
Is Intrinsic Cardioprotection a Laboratory Phenomenon or a Clinically Relevant Tool to Salvage the Failing Heart?

 , , ,
, , ,  ,
,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
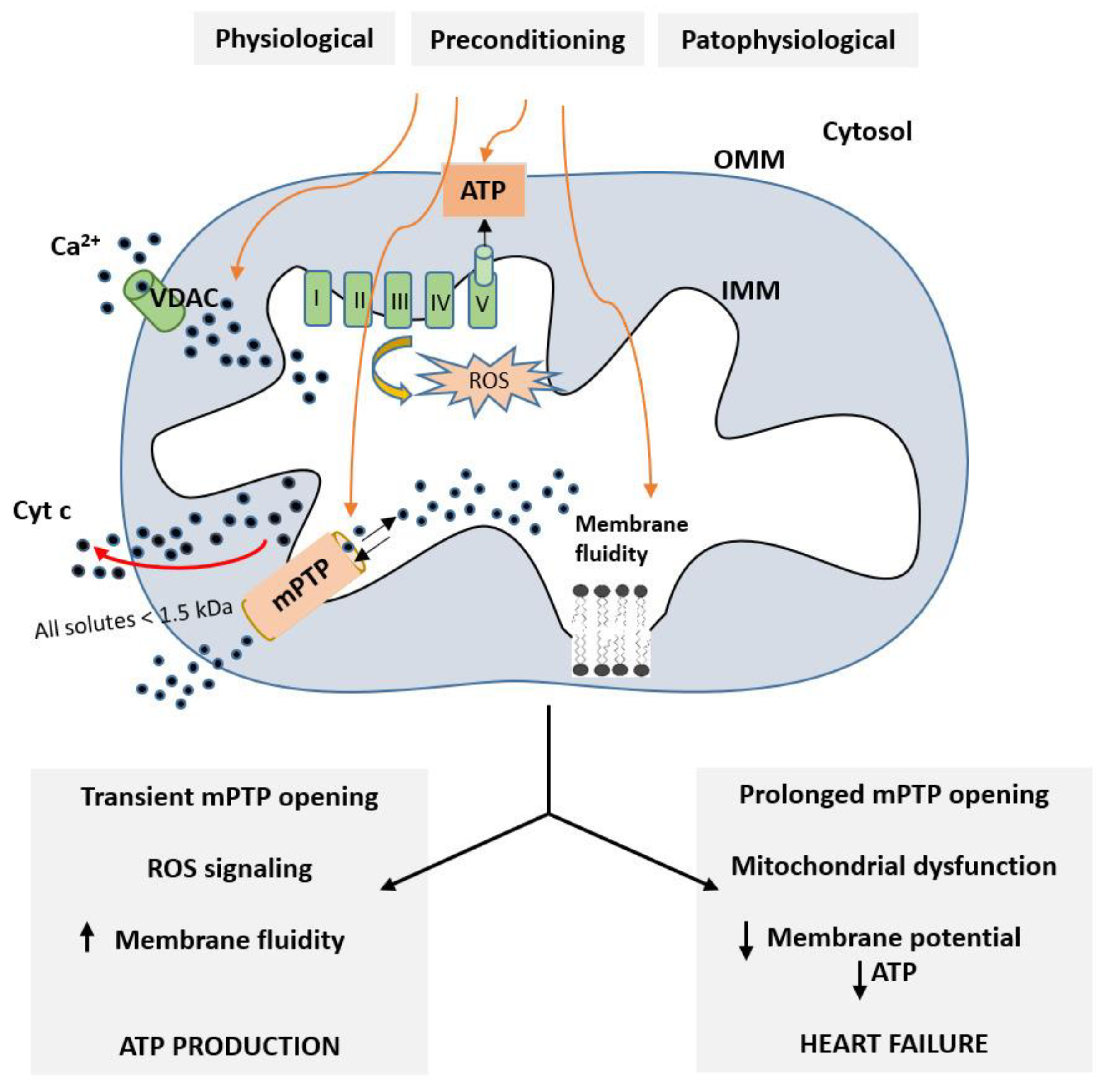{kind=link}
Abstract
:1. Introduction
2. Development of Heart Failure
2.1. Severity of Heart Failure Is Sex- and Aging-Related
2.2. Lifestyle Risk Factors
3. Pathophysiological Mechanisms of Heart Failure
Cell Death Mechanisms
4. Management of Heart Failure
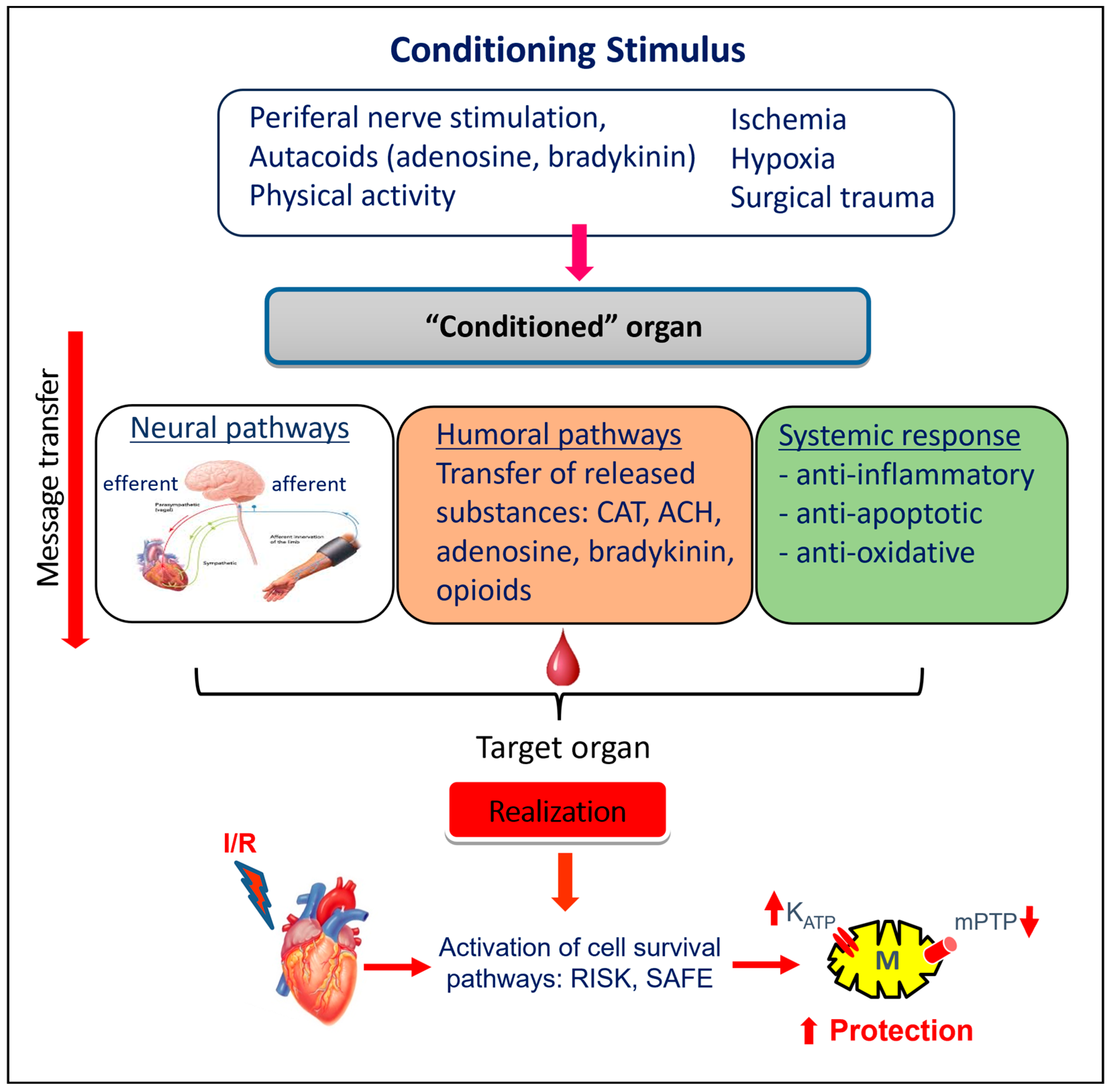
5. Innate Cardioprotection
5.1. Short-Term Cardiac Endogenous Protection—Ischemic “Preconditioning”
Ischemic “Postconditioning”
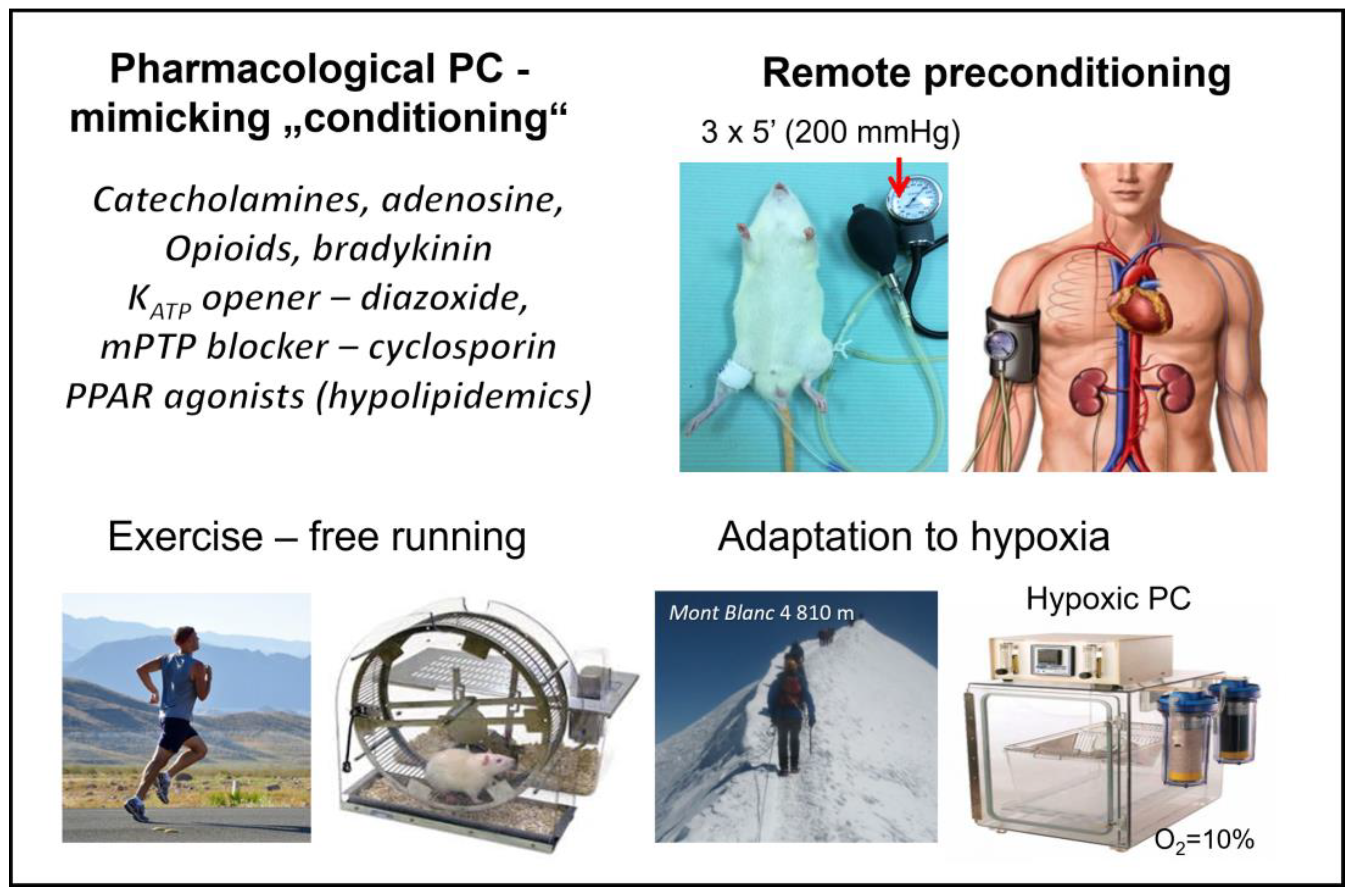
5.2. Other “Conditioning” Interventions
5.2.1. Remote Ischemic “Preconditioning”
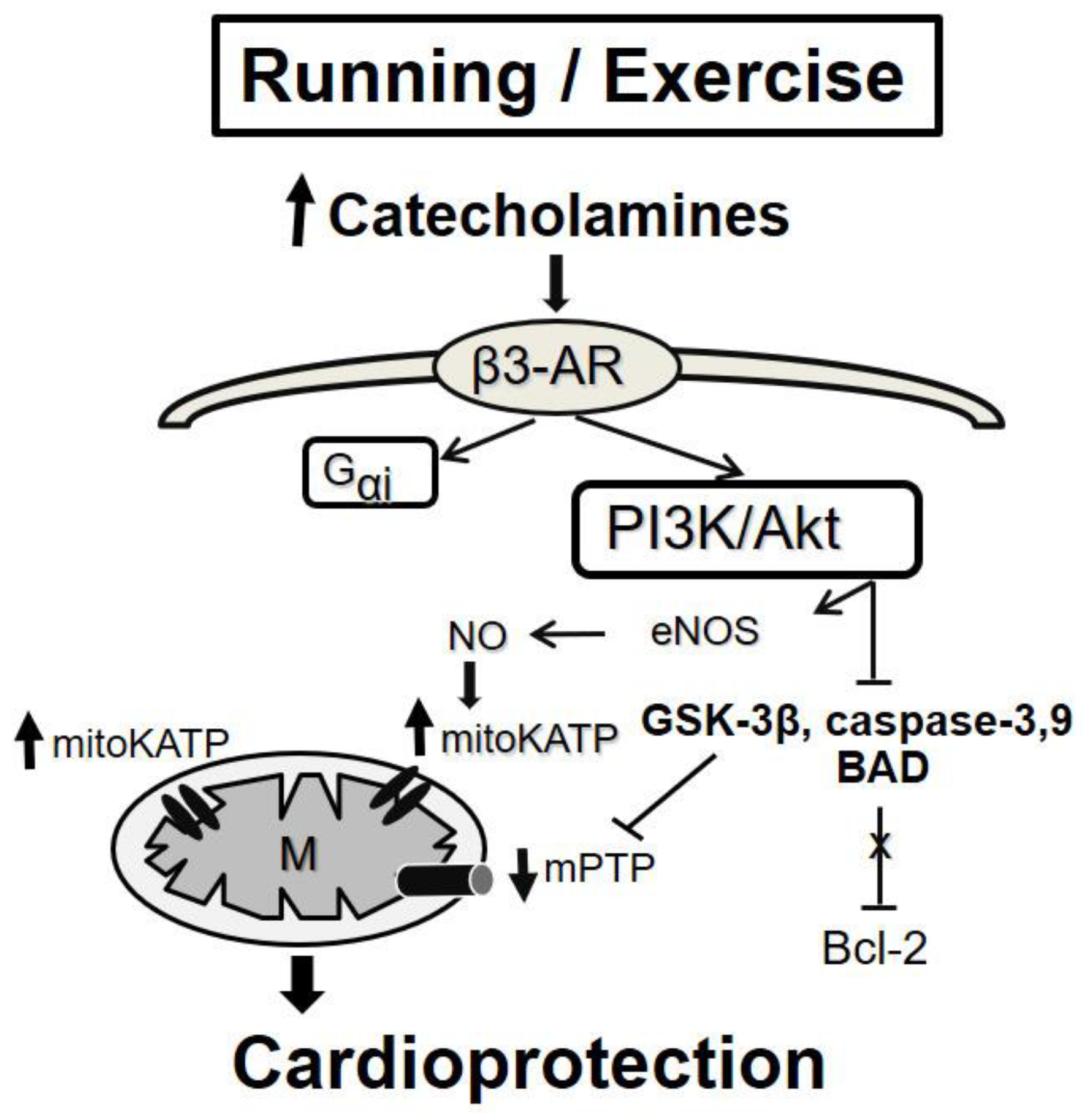
5.2.2. Exercise-Induced “Conditioning”
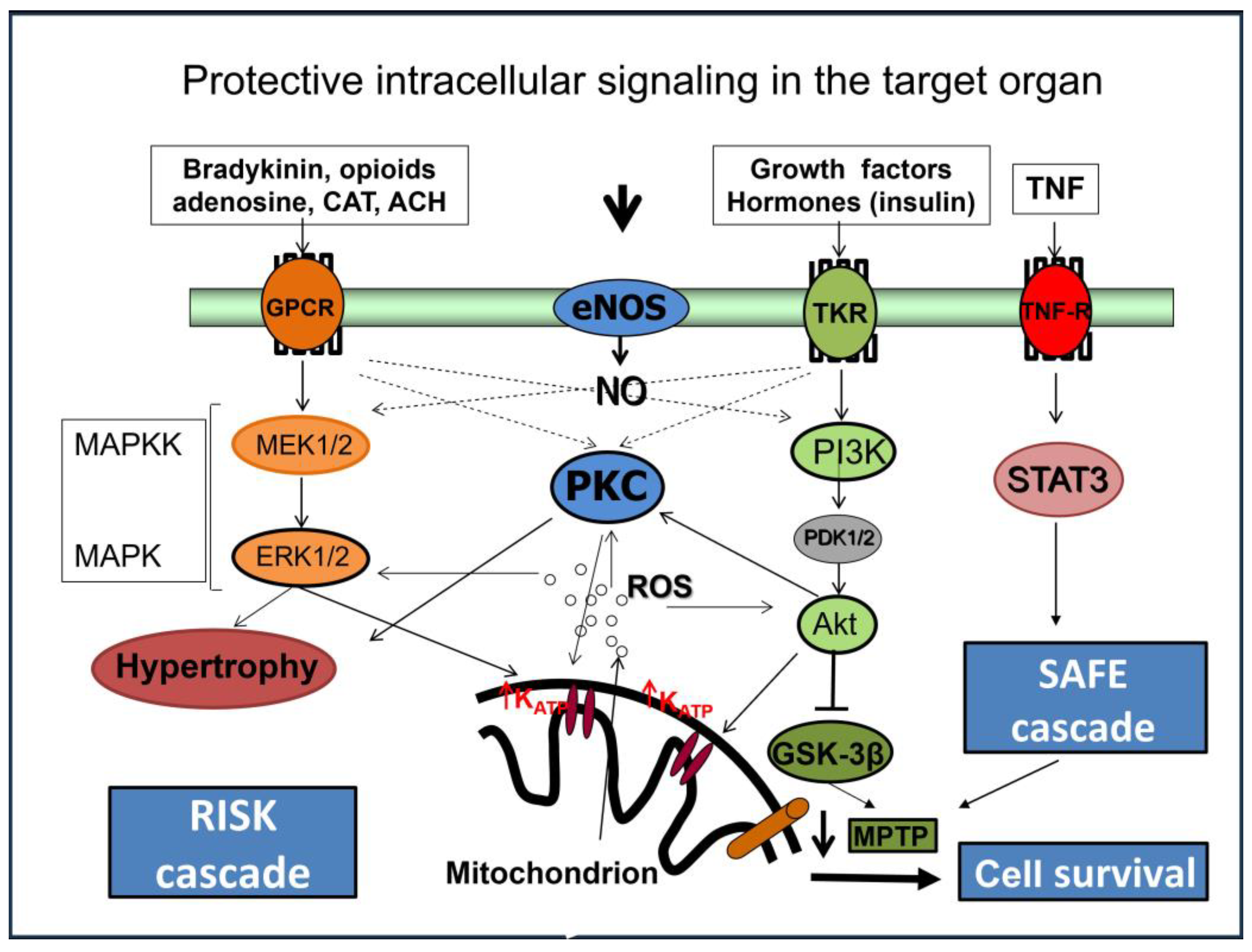
6. Intracellular Mechanisms Involved in Cardioprotection of “Conditioning”
6.1. Micro-RNAs
Long Noncoding RNAs
6.2. Peroxisome Proliferator-Activated Receptors
Role of Mitochondria in Cardioprotective Mechanisms
7. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- World Health Organization. Cardiovascular Diseases—Fact Sheet Number 317; WHO: Geneva, Switzerland, 2021; Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 13 October 2023).
- Braunwald, E. The War against Heart Failure: The Lancet Lecture. Lancet 2015, 385, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Bøtker, H.E.; Schmidt, M.R. The Potential for Remote Ischemic Conditioning to Improve Outcomes in Heart Failure. Expert. Rev. Cardiovasc. Ther. 2015, 13, 1173–1176. [Google Scholar] [CrossRef] [PubMed]
- Askoxylakis, V.; Thieke, C.; Pleger, S.T.; Most, P.; Tanner, J.; Lindel, K.; Katus, H.A.; Debus, J.; Bischof, M. Long-Term Survival of Cancer Patients Compared to Heart Failure and Stroke: A Systematic Review. BMC Cancer 2010, 10, 105. [Google Scholar] [CrossRef]
- Heusch, G. Myocardial Ischaemia-Reperfusion Injury and Cardioprotection in Perspective. Nat. Rev. Cardiol. 2020, 17, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Bøtker, H.E.; Heusch, G.; Ibáñez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Bulluck, H.; Yellon, D.M.; Hausenloy, D.J. Reducing Myocardial Infarct Size: Challenges and Future Opportunities. Heart 2016, 102, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with Ischemia: A Delay of Lethal Cell Injury in Ischemic Myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef]
- Liang, Y.; Li, Y.P.; He, F.; Liu, X.Q.; Zhang, J.Y. Long-Term, Regular Remote Ischemic Preconditioning Improves Endothelial Function in Patients with Coronary Heart Disease. Braz. J. Med. Biol. Res. 2015, 48, 568–576. [Google Scholar] [CrossRef]
- Sutton, M.G.; Sharpe, N. Left Ventricular Remodeling after Myocardial Infarction: Pathophysiology and Therapy. Circulation 2000, 101, 2981–2988. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Saini-Chohan, H.K.; Rodriguez-Leyva, D.; Elimban, V.; Dent, M.R.; Tappia, P.S. Subcellular Remodelling May Induce Cardiac Dysfunction in Congestive Heart Failure. Cardiovasc. Res. 2008, 81, 429–438. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Shah, A.K.; Adameova, A.; Bartekova, M. Role of Oxidative Stress in Cardiac Dysfunction and Subcellular Defects Due to Ischemia-Reperfusion Injury. Biomedicines 2022, 10, 1473. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Qi, Y.; Xu, L.; Tao, X.; Han, X.; Yin, L.; Peng, J. MicroRNA-140-5p Aggravates Doxorubicin-Induced Cardiotoxicity by Promoting Myocardial Oxidative Stress via Targeting Nrf2 and Sirt2. Redox Biol. 2018, 15, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Piroth, M.D.; Baumann, R.; Budach, W.; Dunst, J.; Feyer, P.; Fietkau, R.; Haase, W.; Harms, W.; Hehr, T.; Krug, D.; et al. Heart Toxicity from Breast Cancer Radiotherapy: Current Findings, Assessment, and Prevention. Strahlenther. Onkol. 2019, 195, 1–12. [Google Scholar] [CrossRef]
- Xie, Y.; Collins, W.J.; Audeh, M.W.; Shiao, S.L.; Gottlieb, R.A.; Goodman, M.T.; Merz, C.N.B.; Mehta, P.K. Breast Cancer Survivorship and Cardiovascular Disease: Emerging Approaches in Cardio-Oncology. Curr. Treat. Options Cardiovasc. Med. 2015, 17, 60. [Google Scholar] [CrossRef] [PubMed]
- Oldman, A.H.; Martin, D.S.; Feelisch, M.; Grocott, M.P.W.; Cumpstey, A.F. Effects of Perioperative Oxygen Concentration on Oxidative Stress in Adult Surgical Patients: A Systematic Review. Br. J. Anaesth. 2021, 126, 622–632. [Google Scholar] [CrossRef]
- Lan, H.; Zheng, Q.; Wang, K.; Li, C.; Xiong, T.; Shi, J.; Dong, N. Cinnamaldehyde Protects Donor Heart from Cold Ischemia–Reperfusion Injury via the PI3K/AKT/MTOR Pathway. Biomed. Pharmacother. 2023, 165, 114867. [Google Scholar] [CrossRef] [PubMed]
- Abete, P.; Testa, G.; Ferrara, N.; De Santis, D.; Capaccio, P.; Viati, L.; Calabrese, C.; Cacciatore, F.; Longobardi, G.; Condorelli, M.; et al. Cardioprotective Effect of Ischemic Preconditioning Is Preserved in Food-Restricted Senescent Rats. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1978–H1987. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, H.; Xue, G.; Zhang, L.; Zhang, W.; Wang, L.; Lu, F.; Li, H.; Bai, S.; Lin, Y.; et al. Exercise Training Preserves Ischemic Preconditioning in Aged Rat Hearts by Restoring the Myocardial Polyamine Pool. Oxid. Med. Cell Longev. 2014, 2014, 457429. [Google Scholar] [CrossRef]
- Duan, X.; Ji, B.; Wang, X.; Liu, J.; Zheng, Z.; Long, C.; Tang, Y.; Hu, S. Expression of MicroRNA-1 and MicroRNA-21 in Different Protocols of Ischemic Conditioning in an Isolated Rat Heart Model. Cardiology 2012, 122, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Ostadal, B.; Ostadalova, I.; Szarszoi, O.; Netuka, I.; Olejnickova, V.; Hlavackova, M. Sex-Dependent Effect of Perinatal Hypoxia on Cardiac Tolerance to Oxygen Deprivation in Adults. Can. J. Physiol. Pharmacol. 2021, 99, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Olivetti, G.; Melissari, M.; Balbi, T.; Quaini, F.; Cigola, E.; Sonnenblick, E.H.; Anversa, P. Myocyte Cellular Hypertrophy Is Responsible for Ventricular Remodelling in the Hypertrophied Heart of Middle Aged Individuals in the Absence of Cardiac Failure. Cardiovasc. Res. 1994, 28, 1199–1208. [Google Scholar] [CrossRef]
- Olivetti, G.; Melissari, M.; Capasso, J.M.; Anversa, P. Cardiomyopathy of the Aging Human Heart. Myocyte Loss and Reactive Cellular Hypertrophy. Circ. Res. 1991, 68, 1560–1568. [Google Scholar] [CrossRef]
- Gao, Z.; Chen, Z.; Sun, A.; Deng, X. Gender Differences in Cardiovascular Disease. Med. Nov. Technol. Devices 2019, 4, 100025. [Google Scholar] [CrossRef]
- Chen, J.; Liu, Y.; Pan, D.; Xu, T.; Luo, Y.; Wu, W.; Wu, P.; Zhu, H.; Li, D. Estrogen Inhibits Endoplasmic Reticulum Stress and Ameliorates Myocardial Ischemia/Reperfusion Injury in Rats by Upregulating SERCA2a. Cell Commun. Signal. 2022, 20, 38. [Google Scholar] [CrossRef] [PubMed]
- Vaina, S.; Milkas, A.; Crysohoou, C.; Stefanadis, C. Coronary Artery Disease in Women: From the Yentl Syndrome to Contemporary Treatment. World J. Cardiol. 2015, 7, 10–18. [Google Scholar] [CrossRef]
- Obas, V.; Vasan, R.S. The Aging Heart. Clin. Sci. 2018, 132, 1367–1382. [Google Scholar] [CrossRef] [PubMed]
- Kindernay, L.; Farkasova, V.; Neckar, J.; Hrdlicka, J.; Ytrehus, K.; Ravingerova, T. Impact of Maturation on Myocardial Response to Ischemia and the Effectiveness of Remote Preconditioning in Male Rats. Int. J. Mol. Sci. 2021, 22, 11009. [Google Scholar] [CrossRef] [PubMed]
- Ledvenyiova, V.; Pancza, D.; Matejiková, J.; Ferko, M.; Bernatova, I.; Ravingerova, T. Impact of Age and Sex on Response to Ischemic Preconditioning in the Rat Heart: Differential Role of the PI3K-AKT Pathway. Can. J. Physiol. Pharmacol. 2013, 91, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Griecsová, L.; Farkašová, V.; Gáblovskỳ, I.; Khandelwal, V.K.M.; Bernátová, I.; Tatarková, Z.; Kaplan, P.; Ravingerová, T. Effect of Maturation on the Resistance of Rat Hearts against Ischemia. Study of Potential Molecular Mechanisms. Physiol. Res. 2015, 64, S685–S696. [Google Scholar] [CrossRef]
- Andersson, C.; Gislason, G.H.; Weeke, P.; Hoffmann, S.; Hansen, P.R.; Torp-Pedersen, C.; Søgaard, P. Diabetes Is Associated with Impaired Myocardial Performance in Patients without Significant Coronary Artery Disease. Cardiovasc. Diabetol. 2010, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandy, P.; Andreadou, I.; Baxter, G.F.; Bøtker, H.E.; Davidson, S.M.; Dobrev, D.; Gersh, B.J.; Heusch, G.; Lecour, S.; Ruiz-Meana, M.; et al. Interaction of Cardiovascular Nonmodifiable Risk Factors, Comorbidities and Comedications With Ischemia/Reperfusion Injury and Cardioprotection by Pharmacological Treatments and Ischemic Conditioning. Pharmacol. Rev. 2023, 75, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Giricz, Z.; Koncsos, G.; Rajtík, T.; Varga, Z.V.; Baranyai, T.; Csonka, C.; Szobi, A.; Adameová, A.; Gottlieb, R.A.; Ferdinandy, P. Hypercholesterolemia Downregulates Autophagy in the Rat Heart. Lipids Health Dis. 2017, 16, 60. [Google Scholar] [CrossRef] [PubMed]
- Perreault, S.; Dragomir, A.; Roy, L.; White, M.; Blais, L.; Lalonde, L.; Bérard, A. Adherence Level of Antihypertensive Agents in Coronary Artery Disease. Br. J. Clin. Pharmacol. 2010, 69, 74–84. [Google Scholar] [CrossRef]
- Deedwania, P.; Singh, V.; Davidson, M.H. Low High-Density Lipoprotein Cholesterol and Increased Cardiovascular Disease Risk: An Analysis of Statin Clinical Trials. Am. J. Cardiol. 2009, 104, 3E–9E. [Google Scholar] [CrossRef] [PubMed]
- Zálešák, M.; Blažíček, P.; Gablovský, I.; Ledvényiová, V.; Barteková, M.; Ziegelhöffer, A.; Ravingerová, T. Impaired PI3K/Akt Signaling as a Potential Cause of Failure to Precondition Rat Hearts under Conditions of Simulated Hyperglycemia. Physiol. Res. 2015, 64, 633–641. [Google Scholar] [CrossRef]
- Zálešák, M.; BlaŽíček, P.; Pancza, D.; Gablovský, I.; Štrbák, V.; Ravingerová, T. Hyperosmotic Environment Blunts Effectivity of Ischemic Preconditioning against Ischemia-Reperfusion Injury and Improves Ischemic Tolerance in Non-Preconditioned Isolated Rat Hearts. Physiol. Res. 2016, 65, 1045–1051. [Google Scholar] [CrossRef]
- Farkašová, F.; Kindernay, L.; Ferko, M.; Rajtík, T.; Szobi, A.; Ravingerová, T. Age-Dependent Effects of Remote Preconditioning in Hypertensive Rat Hearts Are Associated With Activation of RISK Signaling. Physiol. Res. 2023, 72, S11–S22. [Google Scholar] [CrossRef]
- Friehs, I.; del Nido, P.J. Increased Susceptibility of Hypertrophied Hearts to Ischemic Injury. Ann. Thorac. Surg. 2003, 75, S678–S684. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Elimban, V.; Bartekova, M.; Adameova, A. Involvement of Oxidative Stress in the Development of Subcellular Defects and Heart Disease. Biomedicines 2022, 10, 393. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G. Molecular Basis of Cardioprotection. Circ. Res. 2015, 116, 674–699. [Google Scholar] [CrossRef]
- Ravingerová, T.; Kindernay, L.; Barteková, M.; Ferko, M.; Adameová, A.; Zohdi, V.; Bernátová, I.; Ferenczyová, K.; Lazou, A. The Molecular Mechanisms of Iron Metabolism and Its Role in Cardiac Dysfunction and Cardioprotection. Int. J. Mol. Sci. 2020, 21, 7889. [Google Scholar] [CrossRef] [PubMed]
- Szobi, A.; Gonçalvesová, E.; Varga, Z.V.; Leszek, P.; Kuśmierczyk, M.; Hulman, M.; Kyselovič, J.; Ferdinandy, P.; Adameová, A. Analysis of Necroptotic Proteins in Failing Human Hearts. J. Transl. Med. 2017, 15, 86. [Google Scholar] [CrossRef]
- Lichý, M.; Szobi, A.; Hrdlička, J.; Horváth, C.; Kormanová, V.; Rajtík, T.; Neckář, J.; Kolář, F.; Adameová, A. Different Signalling in Infarcted and Non-Infarcted Areas of Rat Failing Hearts: A Role of Necroptosis and Inflammation. J. Cell Mol. Med. 2019, 23, 6429–6441. [Google Scholar] [CrossRef]
- Wang, J.; Deng, B.; Liu, Q.; Huang, Y.; Chen, W.; Li, J.; Zhou, Z.; Zhang, L.; Liang, B.; He, J.; et al. Pyroptosis and Ferroptosis Induced by Mixed Lineage Kinase 3 (MLK3) Signaling in Cardiomyocytes Are Essential for Myocardial Fibrosis in Response to Pressure Overload. Cell Death Dis. 2020, 11, 574. [Google Scholar] [CrossRef]
- Han, X.; Zhao, Z.-A.; Yan, S.; Lei, W.; Wu, H.; Lu, X.-A.; Chen, Y.; Li, J.; Wang, Y.; Yu, M.; et al. CXADR-like Membrane Protein Protects against Heart Injury by Preventing Excessive Pyroptosis after Myocardial Infarction. J. Cell Mol. Med. 2020, 24, 13775–13788. [Google Scholar] [CrossRef]
- Lesauskaite, V.; Epistolato, M.C.; Ivanoviene, L.; Tanganelli, P. Apoptosis of Cardiomyocytes in Explanted and Transplanted Hearts. Comparison of Results from in Situ TUNEL, ISEL, and ISOL Reactions. Am. J. Clin. Pathol. 2004, 121, 108–116. [Google Scholar] [CrossRef] [PubMed]
- de Boer, R.A.; van Veldhuisen, D.J.; van der Wijk, J.; Brouwer, R.M.; de Jonge, N.; Cole, G.M.; Suurmeijer, A.J. Additional Use of Immunostaining for Active Caspase 3 and Cleaved Actin and PARP Fragments to Detect Apoptosis in Patients with Chronic Heart Failure. J. Card. Fail. 2000, 6, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Guo, Z.; Ding, Z.; Mehta, J.L. Inflammation, Autophagy, and Apoptosis After Myocardial Infarction. J. Am. Heart Assoc. 2018, 7, e008024. [Google Scholar] [CrossRef] [PubMed]
- Corsetti, G.; Chen-Scarabelli, C.; Romano, C.; Pasini, E.; Dioguardi, F.S.; Onorati, F.; Knight, R.; Patel, H.; Saravolatz, L.; Faggian, G.; et al. Autophagy and Oncosis/Necroptosis Are Enhanced in Cardiomyocytes from Heart Failure Patients. Med. Sci. Monit. Basic. Res. 2019, 25, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Tannous, P.; Johnstone, J.L.; Kong, Y.; Shelton, J.M.; Richardson, J.A.; Le, V.; Levine, B.; Rothermel, B.A.; Hill, J.A. Cardiac Autophagy Is a Maladaptive Response to Hemodynamic Stress. J. Clin. Invest. 2007, 117, 1782–1793. [Google Scholar] [CrossRef]
- Liu, J.; Wu, P.; Wang, Y.; Du, Y.; Nan, A.; Liu, S.; Zhang, Y.; Zhou, N.; Xu, Z.; Yang, Z. Ad-HGF Improves the Cardiac Remodeling of Rat Following Myocardial Infarction by Upregulating Autophagy and Necroptosis and Inhibiting Apoptosis. Am. J. Transl. Res. 2016, 8, 4605–4627. [Google Scholar]
- Zhang, H.; Yin, Y.; Liu, Y.; Zou, G.; Huang, H.; Qian, P.; Zhang, G.; Zhang, J. Necroptosis Mediated by Impaired Autophagy Flux Contributes to Adverse Ventricular Remodeling after Myocardial Infarction. Biochem. Pharmacol. 2020, 175, 113915. [Google Scholar] [CrossRef]
- Horvath, C.; Young, M.; Jarabicova, I.; Kindernay, L.; Ferenczyova, K.; Ravingerova, T.; Lewis, M.; Suleiman, M.S.; Adameova, A. Inhibition of Cardiac RIP3 Mitigates Early Reperfusion Injury and Calcium-Induced Mitochondrial Swelling without Altering Necroptotic Signalling. Int. J. Mol. Sci. 2021, 22, 7983. [Google Scholar] [CrossRef] [PubMed]
- Ramachandra, C.J.A.; Hernandez-Resendiz, S.; Crespo-Avilan, G.E.; Lin, Y.-H.; Hausenloy, D.J. Mitochondria in Acute Myocardial Infarction and Cardioprotection. EBioMedicine 2020, 57, 102884. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Ueda, K.; Goto, C.; Jitsuiki, D.; Nishioka, K.; Umemura, T.; Noma, K.; Yoshizumi, M.; Chayama, K.; Higashi, Y. Repetition of Ischemic Preconditioning Augments Endothelium-Dependent Vasodilation in Humans. Arter. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1403–1410. [Google Scholar] [CrossRef] [PubMed]
- Heallen, T.R.; Kadow, Z.A.; Kim, J.H.; Wang, J.; Martin, J.F. Stimulating Cardiogenesis as a Treatment for Heart Failure. Circ. Res. 2019, 124, 1647–1657. [Google Scholar] [CrossRef] [PubMed]
- Iliodromitis, E.K.; Lazou, A.; Kremastinos, D.T. Ischemic Preconditioning: Protection against Myocardial Necrosis and Apoptosis. Vasc. Health Risk Manag. 2007, 3, 629–637. [Google Scholar]
- Spannbauer, A.; Traxler, D.; Lukovic, D.; Zlabinger, K.; Winkler, J.; Gugerell, A.; Ferdinandy, P.; Hausenloy, D.J.; Pavo, N.; Emmert, M.Y.; et al. Effect of Ischemic Preconditioning and Postconditioning on Exosome-Rich Fraction MicroRNA Levels, in Relation with Electrophysiological Parameters and Ventricular Arrhythmia in Experimental Closed-Chest Reperfused Myocardial Infarction. Int. J. Mol. Sci. 2019, 20, 2140. [Google Scholar] [CrossRef]
- Vélez, D.E.; Hermann, R.; Frank, M.B.; Cordero, V.E.M.; Savino, E.A.; Varela, A.; Marina Prendes, M.G. Effects of Wortmannin on Cardioprotection Exerted by Ischemic Preconditioning in Rat Hearts Subjected to Ischemia-Reperfusion. J. Physiol. Biochem. 2016, 72, 83–91. [Google Scholar] [CrossRef]
- Shizukuda, Y.; Mallet, R.T.; Lee, S.C.; Downey, H.F. Hypoxic Preconditioning of Ischaemic Canine Myocardium. Cardiovasc. Res. 1992, 26, 534–542. [Google Scholar] [CrossRef]
- Papadopoulos, C.E.; Zioutas, D.G.; Giannakoulas, G.A.; Matsiras, S.; Karamitsos, T.D.; Karvounis, H.I.; Geleris, P.; Stiliadis, I. Beneficial Effect of Ischemic Preconditioning on Post-Infarction Left Ventricular Remodeling and Global Left Ventricular Function. Cardiovasc. Revasc Med. 2011, 12, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Ischaemic Conditioning and Reperfusion Injury. Nat. Rev. Cardiol. 2016, 13, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Marber, M.S.; Latchman, D.S.; Walker, J.M.; Yellon, D.M. Cardiac Stress Protein Elevation 24 Hours after Brief Ischemia or Heat Stress Is Associated with Resistance to Myocardial Infarction. Circulation 1993, 88, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. The Second Window of Preconditioning (SWOP) Where Are We Now? Cardiovasc. Drugs Ther. 2010, 24, 235–254. [Google Scholar] [CrossRef]
- Vinten-Johansen, J.; Yellon, D.M.; Opie, L.H. Postconditioning: A Simple, Clinically Applicable Procedure to Improve Revascularization in Acute Myocardial Infarction. Circulation 2005, 112, 2085–2088. [Google Scholar] [CrossRef]
- Wang, H.-C.; Zhang, H.-F.; Guo, W.-Y.; Su, H.; Zhang, K.-R.; Li, Q.-X.; Yan, W.; Ma, X.L.; Lopez, B.L.; Christopher, T.A.; et al. Hypoxic Postconditioning Enhances the Survival and Inhibits Apoptosis of Cardiomyocytes Following Reoxygenation: Role of Peroxynitrite Formation. Apoptosis 2006, 11, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Zálešák, M.; Kura, B.; Graban, J.; Farkašová, V.; Slezák, J.; Ravingerová, T. Molecular Hydrogen Potentiates Beneficial Anti-Infarct Effect of Hypoxic Postconditioning in Isolated Rat Hearts: A Novel Cardioprotective Intervention. Can. J. Physiol. Pharmacol. 2017, 95, 888–893. [Google Scholar] [CrossRef]
- Downey, J.M.; Davis, A.M.; Cohen, M. V Signaling Pathways in Ischemic Preconditioning. Heart Fail. Rev. 2007, 12, 181–188. [Google Scholar] [CrossRef]
- Sun, H.-Y.; Wang, N.-P.; Kerendi, F.; Halkos, M.; Kin, H.; Guyton, R.A.; Vinten-Johansen, J.; Zhao, Z.-Q. Hypoxic Postconditioning Reduces Cardiomyocyte Loss by Inhibiting ROS Generation and Intracellular Ca2+ Overload. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1900–H1908. [Google Scholar] [CrossRef] [PubMed]
- Tissier, R.; Ghaleh, B.; Cohen, M.V.; Downey, J.M.; Berdeaux, A. Myocardial Protection with Mild Hypothermia. Cardiovasc. Res. 2012, 94, 217–225. [Google Scholar] [CrossRef]
- Tissier, R.; Chenoune, M.; Pons, S.; Zini, R.; Darbera, L.; Lidouren, F.; Ghaleh, B.; Berdeaux, A.; Morin, D. Mild Hypothermia Reduces Per-Ischemic Reactive Oxygen Species Production and Preserves Mitochondrial Respiratory Complexes. Resuscitation 2013, 84, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Niemann, B.; Pan, R.; Issa, H.; Simm, A.; Schulz, R.; Rohrbach, S. AMPK Activation Is Indispensable for the Protective Effects of Caloric Restriction on Left Ventricular Function in Postinfarct Myocardium. Biology 2022, 11, 448. [Google Scholar] [CrossRef] [PubMed]
- Slagsvold, K.H.; Rognmo, O.; Høydal, M.; Wisløff, U.; Wahba, A. Remote Ischemic Preconditioning Preserves Mitochondrial Function and Influences Myocardial MicroRNA Expression in Atrial Myocardium during Coronary Bypass Surgery. Circ. Res. 2014, 114, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-N.; Yu, H.; Zhu, X.-H.; Yuan, H.-J.; Kang, Y.; Jiao, J.-J.; Gao, W.-Z.; Liu, Y.-X.; Lou, J.-S. Noninvasive Delayed Limb Ischemic Preconditioning Attenuates Myocardial Ischemia-Reperfusion Injury in Rats by a Mitochondrial K(ATP) Channel-Dependent Mechanism. Physiol. Res. 2011, 60, 271–279. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Boston-Griffiths, E.A.; Yellon, D.M. Cyclosporin A and Cardioprotection: From Investigative Tool to Therapeutic Agent. Br. J. Pharmacol. 2012, 165, 1235–1245. [Google Scholar] [CrossRef]
- Lotz, C.; Lazariotto, M.; Redel, A.; Smul, T.M.; Stumpner, J.; Blomeyer, C.; Tischer-Zeitz, T.; Schmidt, J.; Pociej, J.; Roewer, N.; et al. Activation of Peroxisome-Proliferator-Activated Receptors α and γ Mediates Remote Ischemic Preconditioning against Myocardial Infarction in Vivo. Exp. Biol. Med. 2011, 236, 113–122. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, L.; Hong, D.; Gao, J. Remote Ischaemic Preconditioning Reduces Myocardial Ischaemic Reperfusion Injury in Patients with ST-Elevation Myocardial Infarction Undergoing Primary Percutaneous Coronary Intervention. Acta Cardiol. 2016, 71, 596–603. [Google Scholar] [CrossRef]
- Ma, N.; Bai, J.; Zhang, W.; Luo, H.; Zhang, X.; Liu, D.; Qiao, C. Trimetazidine Protects against Cardiac Ischemia/Reperfusion Injury via Effects on Cardiac MiRNA-21 Expression, Akt and the Bcl-2/Bax Pathway. Mol. Med. Rep. 2016, 14, 4216–4222. [Google Scholar] [CrossRef]
- Pickard, J.M.J.; Davidson, S.M.; Hausenloy, D.J.; Yellon, D.M. Co-Dependence of the Neural and Humoral Pathways in the Mechanism of Remote Ischemic Conditioning. Basic. Res. Cardiol. 2016, 111, 50. [Google Scholar] [CrossRef]
- Gong, R.; Wu, Y.-Q. Remote Ischemic Conditioning during Primary Percutaneous Coronary Intervention in Patients with ST-Segment Elevation Myocardial Infarction: A Systematic Review and Meta-Analysis. J. Cardiothorac. Surg. 2019, 14, 14. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, T.; Chen, S.; Zhou, Q.; Li, H.; Hu, N.; Feng, Y.; Dong, N.; Yao, S.; Xia, Z. Cardiac Protective Effects of Remote Ischaemic Preconditioning in Children Undergoing Tetralogy of Fallot Repair Surgery: A Randomized Controlled Trial. Eur. Heart J. 2018, 39, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Drury, N.E.; Bi, R.; Woolley, R.L.; Stickley, J.; Morris, K.P.; Montgomerie, J.; van Doorn, C.; Dunn, W.B.; Madhani, M.; Ives, N.J.; et al. Bilateral Remote Ischaemic Conditioning in Children (BRICC) Trial: Protocol for a Two-Centre, Double-Blind, Randomised Controlled Trial in Young Children Undergoing Cardiac Surgery. BMJ Open 2020, 10, e042176. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, R.K.; Peters, M.; Walton, B.; Kattenhorn, M.; Mullen, M.; Klein, N.; Vallance, P.; Deanfield, J.; MacAllister, R. Ischemic Preconditioning Prevents Endothelial Injury and Systemic Neutrophil Activation During Ischemia-Reperfusion in Humans In Vivo. Circulation 2001, 103, 1624–1630. [Google Scholar] [CrossRef]
- Wei, M.; Xin, P.; Li, S.; Tao, J.; Li, Y.; Li, J.; Liu, M.; Li, J.; Zhu, W.; Redington, A.N. Repeated Remote Ischemic Postconditioning Protects Against Adverse Left Ventricular Remodeling and Improves Survival in a Rat Model of Myocardial Infarction. Circ. Res. 2011, 108, 1220–1225. [Google Scholar] [CrossRef]
- Ferko, M.; Kancirová, I.; Jašová, M.; Čarnická, S.; Muráriková, M.; Waczulíková, I.; Sumbalová, Z.; Kucharská, J.; Uličná, O.; Ravingerová, T.; et al. Remote Ischemic Preconditioning of the Heart: Protective Responses in Functional and Biophysical Properties of Cardiac Mitochondria. Physiol. Res. 2014, 63, S469–S478. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Garcia-Dorado, D.; Bøtker, H.E.; Davidson, S.M.; Downey, J.; Engel, F.B.; Jennings, R.; Lecour, S.; Leor, J.; Madonna, R.; et al. Novel Targets and Future Strategies for Acute Cardioprotection: Position Paper of the European Society of Cardiology Working Group on Cellular Biology of the Heart. Cardiovasc. Res. 2017, 113, 564–585. [Google Scholar] [CrossRef]
- Ruiz-Meana, M.; Boengler, K.; Garcia-Dorado, D.; Hausenloy, D.J.; Kaambre, T.; Kararigas, G.; Perrino, C.; Schulz, R.; Ytrehus, K. Ageing, Sex, and Cardioprotection. Br. J. Pharmacol. 2020, 177, 5270–5286. [Google Scholar] [CrossRef] [PubMed]
- Kolár, F.; Jezková, J.; Balková, P.; Breh, J.; Neckár, J.; Novák, F.; Nováková, O.; Tomásová, H.; Srbová, M.; Ost’ádal, B.; et al. Role of Oxidative Stress in PKC-Delta Upregulation and Cardioprotection Induced by Chronic Intermittent Hypoxia. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H224–H230. [Google Scholar] [CrossRef]
- Cai, Z.P.; Parajuli, N.; Zheng, X.; Becker, L. Remote Ischemic Preconditioning Confers Late Protection against Myocardial Ischemia-Reperfusion Injury in Mice by Upregulating Interleukin-10. Basic. Res. Cardiol. 2012, 107, 277. [Google Scholar] [CrossRef]
- Abete, P.; Calabrese, C.; Ferrara, N.; Cioppa, A.; Pisanelli, P.; Cacciatore, F.; Longobardi, G.; Napoli, C.; Rengo, F. Exercise Training Restores Ischemic Preconditioning in the Aging Heart. J. Am. Coll. Cardiol. 2000, 36, 643–650. [Google Scholar] [CrossRef]
- Alleman, R.J.; Stewart, L.M.; Tsang, A.M.; Brown, D.A. Why Does Exercise “Trigger” Adaptive Protective Responses in the Heart? Dose Response 2015, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Lonek, L.; Puhova, A.; Griecsova-Kindernay, L.; Patel, S.P.; Zohdi, V.; Jezova, D.; Ravingerova, T. Voluntary Exercise May Activate Components of Pro-Survival Risk Pathway in the Rat Heart and Potentially Modify Cell Proliferation in the Myocardium. Physiol. Res. 2019, 68, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Michelsen, M.M.; Støttrup, N.B.; Schmidt, M.R.; Løfgren, B.; Jensen, R.V.; Tropak, M.; St-Michel, E.J.; Redington, A.N.; Bøtker, H.E. Exercise-Induced Cardioprotection Is Mediated by a Bloodborne, Transferable Factor. Basic. Res. Cardiol. 2012, 107, 260. [Google Scholar] [CrossRef] [PubMed]
- Suleiman, M.S.; Halestrap, A.P.; Griffiths, E.J. Mitochondria: A Target for Myocardial Protection. Pharmacol. Ther. 2001, 89, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Ham, P.B.; Raju, R. Mitochondrial Function in Hypoxic Ischemic Injury and Influence of Aging. Prog. Neurobiol. 2017, 157, 92–116. [Google Scholar] [CrossRef] [PubMed]
- Donato, M.; Goyeneche, M.A.; Garces, M.; Marchini, T.; Pérez, V.; Del Mauro, J.; Höcht, C.; Rodríguez, M.; Evelson, P.; Gelpi, R.J. Myocardial Triggers Involved in Activation of Remote Ischaemic Preconditioning. Exp. Physiol. 2016, 101, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Lecour, S. Activation of the Protective Survivor Activating Factor Enhancement (SAFE) Pathway against Reperfusion Injury: Does It Go beyond the RISK Pathway? J. Mol. Cell Cardiol. 2009, 47, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Jia, Z.; Yang, L.; Zhu, M.; Zhang, J.; Liu, J.; Wu, P.; Tian, W.; Li, J.; Qi, Z.; et al. Exercise Protects against Chronic β-Adrenergic Remodeling of the Heart by Activation of Endothelial Nitric Oxide Synthase. PLoS ONE 2014, 9, e96892. [Google Scholar] [CrossRef]
- Calvert, J.W.; Condit, M.E.; Aragón, J.P.; Nicholson, C.K.; Moody, B.F.; Hood, R.L.; Sindler, A.L.; Gundewar, S.; Seals, D.R.; Barouch, L.A.; et al. Exercise Protects against Myocardial Ischemia-Reperfusion Injury via Stimulation of β(3)-Adrenergic Receptors and Increased Nitric Oxide Signaling: Role of Nitrite and Nitrosothiols. Circ. Res. 2011, 108, 1448–1458. [Google Scholar] [CrossRef]
- Wang, B.; Xu, M.; Li, W.; Li, X.; Zheng, Q.; Niu, X. Aerobic Exercise Protects against Pressure Overload-Induced Cardiac Dysfunction and Hypertrophy via Β3-AR-NNOS-NO Activation. PLoS ONE 2017, 12, e0179648. [Google Scholar] [CrossRef]
- Kleindienst, A.; Battault, S.; Belaidi, E.; Tanguy, S.; Rosselin, M.; Boulghobra, D.; Meyer, G.; Gayrard, S.; Walther, G.; Geny, B.; et al. Exercise Does Not Activate the Β3 Adrenergic Receptor–ENOS Pathway, but Reduces Inducible NOS Expression to Protect the Heart of Obese Diabetic Mice. Basic. Res. Cardiol. 2016, 111, 40. [Google Scholar] [CrossRef] [PubMed]
- McMullen, J.R.; Shioi, T.; Zhang, L.; Tarnavski, O.; Sherwood, M.C.; Kang, P.M.; Izumo, S. Phosphoinositide 3-Kinase(P110alpha) Plays a Critical Role for the Induction of Physiological, but Not Pathological, Cardiac Hypertrophy. Proc. Natl. Acad. Sci. USA 2003, 100, 12355–12360. [Google Scholar] [CrossRef] [PubMed]
- Weeks, K.L.; Gao, X.; Du, X.-J.; Boey, E.J.H.; Matsumoto, A.; Bernardo, B.C.; Kiriazis, H.; Cemerlang, N.; Tan, J.W.; Tham, Y.K.; et al. Phosphoinositide 3-Kinase P110α Is a Master Regulator of Exercise-Induced Cardioprotection and PI3K Gene Therapy Rescues Cardiac Dysfunction. Circ. Heart Fail. 2012, 5, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Machuki, J.O.; Zhang, H.Y.; Harding, S.E.; Sun, H. Molecular Pathways of Oestrogen Receptors and β-Adrenergic Receptors in Cardiac Cells: Recognition of Their Similarities, Interactions and Therapeutic Value. Acta Physiol. 2018, 222, e12978. [Google Scholar] [CrossRef] [PubMed]
- Heiat, F.; Ahmadi, A.; Shojaeifard, M. The Exercise Preconditioning Effect on Cardiac Tissue Injury Following Induction of Myocardial Infarction in Male Rats. Biomed. Res. Int. 2023, 2023, 3631458. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, M.F.; Pelúzio, M. do C.G.; Amorim, P.R. dos S.; Lavorato, V.N.; Santos, N.P. do; Bozi, L.H.M.; Penitente, A.R.; Falkoski, D.L.; Berfort, F.G.; Natali, A.J. Swimming Training Attenuates Contractile Dysfunction in Diabetic Rat Cardiomyocytes. Arq. Bras. Cardiol. 2011, 97, 33–39. [Google Scholar] [CrossRef]
- da Silva, E.; Natali, A.J.; da Silva, M.F.; de Jesus Gomes, G.; da Cunha, D.N.Q.; Toledo, M.M.; Drummond, F.R.; Ramos, R.M.S.; Dos Santos, E.C.; Novaes, R.D.; et al. Swimming Training Attenuates the Morphological Reorganization of the Myocardium and Local Inflammation in the Left Ventricle of Growing Rats with Untreated Experimental Diabetes. Pathol. Res. Pr. Pract. 2016, 212, 325–334. [Google Scholar] [CrossRef]
- Cugusi, L.; Cadeddu, C.; Nocco, S.; Orrù, F.; Bandino, S.; Deidda, M.; Caria, A.; Bassareo, P.P.; Piras, A.; Cabras, S.; et al. Effects of an Aquatic-Based Exercise Program to Improve Cardiometabolic Profile, Quality of Life, and Physical Activity Levels in Men with Type 2 Diabetes Mellitus. PM&R 2015, 7, 141–148, quiz 148. [Google Scholar] [CrossRef]
- Börzsei, D.; Priksz, D.; Szabó, R.; Bombicz, M.; Karácsonyi, Z.; Puskás, L.G.; Fehér, L.Z.; Radák, Z.; Kupai, K.; Berkó, A.M.; et al. Exercise-Mitigated Sex-Based Differences in Aging: From Genetic Alterations to Heart Performance. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H854–H866. [Google Scholar] [CrossRef]
- Kura, B.; Szeiffova Bacova, B.; Kalocayova, B.; Sykora, M.; Slezak, J. Oxidative Stress-Responsive MicroRNAs in Heart Injury. Int. J. Mol. Sci. 2020, 21, 358. [Google Scholar] [CrossRef]
- Huntzinger, E.; Izaurralde, E. Gene Silencing by MicroRNAs: Contributions of Translational Repression and MRNA Decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Skommer, J.; Rana, I.; Marques, F.Z.; Zhu, W.; Du, Z.; Charchar, F.J. Small Molecules, Big Effects: The Role of MicroRNAs in Regulation of Cardiomyocyte Death. Cell Death Dis. 2014, 5, e1325. [Google Scholar] [CrossRef] [PubMed]
- Diehl, P.; Fricke, A.; Sander, L.; Stamm, J.; Bassler, N.; Htun, N.; Ziemann, M.; Helbing, T.; El-Osta, A.; Jowett, J.B.M.; et al. Microparticles: Major Transport Vehicles for Distinct MicroRNAs in Circulation. Cardiovasc. Res. 2012, 93, 633–644. [Google Scholar] [CrossRef]
- Weiss, J.B.; Eisenhardt, S.U.; Stark, G.B.; Bode, C.; Moser, M.; Grundmann, S. MicroRNAs in Ischemia-Reperfusion Injury. Am. J. Cardiovasc. Dis. 2012, 2, 237–247. [Google Scholar] [PubMed]
- Li, J.; Xuan, W.; Yan, R.; Tropak, M.B.; Jean-St-Michel, E.; Liang, W.; Gladstone, R.; Backx, P.H.; Kharbanda, R.K.; Redington, A.N. Remote Preconditioning Provides Potent Cardioprotection via PI3K/Akt Activation and Is Associated with Nuclear Accumulation of β-Catenin. Clin. Sci. 2011, 120, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Wen, M.; Gong, Z.; Huang, C.; Liang, Q.; Xu, M.; Wang, L.; Zhang, W.; Lu, P.; Zhan, B.; Yu, L.; et al. Plasma Exosomes Induced by Remote Ischaemic Preconditioning Attenuate Myocardial Ischaemia/Reperfusion Injury by Transferring MiR-24. Cell Death Dis. 2018, 9, 320. [Google Scholar] [CrossRef]
- Varga, Z.V.; Ferdinandy, P.; Liaudet, L.; Pacher, P. Drug-Induced Mitochondrial Dysfunction and Cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1453–H1467. [Google Scholar] [CrossRef]
- Bär, C.; Chatterjee, S.; Thum, T. Long Noncoding RNAs in Cardiovascular Pathology, Diagnosis, and Therapy. Circulation 2016, 134, 1484–1499. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Wu, D.; Wu, Q.; Zou, B.; Huang, X.; Cheng, X.; Wu, Y.; Hong, K.; Li, P.; Yang, R.; et al. Knockdown of Long Non-Coding RNA-ZFAS1 Protects Cardiomyocytes Against Acute Myocardial Infarction Via Anti-Apoptosis by Regulating MiR-150/CRP. J. Cell Biochem. 2017, 118, 3281–3289. [Google Scholar] [CrossRef]
- Li, X.; Luo, S.; Zhang, J.; Yuan, Y.; Jiang, W.; Zhu, H.; Ding, X.; Zhan, L.; Wu, H.; Xie, Y.; et al. LncRNA H19 Alleviated Myocardial I/RI via Suppressing MiR-877-3p/Bcl-2-Mediated Mitochondrial Apoptosis. Mol. Ther. Nucleic Acids 2019, 17, 297–309. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, R.; Niu, Q.; Wang, H.; Yang, Z.; Bao, Y. Morphine Postconditioning Alleviates Autophage in Ischemia-Reperfusion Induced Cardiac Injury through up-Regulating LncRNA UCA1. Biomed. Pharmacother. 2018, 108, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-X.; Zhang, X.-J.; Li, Q.; Wang, K.; Wang, Y.; Jiao, J.-Q.; Feng, C.; Teng, S.; Zhou, L.-Y.; Gong, Y.; et al. MicroRNA-103/107 Regulate Programmed Necrosis and Myocardial Ischemia/Reperfusion Injury Through Targeting FADD. Circ. Res. 2015, 117, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, M.; Tang, Y.; Sun, H.; Lin, X.; Liang, P.; Jiang, B. LncRNA H19 Is Involved in Myocardial Ischemic Preconditioning via Increasing the Stability of Nucleolin Protein. J. Cell Physiol. 2020, 235, 5985–5994. [Google Scholar] [CrossRef] [PubMed]
- Vega, R.B.; Kelly, D.P. Cardiac Nuclear Receptors: Architects of Mitochondrial Structure and Function. J. Clin. Investig. 2017, 127, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Ravingerová, T.; Farkašová, V.; Griecsová, L.; Muráriková, M.; Carnická, S.; Lonek, L.; Ferko, M.; Slezak, J.; Zálešák, M.; Adameova, A.; et al. Noninvasive Approach to Mend the Broken Heart: Is “Remote Conditioning” a Promising Strategy for Application in Humans? Can. J. Physiol. Pharmacol. 2017, 95, 1204–1212. [Google Scholar] [CrossRef]
- Barlaka, E.; Galatou, E.; Mellidis, K.; Ravingerova, T.; Lazou, A. Role of Pleiotropic Properties of Peroxisome Proliferator-Activated Receptors in the Heart: Focus on the Nonmetabolic Effects in Cardiac Protection. Cardiovasc. Ther. 2016, 34, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Papatheodorou, I.; Galatou, E.; Panagiotidis, G.-D.; Ravingerová, T.; Lazou, A. Cardioprotective Effects of PPARβ/δ Activation against Ischemia/Reperfusion Injury in Rat Heart Are Associated with ALDH2 Upregulation, Amelioration of Oxidative Stress and Preservation of Mitochondrial Energy Production. Int. J. Mol. Sci. 2021, 22, 6399. [Google Scholar] [CrossRef]
- Jašová, M.; Kancirová, I.; Waczulíková, I.; Ferko, M. Mitochondria as a Target of Cardioprotection in Models of Preconditioning. J. Bioenerg. Biomembr. 2017, 49, 357–368. [Google Scholar] [CrossRef]
- Ramaccini, D.; Montoya-Uribe, V.; Aan, F.J.; Modesti, L.; Potes, Y.; Wieckowski, M.R.; Krga, I.; Glibetić, M.; Pinton, P.; Giorgi, C.; et al. Mitochondrial Function and Dysfunction in Dilated Cardiomyopathy. Front. Cell Dev. Biol. 2021, 8, 624216. [Google Scholar] [CrossRef]
- Manolis, A.S.; Manolis, A.A.; Manolis, T.A.; Apostolaki, N.E.; Apostolopoulos, E.J.; Melita, H.; Katsiki, N. Mitochondrial Dysfunction in Cardiovascular Disease: Current Status of Translational Research/Clinical and Therapeutic Implications. Med. Res. Rev. 2021, 41, 275–313. [Google Scholar] [CrossRef]
- Ferko, M.; Andelová, N.; Szeiffová Bačová, B.; Jašová, M. Myocardial Adaptation in Pseudohypoxia: Signaling and Regulation of MPTP via Mitochondrial Connexin 43 and Cardiolipin. Cells 2019, 8, 1449. [Google Scholar] [CrossRef]
- Mishra, K.; Luo, M. Mitochondrial Channels and Their Role in Cardioprotection. In Ion Transporters; Tomaskova, Z.S., Ed.; IntechOpen: Rijeka, Croatia, 2022. [Google Scholar]
- Paradies, G.; Paradies, V.; De Benedictis, V.; Ruggiero, F.M.; Petrosillo, G. Functional Role of Cardiolipin in Mitochondrial Bioenergetics. Biochim. Biophys. Acta (BBA)-Bioenerg. 2014, 1837, 408–417. [Google Scholar] [CrossRef]
- Seidlmayer, L.K.; Juettner, V.V.; Kettlewell, S.; Pavlov, E.V.; Blatter, L.A.; Dedkova, E.N. Distinct MPTP Activation Mechanisms in Ischaemia–Reperfusion: Contributions of Ca2+, ROS, PH, and Inorganic Polyphosphate. Cardiovasc. Res. 2015, 106, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Ghosh, P.; Wan, R.; Ouyang, X.; Cheng, H.; Mattson, M.P.; Cheng, A. Permeability Transition Pore-Mediated Mitochondrial Superoxide Flashes Mediate an Early Inhibitory Effect of Amyloid Beta1−42 on Neural Progenitor Cell Proliferation. Neurobiol. Aging 2014, 35, 975–989. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.J.; Quintanilla, R.A. Development or Disease: Duality of the Mitochondrial Permeability Transition Pore. Dev. Biol. 2017, 426, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mnatsakanyan, N.; Beutner, G.; Porter, G.A.; Alavian, K.N.; Jonas, E.A. Physiological Roles of the Mitochondrial Permeability Transition Pore. J. Bioenerg. Biomembr. 2017, 49, 13–25. [Google Scholar] [CrossRef]
- Andrienko, T.; Pasdois, P.; Rossbach, A.; Halestrap, A.P. Real-Time Fluorescence Measurements of ROS and [Ca2+] in Ischemic / Reperfused Rat Hearts: Detectable Increases Occur Only after Mitochondrial Pore Opening and Are Attenuated by Ischemic Preconditioning. PLoS ONE 2016, 11, e0167300. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-B.; Dongworth, R.K.; Cabrera-Fuentes, H.A.; Hausenloy, D.J. Role of the MPTP in Conditioning the Heart—Translatability and Mechanism. Br. J. Pharmacol. 2015, 172, 2074–2084. [Google Scholar] [CrossRef] [PubMed]
- Elrod, J.W.; Molkentin, J.D. Physiologic Functions of Cyclophilin D and the Mitochondrial Permeability Transition Pore. Circ. J. 2013, 77, 1111–1122. [Google Scholar] [CrossRef]
- Lu, X.; Kwong, J.Q.; Molkentin, J.D.; Bers, D.M. Individual Cardiac Mitochondria Undergo Rare Transient Permeability Transition Pore Openings. Circ. Res. 2016, 118, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Paggio, A.; Checchetto, V.; Campo, A.; Menabò, R.; Di Marco, G.; Di Lisa, F.; Szabo, I.; Rizzuto, R.; De Stefani, D. Identification of an ATP-Sensitive Potassium Channel in Mitochondria. Nature 2019, 572, 609–613. [Google Scholar] [CrossRef]
- Briston, T.; Selwood, D.L.; Szabadkai, G.; Duchen, M.R. Mitochondrial Permeability Transition: A Molecular Lesion with Multiple Drug Targets. Trends Pharmacol. Sci. 2019, 40, 50–70. [Google Scholar] [CrossRef] [PubMed]
- Piot, C.; Croisille, P.; Staat, P.; Thibault, H.; Rioufol, G.; Mewton, N.; Elbelghiti, R.; Cung, T.T.; Bonnefoy, E.; Angoulvant, D.; et al. Effect of Cyclosporine on Reperfusion Injury in Acute Myocardial Infarction. N. Engl. J. Med. 2008, 359, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Cung, T.-T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guérin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N. Engl. J. Med. 2015, 373, 1021–1031. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Zang, J.; Li, Y.; Wu, X. Pharmaceutical Therapies for Necroptosis in Myocardial Ischemia–Reperfusion Injury. J. Cardiovasc. Dev. Dis. 2023, 10, 303. [Google Scholar] [CrossRef] [PubMed]
- Szobi, A.; Farkašová-Ledvényiová, V.; Lichý, M.; Muráriková, M.; Čarnická, S.; Ravingerová, T.; Adameová, A. Cardioprotection of Ischaemic Preconditioning Is Associated with Inhibition of Translocation of MLKL within the Plasma Membrane. J. Cell Mol. Med. 2018, 22, 4183–4196. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravingerova, T.; Adameova, A.; Lonek, L.; Farkasova, V.; Ferko, M.; Andelova, N.; Kura, B.; Slezak, J.; Galatou, E.; Lazou, A.; et al. Is Intrinsic Cardioprotection a Laboratory Phenomenon or a Clinically Relevant Tool to Salvage the Failing Heart? Int. J. Mol. Sci. 2023, 24, 16497. https://doi.org/10.3390/ijms242216497
Ravingerova T, Adameova A, Lonek L, Farkasova V, Ferko M, Andelova N, Kura B, Slezak J, Galatou E, Lazou A, et al. Is Intrinsic Cardioprotection a Laboratory Phenomenon or a Clinically Relevant Tool to Salvage the Failing Heart? International Journal of Molecular Sciences. 2023; 24(22):16497. https://doi.org/10.3390/ijms242216497
Chicago/Turabian StyleRavingerova, Tanya, Adriana Adameova, Lubomir Lonek, Veronika Farkasova, Miroslav Ferko, Natalia Andelova, Branislav Kura, Jan Slezak, Eleftheria Galatou, Antigone Lazou, and et al. 2023. "Is Intrinsic Cardioprotection a Laboratory Phenomenon or a Clinically Relevant Tool to Salvage the Failing Heart?" International Journal of Molecular Sciences 24, no. 22: 16497. https://doi.org/10.3390/ijms242216497
APA StyleRavingerova, T., Adameova, A., Lonek, L., Farkasova, V., Ferko, M., Andelova, N., Kura, B., Slezak, J., Galatou, E., Lazou, A., Zohdi, V., & Dhalla, N. S. (2023). Is Intrinsic Cardioprotection a Laboratory Phenomenon or a Clinically Relevant Tool to Salvage the Failing Heart? International Journal of Molecular Sciences, 24(22), 16497. https://doi.org/10.3390/ijms242216497