. Life Sci. 2023, 317, 121411. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.C.; Chang, W.H.; Tung, M.C.; Chen, H.L.; Liu, H.C.; Liao, C.H.; Lan, Y.W.; Chong, K.Y.; Yang, S.H.; Chen, C.M. Lactoferrin protects hyperoxia-induced lung and kidney systemic inflammation in an in vivo imaging model of NF-κB/luciferase transgenic mice. Mol. Imaging Biol. 2020, 22, 526–538. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.W.; Chen, C.M.; Chong, K.Y. In vitro methods to evaluate the effects of mesenchymal stem cells on TGF-β1-induced pulmonary fibrosis. Methods Mol. Biol. 2021, 2269, 83–92. [Google Scholar] [CrossRef]
- Kuo, C.H.; Lee, I.C.; Huang, B.J.; Chen, C.M.; Liou, Y.M. Effects of aldo-keto reductase family 1 member A on osteoblast differentiation associated with lactate production in MC3T3-E1 preosteoblastic cells. Biochem. Cell Biol. 2022, 100, 413–424. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kao, Y.-T.; Yen, C.-C.; Fan, H.-C.; Chen, J.-K.; Chen, M.-S.; Lan, Y.-W.; Yang, S.-H.; Chen, C.-M. In Utero Cell Treatment of Hemophilia A Mice via Human Amniotic Fluid Mesenchymal Stromal Cell Engraftment. Int. J. Mol. Sci. 2023, 24, 16411. https://doi.org/10.3390/ijms242216411
Kao Y-T, Yen C-C, Fan H-C, Chen J-K, Chen M-S, Lan Y-W, Yang S-H, Chen C-M. In Utero Cell Treatment of Hemophilia A Mice via Human Amniotic Fluid Mesenchymal Stromal Cell Engraftment. International Journal of Molecular Sciences. 2023; 24(22):16411. https://doi.org/10.3390/ijms242216411
Chicago/Turabian StyleKao, Yung-Tsung, Chih-Ching Yen, Hueng-Chuen Fan, Jen-Kun Chen, Ming-Shan Chen, Ying-Wei Lan, Shang-Hsun Yang, and Chuan-Mu Chen. 2023. "In Utero Cell Treatment of Hemophilia A Mice via Human Amniotic Fluid Mesenchymal Stromal Cell Engraftment" International Journal of Molecular Sciences 24, no. 22: 16411. https://doi.org/10.3390/ijms242216411
APA StyleKao, Y. -T., Yen, C. -C., Fan, H. -C., Chen, J. -K., Chen, M. -S., Lan, Y. -W., Yang, S. -H., & Chen, C. -M. (2023). In Utero Cell Treatment of Hemophilia A Mice via Human Amniotic Fluid Mesenchymal Stromal Cell Engraftment. International Journal of Molecular Sciences, 24(22), 16411. https://doi.org/10.3390/ijms242216411