
Host Transcriptional Regulatory Genes and Microbiome Networks Crosstalk through Immune Receptors Establishing Normal and Tumor Multiomics Metafirm of the Oral-Gut-Lung Axis

 ,
,
Abstract
:1. Introduction
2. The Molecular Metafirm of the Gut-Lung Axis during Tumorigenesis
2.1. Genomics Studies
2.1.1. Colorectum Cancer
2.1.2. Gastric Cancer
2.1.3. Lung Cancer
2.2. Transcriptomics Studies
2.2.1. Colorectum Cancer
2.2.2. Gastric Cancer
2.2.3. Lung Cancer
2.3. Epigenomics Studies
2.3.1. Colorectum Cancer
2.3.2. Gastric Cancer
2.3.3. Lung Cancer
3. Microbiome Implicated in the Oral-Gut-Lung Axis
3.1. Oral Microbiota
3.2. Gut Microbiota
3.3. Lung Microbiota
4. Microbiome Crosstalk in the Oral-Gut-Lung Axis
5. Human Oral Microbiota, Inflammation, and Tumorigenesis in the Gut-Lung Axis
6. Host Immunity–Microbiome Crosstalk in the Oral-Gut-Lung Axis
6.1. Microbiome and Major Histocompatibility Complex (MHC) Crosstalk
6.2. Microbiome and Innate Immune Receptors Crosstalk
6.3. The Activation of Intracellular Transcriptional Regulatory Program
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| KSHV | Kaposi’s sarcoma-associated herpesvirus |
| HHV8 | Human herpesvirus-8 |
| EBV | Epstein–Barr virus |
| TRN | Transcription factor or transcriptional regulatory network |
| IEGs | Immediate-early genes |
| TFs | Transcription factors |
| IL-8 | Interleukin 8 |
| CXCL8 | Chemokine (C-X-C motif) ligand 8 |
| CXCL7 | Chemokine (C-X-C motif) ligand 7 |
| CCL19 | Chemokine (C-C motif) ligand 19 |
| CXCL13 | Chemokine (C-X-C motif) ligand 13 |
| MSI | Microsatellite instability |
| MSS | Microsatellite stability |
| APC | Adenomatous polyposis coli |
| KRAS | Kirsten rat sarcoma virus |
| PIK3CA | Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha |
| SMAD4 | Mothers against decapentaplegic homolog 4 |
| TP53 | Tumor protein P53 |
| DCC | Netrin receptor |
| SMAD | Mothers against decapentaplegic |
| BRAF | B-Raf Proto-Oncogene |
| Wnt | Wingless-related integration site |
| BMP | Bone morphogenetic protein |
| TGFβ | Transforming growth factor-beta |
| Ras | Rat sarcoma virus |
| ST6GALNAC1 | ST6 N-acetylgalactosaminide alpha-2,6-sialyltransferase 1 |
| CDH1 | Cadherin 1 |
| STK11 | Serine/threonine kinase 11 |
| MLH1 | mutL homolog 1 |
| MSH2 | mutS homolog 2 |
| EPCAM | Epithelial cell adhesion molecule |
| MSH6 | mutS homolog 6 |
| PMS2 | PMS1 homolog 2, mismatch repair system component |
| FAP | Familial adenomatous polyposis |
| BRCA2 | BReast CAncer gene 2 |
| PALB2 | Partner and localizer of BRCA2 |
| CLDN18-ARHGAP | Fusion Gene_Claudin 18-Rho GTPase-activating protein |
| ARID1A | AT-rich interaction domain 1A |
| PD-L1/2 | Programmed cell death ligand ½ |
| ICIs | Immune checkpoint inhibitors |
| CTLA-4 | Cytotoxic T-lymphocyte associated protein 4 |
| PD-1 | Programmed cell death 1 |
| LAG-3 | Lymphocyte activating 3 |
| TIM-3 | Hepatitis A virus cellular receptor 2 |
| PD-L1 | CD274 molecule |
| ERBB2 | erb-b2 receptor tyrosine kinase 2 |
| MET | Methoprene-tolerant proto-oncogene, receptor tyrosine kinase |
| NKX2 | Ventral nervous system defective |
| DOK2 | Docking protein 2 |
| ALK | Anaplastic lymphoma receptor tyrosine kinase |
| ROS1 | ROS proto-oncogene 1, receptor tyrosine kinase |
| RET | Ret proto-oncogene |
| ALK/EML4 | Anaplastic lymphoma receptor tyrosine kinase–Echinoderm microtubule-associated protein-like 4 |
| TTN | titin |
| CSMD3 | CUB and Sushi multiple domains 3 |
| MUT16 | MUTator 16 |
| RYR2 | Ryanodine receptor 2 |
| BRINP3 | BMP/retinoic acid inducible neural specific 3 |
| COL11A1 | Collagen type XI alpha 1 chain |
| GRIN2B | Glutamate ionotropic receptor NMDA type subunit 2B |
| MUC5B | Mucin 5B, oligomeric mucus/gel-forming |
| NLRP3 | NLR family pyrin domain containing 3 |
| TENM3 | Teneurin transmembrane protein 3 |
| NSCLC | Non-small cell lung cancer |
| LUAD | Lung adenocarcinoma |
| LUSC | Lung squamous cell carcinoma |
| TCGA-STAD | The Cancer Genome Atlas Stomach Adenocarcinoma |
| RUNX2 | Runt-related transcription factor 2 |
| SOX4 | SRY-box transcription factor 4 |
| SOX17 | SRY-box transcription factor 17 |
| BZW2 | Basic leucine zipper and W2 domains 2 |
| FOXM1 | Forkhead box M1 |
| ZBTB16 | Zinc finger and BTB domain containing 16 |
| TAL1 | T-cell acute lymphocytic leukemia 1 bHLH transcription factor 1, erythroid differentiation factor |
| KLF4 | Kruppel-like transcription factor 4 |
| EPAS1 | Endothelial PAS domain protein 1 |
| HOXC6 | Homeobox C6 |
| ID4 | Inhibitor of DNA binding 4 |
| KLF2 | Kruppel-like transcription factor 2 |
| MEIS1 | Meis homeobox 1 |
| NR2F1 | Nuclear receptor subfamily 2 group F member 1 |
| TBX4 | T-box transcription factor 4 |
| TCF21 | Transcription factor 21 |
| TFAP2C | Transcription factor AP-2 gamma |
| LMO2 | LIM domain only 2 |
| MNDA | Myeloid cell nuclear differentiation antigen |
| FOXF1 | Forkhead box F1 |
| HLF | HLF transcription factor, PAR bZIP family member |
| RFX2 | Regulatory factor X2 |
| DLX5 | Distal-less homeobox 5 |
| MYBL2 | MYB proto-oncogene like 2 |
| NR4A3 | Nuclear receptor subfamily 4 group A member 3 |
| PKNOX2 | PBX/knotted 1 homeobox 2 |
| GPRASP1 | G protein-coupled receptor associated sorting protein 1 |
| PAH | Pulmonary arterial hypertension |
| DEGs | Differentially expressed genes |
| CpG | Cytosine–guanine dinucleotides |
| DNMT1 | DNA methyltransferase 1 |
| DNMT3A | DNA methyltransferase 3 Alpha |
| DNMT3B | DNA methyltransferase 3 Beta |
| TET | Ten-eleven translocation family |
| FIGN | Fidgetin, microtubule severing factor |
| HTRA3 | HtrA serine peptidase 3 |
| BDNF | Brain-derived neurotrophic factor |
| HCN4 | Hyperpolarization activated cyclic nucleotide gated potassium channel 4 |
| STAC2 | SH3- and cysteine-rich domain 2 |
| SEPT9 | Septin 9 |
| SDC2 | Syndecan 2 |
| MGMT | O-6-methylguanine-DNA methyltransferase |
| NDGR4 | N-Myc Downstream-Regulated Gene 4 Protein |
| BMP3 | Bone morphogenetic protein 3 |
| VIM | Vimentin |
| SFRP | Secreted frizzled-related protein |
| p16 | Cyclin-dependent kinase inhibitor 2A |
| LINE-1 | Long interspersed nuclear element 1 |
| BCAT1/IKZF1 | Branched-chain amino acid transaminase 1-IKAROS family zinc finger 1 |
| RASSF1A | Ras-association domain family member 1 |
| Wnt5A | Wnt family member 5A |
| SFRP2 | Secreted frizzled-related protein 2 |
| DKK2 | Dickkopf WNT signaling pathway inhibitor 2 |
| PCDH10 | Protocadherin 10 |
| TMEFF2 | Transmembrane protein with EGF-like and two follistatin-like domains 2 |
| SFRP1 | Secreted frizzled-related protein 1 |
| HS3ST2 | Heparan sulfate-glucosamine 3-sulfotransferase 2 |
| CDH1/RHOA | Cadherin 1-ras homolog family member A |
| RPRM | Reprimo, TP53-dependent G2 arrest mediator homolog |
| ZNF793 | Zinc finger protein 793 |
| RUNX3 | Runt-related transcription factor 3 |
| PTEN | Phosphatase and tensin homolog |
| CXXC4 | CXXC finger protein 4 |
| TIMP2 | TIMP metallopeptidase inhibitor 2 |
| COL9A2 | Collagen type IX alpha 2 chain |
| EYA1 | EYA transcriptional coactivator and phosphatase 1 |
| ZNF365 | Zinc finger protein 365 |
| AMPH | Amphiphysin |
| SORCS3 | Sortilin-related VPS10 domain containing receptor 3 |
| AJAP1 | Adherens junctions-associated protein 1 |
| TGFβ2 | Transforming growth factor beta 2 |
| SHOX2 | Short stature homeobox 2 |
| RASSF1A | Ras-association domain family member 1 |
| RASSF1A-RARβ2 | Ras-association domain family member 1–retinoic acid receptor beta 2 |
| SHOX2-PTGER4 | Short stature homeobox 2-prostaglandin E receptor 4 |
| p16-RARβ2 | Cyclin=dependent kinase inhibitor 2A–retinoic acid receptor beta 2 |
| DAL-1 | Erythrocyte membrane protein band 4.1 like 3 |
| EPHB6 | EPH receptor B6 |
| HS3ST2 | Heparan sulfate-glucosamine 3-sulfotransferase 2 |
| TMEM88 | Transmembrane protein 88 |
| ELMO3 | Engulfment and cell motility 3 |
| HOXA9 | Homeobox A9 |
| RARβ2 | Retinoic acid receptor beta 2 |
| CDH13 | Cadherin 13 |
| CDKN2A/P16 | Cyclin-dependent kinase inhibitor 2A |
| CASP8 | Caspase 8 |
| TNFRSF6/Fas | Fas cell surface death receptor |
| TRAIL-R1/DR4 | TNF receptor superfamily member 10a |
| SCLC | Small-cell carcinoma |
| cfDNA | Cell-free DNA |
| ANK1 | Ankyrin 1 |
| CCND2 | Cyclin D2 |
| GATA3 | GATA-binding protein 3 |
| KCNH5 | Potassium voltage gated channel subfamily H member 5 |
| RARβ | Retinoic acid receptor beta |
| RASSF1 | Ras-association domain family member 1 |
| CLDN1 | Claudin 1 |
| TP63 | Tumor protein p63 |
| TPX5 | T-box transcription factor 5 |
| ADHFE1 | Alcohol dehydrogenase iron containing 1 |
| HNF1B | HNF1 homeobox B |
| EZH2 | Enhancer of zeste 2 polycomb-repressive complex 2 subunit |
| IBD | Inflammatory bowel disease |
| COPD | Chronic obstructive pulmonary disease |
| PAH-LTRS | PAH associated with the congenital left-to-right shunt |
| CSCs | Cancer stem cells |
| NF-kB | Nuclear factor kappaB |
| STAT3 | Signal transducer and activator of transcription 3 |
| SCFAs | Short-chain fatty acids |
| MUC2 | Mucin 2, oligomeric mucus/gel-forming |
| IL-6 | Interleukin 6 |
| MUC5B | Mucin 5B, oligomeric mucus/gel-forming |
| MUC7 | Mucin 7, secreted |
| IL-1 | Interleukin 1 |
| IL-8 | Interleukin 8 |
| TNF α | Tumor necrosis factor alpha-like |
| GRHL2 | Grainyhead-like 2 transcription factor |
| RhoG | Ras homology growth |
| ZEB1 | Zinc finger E-box binding homeobox 1 |
| MHC | Major histocompatibility complex |
| TLR | Toll-like receptors |
| HLA-A | Major histocompatibility complex, class I, A |
| HLA-B | Major histocompatibility complex, class I, B |
| HLA-C | Major histocompatibility complex, class I, C |
| CD8+ T cells | Cytotoxic T lymphocytes |
| HLA-DP | Major histocompatibility complex, class II, DP |
| HLA-DQ | Major histocompatibility complex, class II, DQ |
| HLA-DR | Major histocompatibility complex, class II, DR |
| CD4+ T cells | Helper T lymphocytes |
| TSA | Tumor-specific antigen |
| CTLA4 | Cytotoxic T-lymphocyte-associated protein 4 |
| APOBEC | Apolipoprotein B mRNA-editing enzyme, catalytic polypeptide 3 |
| PAMPs | Pathogen-associated molecular patterns |
| PRRs | Pattern-recognition receptors |
| RLRs | Retinoic acid-inducible gene-I (RIG-I)-like receptors |
| ALRs | Melanoma-2 (AIM2)-like receptors |
| CLRs | C-type lectin receptors |
| NLRs | Nucleotide oligomerization domain (NOD)-like receptors |
| MAVS | Mitochondrial antiviral-signaling protein |
| TBK1 | TANK-binding kinase 1 |
| IKKε | Nuclear factor kappaB kinase-ε |
| IRF3 | Interferon regulatory factor 3 |
| IRF7 | Interferon regulatory factor 7 |
| CRD | Carbohydrate-recognition domain |
| CTLD | C-type lectin-like domain |
| Dectin-1 | C-type lectin 1 |
| ITAM | Immunoreceptor tyrosine-based activation motif |
| DC-SIGN | DC-specific ICAM3-grabbing non-integrin |
| NAIP | NLR family apoptosis inhibitory protein |
| CIITA | Class II major histocompatibility complex transactivator |
| HETE | Cytochrome P450 family 4 subfamily F member 2 |
| TP1 | Transition protein 1 |
| TIR | Toll/IL-1R domain |
References
- Weng, M.T.; Chiu, Y.T.; Wei, P.Y.; Chiang, C.W.; Fang, H.L.; Wei, S.C. Microbiota and Gastrointestinal Cancer. J. Formos. Med. Assoc. 2019, 118, S32–S41. [Google Scholar] [CrossRef] [PubMed]
- de Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global Burden of Cancer Attributable to Infections in 2018: A Worldwide Incidence Analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.; Meehan, M.T.; Burrows, S.R.; Doolan, D.L.; Miles, J.J. Estimating the Global Burden of Epstein-Barr Virus-Related Cancers. J. Cancer Res. Clin. Oncol. 2022, 148, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Hayes, R.B. Environmental Influences on the Human Microbiome and Implications for Noncommunicable Disease. Annu. Rev. Public. Health 2021, 42, 277–292. [Google Scholar] [CrossRef]
- Amato, A. Oral-Systemic Health and Disorders: Latest Advances on Oral–Gut–Lung Microbiome Axis. Appl. Sci. 2022, 12, 8213. [Google Scholar] [CrossRef]
- Belizário, J.E.; Faintuch, J. Microbiome and Gut Dysbiosis. Exp. Suppl. 2018, 109, 459–476. [Google Scholar] [CrossRef]
- Mukherjee, S.; Bassler, B.L. Bacterial Quorum Sensing in Complex and Dynamically Changing Environments. Nat. Rev. Microbiol. 2019, 17, 371–382. [Google Scholar] [CrossRef]
- Bahrami, S.; Drabløs, F. Gene Regulation in the Immediate-Early Response Process. Adv. Biol. Regul. 2016, 62, 37–49. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Bogaert, J.; Prenen, H. Molecular Genetics of Colorectal Cancer. Ann. Gastroenterol. 2014, 27, 9–14. [Google Scholar]
- Nagata, N.; Ohta, H.; Yamada, A.; Teoh, Y.B.; Ichii, O.; Morishita, K.; Sasaki, N.; Takiguchi, M. Activities of Matrix Metalloproteinase-2, Matrix Metalloproteinase-9, and Serine Proteases in Samples of the Colorectal Mucosa of Miniature Dachshunds with Inflammatory Colorectal Polyps. Am. J. Vet. Res. 2020, 81, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Rosa, C.C.D.L.; Ramirez-Acuña, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef]
- Paczek, S.; Łukaszewicz-Zając, M.; Gryko, M.; Mroczko, P.; Kulczyńska-Przybik, A.; Mroczko, B. CXCL-8 in Preoperative Colorectal Cancer Patients: Significance for Diagnosis and Cancer Progression. Int. J. Mol. Sci. 2020, 21, 2040. [Google Scholar] [CrossRef]
- Li, L.; Zhang, L.; Tian, Y.; Zhang, T.; Duan, G.; Liu, Y.; Yin, Y.; Hua, D.; Qi, X.; Mao, Y. Serum Chemokine CXCL7 as a Diagnostic Biomarker for Colorectal Cancer. Front. Oncol. 2019, 9, 00921. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhu, C.; Chen, C.; Zong, Y.; Feng, H.; Liu, D.; Feng, W.; Zhao, J.; Lu, A. CCL19 Suppresses Angiogenesis through Promoting MiR-206 and Inhibiting Met/ERK/Elk-1/HIF-1α/VEGF-A Pathway in Colorectal Cancer. Cell Death Dis. 2018, 9, 974. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Luo, X.; Zhang, W.; Chen, E.; Xu, J.; Wang, F.; Cao, G.; Ju, Z.; Jin, D.; Huang, X.; et al. CXCL-13 Regulates Resistance to 5-Fluorouracil in Colorectal Cancer. Cancer Res. Treat. 2020, 52, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Song, F.; Hu, X.; Guo, D.; Liu, Y.; Wang, J.; Jiang, L.; Huang, P.; Zhang, Y. Analysis of Dynamic Molecular Networks: The Progression from Colorectal Adenoma to Cancer. J. Gastrointest. Oncol. 2021, 12, 2823–2837. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Malki, A.; Elruz, R.A.; Gupta, I.; Allouch, A.; Vranic, S.; Al Moustafa, A.E. Molecular Mechanisms of Colon Cancer Progression and Metastasis: Recent Insights and Advancements. Int. J. Mol. Sci. 2021, 22, 130. [Google Scholar] [CrossRef]
- Komor, M.A.; Bosch, L.J.W.; Bounova, G.; Bolijn, A.S.; Delis-van Diemen, P.M.; Rausch, C.; Hoogstrate, Y.; Stubbs, A.P.; de Jong, M.; Jenster, G.; et al. Consensus Molecular Subtype Classification of Colorectal Adenomas. J. Pathol. 2018, 246, 266–276. [Google Scholar] [CrossRef]
- Hoorn, S.T.; De Back, T.R.; Sommeijer, D.W.; Vermeulen, L. Clinical Value of Consensus Molecular Subtypes in Colorectal Cancer: A Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2022, 114, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Dobre, M.; Salvi, A.; Pelisenco, I.A.; Vasilescu, F.; De Petro, G.; Herlea, V.; Milanesi, E. Crosstalk between DNA Methylation and Gene Mutations in Colorectal Cancer. Front. Oncol. 2021, 11, 697409. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A Genetic Model for Colorectal Tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Afanador, C.H.; Palacio, K.A.; Isaza, L.F.; Ahumada, E.; Ocampo, C.M.; Muñetón, C.M. Caracterización Molecular de Pacientes Con Cáncer Colorrectal. Biomédica 2022, 42, 154–171. [Google Scholar] [CrossRef] [PubMed]
- Cornish, A.J.; Gruber, A.J.; Kinnersley, B.; Chubb, D.; Frangou, A.; Caravagna, G.; Noyvert, B.; Lakatos, E.; Wood, H.M.; Arnedo-Pac, C.; et al. Whole Genome Sequencing of 2023 Colorectal Cancers Reveals Mutational Landscapes, New Driver Genes and Immune Interactions. bioRxiv 2022. [Google Scholar] [CrossRef]
- Slavin, T.P.; Weitzel, J.N.; Neuhausen, S.L.; Schrader, K.A.; Oliveira, C.; Karam, R. Genetics of Gastric Cancer: What Do We Know about the Genetic Risks? Transl. Gastroenterol. Hepatol. 2019, 4, 55. [Google Scholar] [CrossRef]
- Fewings, E.; Larionov, A.; Redman, J.; Goldgraben, M.A.; Scarth, J.; Richardson, S.; Brewer, C.; Davidson, R.; Ellis, I.; Evans, D.G.; et al. Germline Pathogenic Variants in PALB2 and Other Cancer-Predisposing Genes in Families with Hereditary Diffuse Gastric Cancer without CDH1 Mutation: A Whole-Exome Sequencing Study. Lancet Gastroenterol. Hepatol. 2018, 3, 489–498. [Google Scholar] [CrossRef]
- Onoyama, T.; Ishikawa, S.; Isomoto, H. Gastric Cancer and Genomics: Review of Literature. J. Gastroenterol. 2022, 57, 505–516. [Google Scholar] [CrossRef]
- Franzin, R.; Netti, G.S.; Spadaccino, F.; Porta, C.; Gesualdo, L.; Stallone, G.; Castellano, G.; Ranieri, E. The Use of Immune Checkpoint Inhibitors in Oncology and the Occurrence of AKI: Where Do We Stand? Front. Immunol. 2020, 11, 574271. [Google Scholar] [CrossRef]
- Kim, S.T.; Cristescu, R.; Bass, A.J.; Kim, K.-M.; Odegaard, J.I.; Kim, K.; Liu, X.Q.; Sher, X.; Jung, H.; Lee, M.; et al. Comprehensive Molecular Characterization of Clinical Responses to PD-1 Inhibition in Metastatic Gastric Cancer. Nat. Med. 2018, 24, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Passaro, A.; Attili, I.; Rappa, A.; Vacirca, D.; Ranghiero, A.; Fumagalli, C.; Guarize, J.; Spaggiari, L.; de Marinis, F.; Barberis, M.; et al. Genomic Characterization of Concurrent Alterations in Non-Small Cell Lung Cancer (NSCLC) Harboring Actionable Mutations. Cancers 2021, 13, 2172. [Google Scholar] [CrossRef]
- Liu, Y.; Duan, J.; Zhang, F.; Liu, F.; Luo, X.; Shi, Y.; Lei, Y. Mutational and Transcriptional Characterization Establishes Prognostic Models for Resectable Lung Squamous Cell Carcinoma. Cancer Manag. Res. 2023, 15, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Chen, D.; Zhang, X.; Luo, Y.; Li, S. Integrating Genetic Mutations and Expression Profiles for Survival Prediction of Lung Adenocarcinoma. Thorac. Cancer 2019, 10, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Adderley, H.; Blackhall, F.H.; Lindsay, C.R. KRAS-Mutant Non-Small Cell Lung Cancer: Converging Small Molecules and Immune Checkpoint Inhibition. eBioMedicine 2019, 41, 711–716. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Yuan, J.Q.; Wang, K.F.; Fu, X.H.; Han, X.R.; Threapleton, D.; Yang, Z.Y.; Mao, C.; Tang, J.L. The Prevalence of EGFR Mutation in Patients with Non-Small Cell Lung Cancer: A Systematic Review and Meta-Analysis. Oncotarget 2016, 7, 78985–78993. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Cordero, R.; Ma, J.; Khanna, A.; Lyons, G.; Rinsurongkawong, W.; Bassett, R.; Guo, M.; Routbort, M.J.; Zhang, J.; Skoulidis, F.; et al. Simplified Molecular Classification of Lung Adenocarcinomas Based on EGFR, KRAS, and TP53 Mutations. BMC Cancer 2020, 20, 83. [Google Scholar] [CrossRef]
- Otálora-Otálora, B.A.; López-Kleine, L.; Rojas, A. Lung Cancer Gene Regulatory Network of Transcription Factors Related to the Hallmarks of Cancer. Curr. Issues Mol. Biol. 2023, 45, 434–464. [Google Scholar] [CrossRef]
- Otálora-Otálora, B.A.; Florez, M.; López-Kleine, L.; Canas Arboleda, A.; Grajales Urrego, D.M.; Rojas, A. Joint Transcriptomic Analysis of Lung Cancer and Other Lung Diseases. Front. Genet. 2019, 10, 1260. [Google Scholar] [CrossRef] [PubMed]
- Weidemüller, P.; Kholmatov, M.; Petsalaki, E.; Zaugg, J.B. Transcription Factors: Bridge between Cell Signaling and Gene Regulation. Proteomics 2021, 21, 2000034. [Google Scholar] [CrossRef]
- Filtz, T.M.; Vogel, W.K.; Leid, M. Regulation of Transcription Factor Activity by Interconnected Post-Translational Modifications. Trends Pharmacol. Sci. 2014, 35, 76–85. [Google Scholar] [CrossRef]
- Whiteside, S.T.; Goodbourn, S. Signal Transduction and Nuclear Targeting: Regulation of Transcription Factor Activity by Subcellular Localisation. J. Cell Sci. 1993, 104, 949–955. [Google Scholar] [CrossRef]
- Cordero, D.; Solé, X.; Crous-Bou, M.; Sanz-Pamplona, R.; Paré-Brunet, L.; Guinó, E.; Olivares, D.; Berenguer, A.; Santos, C.; Salazar, R.; et al. Large Differences in Global Transcriptional Regulatory Programs of Normal and Tumor Colon Cells. BMC Cancer 2014, 14, 708. [Google Scholar] [CrossRef] [PubMed]
- Mastrogamvraki, N.; Zaravinos, A. Signatures of Co-Deregulated Genes and Their Transcriptional Regulators in Colorectal Cancer. NPJ Syst. Biol. Appl. 2020, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Horaira, M.A.; Islam, M.A.; Kibria, M.K.; Alam, M.J.; Kabir, S.R.; Mollah, M.N.H. Bioinformatics Screening of Colorectal-Cancer Causing Molecular Signatures through Gene Expression Profiles to Discover Therapeutic Targets and Candidate Agents. BMC Med. Genom. 2023, 16, 64. [Google Scholar] [CrossRef]
- Wang, Y. Transcriptional Regulatory Network Analysis for Gastric Cancer Based on MRNA Microarray. Pathol. Oncol. Res. 2017, 23, 785–791. [Google Scholar] [CrossRef]
- Zhao, X.; Wu, S.; Jing, J. Identifying Diagnostic and Prognostic Biomarkers and Candidate Therapeutic Drugs of Gastric Cancer Based on Transcriptomics and Single-Cell Sequencing. Pathol. Oncol. Res. 2021, 27, 1609955. [Google Scholar] [CrossRef]
- Dong, C.; Rao, N.; Du, W.; Gao, F.; Lv, X.; Wang, G.; Zhang, J. MRBioM: An Algorithm for the Identification of Potential MRNA Biomarkers From Complete Transcriptomic Profiles of Gastric Adenocarcinoma. Front. Genet. 2021, 12, 679612. [Google Scholar] [CrossRef]
- Otálora-Otálora, B.A.; González Prieto, C.; Guerrero, L.; Bernal-Forigua, C.; Montecino, M.; Cañas, A.; López-Kleine, L.; Rojas, A. Identification of the Transcriptional Regulatory Role of RUNX2 by Network Analysis in Lung Cancer Cells. Biomedicines 2022, 10, 3122. [Google Scholar] [CrossRef] [PubMed]
- Otálora-Otálora, B.A.; Osuna-Garzón, D.A.; Carvajal-Parra, M.S.; Cañas, A.; Montecino, M.; López-Kleine, L.; Rojas, A. Identifying General Tumor and Specific Lung Cancer Biomarkers by Transcriptomic Analysis. Biology 2022, 11, 1082. [Google Scholar] [CrossRef] [PubMed]
- Cool, C.D.; Kuebler, W.M.; Bogaard, H.J.; Spiekerkoetter, E.; Nicolls, M.R.; Voelkel, N.F. The Hallmarks of Severe Pulmonary Arterial Hypertension: The Cancer Hypothesis-Ten Years Later. Am. J. Physiol.—Lung Cell. Mol. Physiol. 2020, 318, L1115–L1130. [Google Scholar] [CrossRef]
- Tuder, R.M.; Davis, L.A.; Graham, B.B. Targeting Energetic Metabolism: A New Frontier in the Pathogenesis and Treatment of Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 260–266. [Google Scholar] [CrossRef]
- Masri, F.A.; Xu, W.; Comhair, S.A.A.; Asosingh, K.; Koo, M.; Vasanji, A.; Drazba, J.; Anand-Apte, B.; Erzurum, S.C. Hyperproliferative Apoptosis-Resistant Endothelial Cells in Idiopathic Pulmonary Arterial Hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L548–L554. [Google Scholar] [CrossRef]
- Chen, C.; Wang, Z.; Ding, Y.; Wang, L.; Wang, S.; Wang, H.; Qin, Y. DNA Methylation: From Cancer Biology to Clinical Perspectives. Front. Biosci.—Landmark 2022, 27, 326. [Google Scholar] [CrossRef]
- Angeloni, A.; Bogdanovic, O. Sequence Determinants, Function, and Evolution of CpG Islands. Biochem. Soc. Trans. 2021, 49, 1109–1119. [Google Scholar] [CrossRef]
- Héberlé, É.; Bardet, A.F. Sensitivity of Transcription Factors to DNA Methylation. Essays Biochem. 2019, 63, 727–741. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Drier, Y.; Johnstone, S.E.; Hemming, M.L.; Tarjan, D.R.; Hegazi, E.; Shareef, S.J.; Javed, N.M.; Raut, C.P.; Eschle, B.K.; et al. Altered Chromosomal Topology Drives Oncogenic Programs in SDH-Deficient GISTs. Nature 2019, 575, 229–233. [Google Scholar] [CrossRef]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef]
- Ili, C.; Buchegger, K.; Demond, H.; Castillo-Fernandez, J.; Kelsey, G.; Zanella, L.; Abanto, M.; Riquelme, I.; López, J.; Viscarra, T.; et al. Landscape of Genome-Wide Dna Methylation of Colorectal Cancer Metastasis. Cancers 2020, 12, 2710. [Google Scholar] [CrossRef]
- Müller, D.; Győrffy, B. DNA Methylation-Based Diagnostic, Prognostic, and Predictive Biomarkers in Colorectal Cancer. Biochim. Biophys. Acta—Rev. Cancer 2022, 1877, 188722. [Google Scholar] [CrossRef]
- Mo, S.; Dai, W.; Wang, H.; Lan, X.; Ma, C.; Su, Z.; Xiang, W.; Han, L.; Luo, W.; Zhang, L.; et al. Early Detection and Prognosis Prediction for Colorectal Cancer by Circulating Tumour DNA Methylation Haplotypes: A Multicentre Cohort Study. eClinicalMedicine 2023, 55, 101717. [Google Scholar] [CrossRef]
- Kong, C.; Fu, T. Value of Methylation Markers in Colorectal Cancer (Review). Oncol. Rep. 2021, 46, 8128. [Google Scholar] [CrossRef]
- Tanaka, N.; Huttenhower, C.; Nosho, K.; Baba, Y.; Shima, K.; Quackenbush, J.; Haigis, K.M.; Giovannucci, E.; Fuchs, C.S.; Ogino, S. Novel Application of Structural Equation Modeling to Correlation Structure Analysis of CpG Island Methylation in Colorectal Cancer. Am. J. Pathol. 2010, 177, 2731–2740. [Google Scholar] [CrossRef] [PubMed]
- Milanesi, E.; Dobre, M.; Bucuroiu, A.I.; Herlea, V.; Manuc, T.E.; Salvi, A.; De Petro, G.; Manuc, M.; Becheanu, G. MiRNAs-Based Molecular Signature for KRAS Mutated and Wild Type Colorectal Cancer: An Explorative Study. J. Immunol. Res. 2020, 2020, 4927120. [Google Scholar] [CrossRef]
- Usui, G.; Matsusaka, K.; Mano, Y.; Urabe, M.; Funata, S.; Fukayama, M.; Ushiku, T.; Kaneda, A. DNA Methylation and Genetic Aberrations in Gastric Cancer. Digestion 2021, 102, 25–32. [Google Scholar] [CrossRef]
- Zeng, Y.; Rong, H.; Xu, J.; Cao, R.; Li, S.; Gao, Y.; Cheng, B.; Zhou, T. DNA Methylation: An Important Biomarker and Therapeutic Target for Gastric Cancer. Front. Genet. 2022, 13, 823905. [Google Scholar] [CrossRef]
- Gómez, A.; Pato, M.L.; Bujanda, L.; Sala, N.; Companioni, O.; Cosme, Á.; Tufano, M.; Hanly, D.J.; García, N.; Sanz-Anquela, J.M.; et al. Follow-up Study Confirms the Presence of Gastric Cancer Dna Methylation Hallmarks in High-Risk Precursor Lesions. Cancers 2021, 13, 2760. [Google Scholar] [CrossRef]
- Muhammad, J.S.; Eladl, M.A.; Khoder, G. Helicobacter Pylori-Induced Dna Methylation as an Epigenetic Modulator of Gastric Cancer: Recent Outcomes and Future Direction. Pathogens 2019, 8, 23. [Google Scholar] [CrossRef]
- Matsusaka, K.; Kaneda, A.; Nagae, G.; Ushiku, T.; Kikuchi, Y.; Hino, R.; Uozaki, H.; Seto, Y.; Takada, K.; Aburatani, H.; et al. Classification of Epstein-Barr Virus-Positive Gastric Cancers by Definition of DNA Methylation Epigenotypes. Cancer Res. 2011, 71, 7187–7197. [Google Scholar] [CrossRef]
- Li, T.; Chen, X.; Gu, M.; Deng, A.; Qian, C. Identification of the Subtypes of Gastric Cancer Based on DNA Methylation and the Prediction of Prognosis. Clin. Epigenetics 2020, 12, 161. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Yang, J.; He, L.; Wang, W.; Liu, Y.; Hu, Y.; Ge, M.; Ding, J.; Ye, Q. The Diagnostic Potential of SHOX2 and RASSF1A DNA Methylation in Early Lung Adenocarcinoma. Front. Oncol. 2022, 12, 849024. [Google Scholar] [CrossRef] [PubMed]
- Hoang, P.H.; Landi, M.T. DNA Methylation in Lung Cancer: Mechanisms and Associations with Histological Subtypes, Molecular Alterations, and Major Epidemiological Factors. Cancers 2022, 14, 961. [Google Scholar] [CrossRef]
- Nunes, S.P.; Diniz, F.; Moreira-Barbosa, C.; Constâncio, V.; Silva, A.V.; Oliveira, J.; Soares, M.; Paulino, S.; Cunha, A.L.; Rodrigues, J.; et al. Subtyping Lung Cancer Using Dna Methylation in Liquid Biopsies. J. Clin. Med. 2019, 8, 1500. [Google Scholar] [CrossRef]
- Shi, Y.X.; Wang, Y.; Li, X.; Zhang, W.; Zhou, H.H.; Yin, J.Y.; Liu, Z.Q. Genome-Wide DNA Methylation Profiling Reveals Novel Epigenetic Signatures in Squamous Cell Lung Cancer. BMC Genomics 2017, 18, 901. [Google Scholar] [CrossRef]
- Tew, B.Y.; Durand, J.K.; Bryant, K.L.; Hayes, T.K.; Peng, S.; Tran, N.L.; Gooden, G.C.; Buckley, D.N.; Der, C.J.; Baldwin, A.S.; et al. Genome-Wide DNA Methylation Analysis of KRAS Mutant Cell Lines. Sci. Rep. 2020, 10, 10149. [Google Scholar] [CrossRef]
- Xu, Z.; Qin, F.; Yuan, L.; Wei, J.; Sun, Y.; Qin, J.; Deng, K.; Zheng, T.; Li, S. EGFR DNA Methylation Correlates With EGFR Expression, Immune Cell Infiltration, and Overall Survival in Lung Adenocarcinoma. Front. Oncol. 2021, 11, 691915. [Google Scholar] [CrossRef]
- Aguiar-Pulido, V.; Huang, W.; Suarez-Ulloa, V.; Cickovski, T.; Mathee, K.; Narasimhan, G. Metagenomics, Metatranscriptomics, and Metabolomics Approaches for Microbiome Analysis. Evol. Bioinform. Online 2016, 12, 5–16. [Google Scholar] [CrossRef]
- Afzaal, M.; Saeed, F.; Shah, Y.A.; Hussain, M.; Rabail, R.; Socol, C.T.; Hassoun, A.; Pateiro, M.; Lorenzo, J.M.; Rusu, A.V.; et al. Human Gut Microbiota in Health and Disease: Unveiling the Relationship. Front. Microbiol. 2022, 13, 999001. [Google Scholar] [CrossRef]
- Hou, K.; Wu, Z.X.; Chen, X.Y.; Wang, J.Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in Health and Diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef]
- Mark Welch, J.L.; Rossetti, B.J.; Rieken, C.W.; Dewhirst, F.E.; Borisy, G.G. Biogeography of a Human Oral Microbiome at the Micron Scale. Proc. Natl. Acad. Sci. USA 2016, 113, E791–E800. [Google Scholar] [CrossRef]
- Peters, B.A.; Wu, J.; Hayes, R.B.; Ahn, J. The Oral Fungal Mycobiome: Characteristics and Relation to Periodontitis in a Pilot Study. BMC Microbiol. 2017, 17, 157. [Google Scholar] [CrossRef]
- Bharatha, A.; Kandamaran, L.; Krishnamurthy, K. Commentary: “Fungal Infections of Oral Cavity: Diagnosis, Management, and Association with COVID-19”. SN Compr. Clin. Med. 2021, 3, 2002–2003. [Google Scholar] [CrossRef]
- Chen, C.; Hemme, C.; Beleno, J.; Shi, Z.J.; Ning, D.; Qin, Y.; Tu, Q.; Jorgensen, M.; He, Z.; Wu, L.; et al. Oral Microbiota of Periodontal Health and Disease and Their Changes after Nonsurgical Periodontal Therapy. ISME J. 2018, 12, 1210–1224. [Google Scholar] [CrossRef] [PubMed]
- Mahendra, J.; Mahendra, L.; Mugri, M.H.; Sayed, M.E.; Bhandi, S.; Alshahrani, R.T.; Balaji, T.M.; Varadarajan, S.; Tanneeru, S.; Abirami Nayaki Rao, P.; et al. Role of Periodontal Bacteria, Viruses, and Placental Mir155 in Chronic Periodontitis and Preeclampsia—A Genetic Microbiological Study. Curr. Issues Mol. Biol. 2021, 43, 831–844. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, G.R.M.; Libra, M.; De Pasquale, R.; Ferlito, S.; Pedullà, E. Association of Viral Infections With Oral Cavity Lesions: Role of SARS-CoV-2 Infection. Front. Med. 2021, 7, 571214. [Google Scholar] [CrossRef]
- Bui, F.Q.; Almeida-da-Silva, C.L.C.; Huynh, B.; Trinh, A.; Liu, J.; Woodward, J.; Asadi, H.; Ojcius, D.M. Association between Periodontal Pathogens and Systemic Disease. Biomed. J. 2019, 42, 27–35. [Google Scholar] [CrossRef]
- Abdelbary, M.M.H.; Hatting, M.; Bott, A.; Dahlhausen, A.; Keller, D.; Trautwein, C.; Conrads, G. The Oral-Gut Axis: Salivary and Fecal Microbiome Dysbiosis in Patients with Inflammatory Bowel Disease. Front. Cell. Infect. Microbiol. 2022, 12, 1010853. [Google Scholar] [CrossRef]
- Reitano, E.; De’angelis, N.; Gavriilidis, P.; Gaiani, F.; Memeo, R.; Inchingolo, R.; Bianchi, G.; De’Angelis, G.L.; Carra, M.C. Oral Bacterial Microbiota in Digestive Cancer Patients: A Systematic Review. Microorganisms 2021, 9, 2585. [Google Scholar] [CrossRef]
- Yang, Y.; Long, J.; Wang, C.; Blot, W.J.; Pei, Z.; Shu, X.; Wu, F.; Rothman, N.; Wu, J.; Lan, Q.; et al. Prospective Study of Oral Microbiome and Gastric Cancer Risk among Asian, African American and European American Populations. Int. J. Cancer 2022, 150, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Li, W.; Wang, Q.; Chen, J.; Zu, Y.; Zhou, X.; Guo, Q. Relationships Between Oral Microecosystem and Respiratory Diseases. Front. Mol. Biosci. 2022, 8, 718222. [Google Scholar] [CrossRef]
- Bhuyan, R.; Bhuyan, S.K.; Mohanty, J.N.; Das, S.; Juliana, N.; Abu, I.F. Periodontitis and Its Inflammatory Changes Linked to Various Systemic Diseases: A Review of Its Underlying Mechanisms. Biomedicines 2022, 10, 2659. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, T.; Lu, W.; Duan, X.; Luo, X.; Liu, S.; Chen, Y.; Li, Y.; Chen, J.; Liao, J.; et al. Altered Airway Microbiota Composition in Patients With Pulmonary Hypertension. Hypertension 2020, 76, 1589–1599. [Google Scholar] [CrossRef] [PubMed]
- Fauroux, B.; Hart, N.; Belfar, S.; Boulé, M.; Tillous-Borde, I.; Bonnet, D.; Bingen, E.; Clément, A. Burkholderia Cepacia Is Associated with Pulmonary Hypertension and Increased Mortality among Cystic Fibrosis Patients. J. Clin. Microbiol. 2004, 42, 5537–5541. [Google Scholar] [CrossRef]
- Vogtmann, E.; Hua, X.; Yu, G.; Purandare, V.; Hullings, A.G.; Shao, D.; Wan, Y.; Li, S.; Dagnall, C.L.; Jones, K.; et al. The Oral Microbiome and Lung Cancer Risk: An Analysis of 3 Prospective Cohort Studies. J. Natl. Cancer Inst. 2022, 114, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Ağagündüz, D.; Cocozza, E.; Cemali, Ö.; Bayazıt, A.D.; Nanì, M.F.; Cerqua, I.; Morgillo, F.; Saygılı, S.K.; Berni Canani, R.; Amero, P.; et al. Understanding the Role of the Gut Microbiome in Gastrointestinal Cancer: A Review. Front. Pharmacol. 2023, 14, 1130562. [Google Scholar] [CrossRef]
- Groer, M.W.; Luciano, A.A.; Dishaw, L.J.; Ashmeade, T.L.; Miller, E.; Gilbert, J.A. Development of the Preterm Infant Gut Microbiome: A Research Priority. Microbiome 2014, 2, 38. [Google Scholar] [CrossRef] [PubMed]
- Mailhe, M.; Ricaboni, D.; Vitton, V.; Gonzalez, J.M.; Bachar, D.; Dubourg, G.; Cadoret, F.; Robert, C.; Delerce, J.; Levasseur, A.; et al. Repertoire of the Gut Microbiota from Stomach to Colon Using Culturomics and Next-Generation Sequencing. BMC Microbiol. 2018, 18, 157. [Google Scholar] [CrossRef]
- Ramos, S.; Martín, M.Á. Impact of Diet on Gut Microbiota. Curr. Opin. Food Sci. 2021, 37, 83–90. [Google Scholar] [CrossRef]
- Zhang, P. Influence of Foods and Nutrition on the Gut Microbiome and Implications for Intestinal Health. Int. J. Mol. Sci. 2022, 23, 9588. [Google Scholar] [CrossRef]
- Wu, S.; Xie, S.; Yuan, C.; Yang, Z.; Liu, S.; Zhang, Q.; Sun, F.; Wu, J.; Zhan, S.; Zhu, S.; et al. Inflammatory Bowel Disease and Long-Term Risk of Cancer: A Prospective Cohort Study Among Half a Million Adults in UK Biobank. Inflamm. Bowel Dis. 2023, 29, 384–395. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, F.; Xue, J.; Lee, S.A.; Liu, L.; Riordan, S.M. Bacterial Species Associated with Human Inflammatory Bowel Disease and Their Pathogenic Mechanisms. Front. Microbiol. 2022, 13, 801892. [Google Scholar] [CrossRef]
- Cai, Z.; Zhu, T.; Liu, F.; Zhuang, Z.; Zhao, L. Co-Pathogens in Periodontitis and Inflammatory Bowel Disease. Front. Med. 2021, 8, 723719. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Qin, H.; Liu, X.; Zhang, X.; Chen, Z.; Qin, T.; Chang, L.; Zhang, W. The Emerging Roles of Human Gut Microbiota in Gastrointestinal Cancer. Front. Immunol. 2022, 13, 915047. [Google Scholar] [CrossRef] [PubMed]
- Wirbel, J.; Pyl, P.T.; Kartal, E.; Zych, K.; Kashani, A.; Milanese, A.; Fleck, J.S.; Voigt, A.Y.; Palleja, A.; Ponnudurai, R.; et al. Global Microbial Signatures That Are Specific for Colorectal Cancer. Nat. Med. 2019, 25, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Chung, J.; Cho, M.L.; Park, D.; Choi, S.S. Analysis of Changes in Microbiome Compositions Related to the Prognosis of Colorectal Cancer Patients Based on Tissue-Derived 16S RRNA Sequences. J. Transl. Med. 2021, 19, 485. [Google Scholar] [CrossRef]
- Iarc, L. IARC Schistosomes, Liver Flukes and Helicobacter Pylori. IARC Monogr. Eval. Carcinog. Risks Hum. 1994, 61, 1–241. [Google Scholar]
- Capurro, M.I.; Greenfield, L.K.; Prashar, A.; Xia, S.; Abdullah, M.; Wong, H.; Zhong, X.Z.; Bertaux-skeirik, N.; Chakrabarti, J.; Siddiqui, I.; et al. VacA Generates a Protective Intracellular Reservoir for Helicobacter Pylori That Is Eliminated by Activation of the Lysosomal Calcium Channel TRPML1. Nat. Microbiol. 2019, 4, 1411–1423. [Google Scholar] [CrossRef]
- Buti, L.; Ruiz-Puig, C.; Sangberg, D.; Leissing, T.M.; Brewer, R.C.; Owen, R.P.; Sgromo, B.; Royer, C.; Ebner, D.; Lu, X. CagA–ASPP2 Complex Mediates Loss of Cell Polarity and Favors H. Pylori Colonization of Human Gastric Organoids. Proc. Natl. Acad. Sci. USA 2020, 117, 2645–2655. [Google Scholar] [CrossRef]
- Schulz, C.; Schütte, K.; Mayerle, J.; Malfertheiner, P. The Role of the Gastric Bacterial Microbiome in Gastric Cancer: Helicobacter Pylori and Beyond. Therap. Adv. Gastroenterol. 2019, 12, 1756284819894062. [Google Scholar] [CrossRef]
- Mejia-Ortiz, L.; Rosero-Galindo, C.Y.; Carlosama-Rosero, Y.H.; Castillo-Giraldo, A. Estimación de La Frecuencia de Infección Por Helicobacter Pylori En Pacientes Con Lesiones Potencialmente Malignas Gástricas Del Municipio de Pasto-Nariño, 2016–2019. Infectio 2021, 26, 46. [Google Scholar] [CrossRef]
- Dai, D.; Yang, Y.; Yu, J.; Dang, T.; Qin, W.; Teng, L.; Ye, J.; Jiang, H. Interactions between Gastric Microbiota and Metabolites in Gastric Cancer. Cell Death Dis. 2021, 12, 1104. [Google Scholar] [CrossRef]
- Nash, A.K.; Auchtung, T.A.; Wong, M.C.; Smith, D.P.; Gesell, J.R.; Ross, M.C.; Stewart, C.J.; Metcalf, G.A.; Muzny, D.M.; Gibbs, R.A.; et al. The Gut Mycobiome of the Human Microbiome Project Healthy Cohort. Microbiome 2017, 5, 153. [Google Scholar] [CrossRef]
- Zhang, F.; Aschenbrenner, D.; Yoo, J.Y.; Zuo, T. The Gut Mycobiome in Health, Disease, and Clinical Applications in Association with the Gut Bacterial Microbiome Assembly. Lancet Microbe 2022, 3, e969–e983. [Google Scholar] [CrossRef] [PubMed]
- Scarpellini, E.; Ianiro, G.; Attili, F.; Bassanelli, C.; De Santis, A.; Antonio, G. The Human Gut Microbiota and Virome: Potential Therapeutic Implications. Dig. Liver Dis. 2015, 47, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Sugimura, N.; Burgermeister, E.; Ebert, M.P.; Zuo, T.; Lan, P. The Gut Virome: A New Microbiome Component in Health and Disease. eBioMedicine 2022, 81, 104113. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Zhao, C.; Zhang, H.; Mattei, L.; Sherrill-Mix, S.; Bittinger, K.; Kessler, L.R.; Wu, G.D.; Baldassano, R.N.; DeRusso, P.; et al. The Stepwise Assembly of the Neonatal Virome Is Modulated by Breastfeeding. Nature 2020, 581, 470–474. [Google Scholar] [CrossRef]
- Zhang, T.; Breitbart, M.; Lee, W.H.; Run, J.Q.; Wei, C.L.; Soh, S.W.L.; Hibberd, M.L.; Liu, E.T.; Rohwer, F.; Ruan, Y. RNA Viral Community in Human Feces: Prevalence of Plant Pathogenic Viruses. PLoS Biol. 2006, 4, 0108–0118. [Google Scholar] [CrossRef]
- Ungaro, F.; Massimino, L.; Furfaro, F.; Rimoldi, V.; Peyrin-Biroulet, L.; D’Alessio, S.; Danese, S. Metagenomic Analysis of Intestinal Mucosa Revealed a Specific Eukaryotic Gut Virome Signature in Early-Diagnosed Inflammatory Bowel Disease. Gut Microbes 2019, 10, 149–158. [Google Scholar] [CrossRef]
- Chen, X.; Kost, J.; Sulovari, A.; Wong, N.; Liang, W.S.; Cao, J.; Li, D. A Virome-Wide Clonal Integration Analysis Platform for Discovering Cancer Viral Etiology. Genome Res. 2019, 29, 819–830. [Google Scholar] [CrossRef]
- Nakatsu, G.; Zhou, H.; Wu, W.K.K.; Wong, S.H.; Coker, O.O.; Dai, Z.; Li, X.; Szeto, C.H.; Sugimura, N.; Lam, T.Y.T.; et al. Alterations in Enteric Virome Are Associated With Colorectal Cancer and Survival Outcomes. Gastroenterology 2018, 155, 529–541.e5. [Google Scholar] [CrossRef]
- Bou Zerdan, M.; Kassab, J.; Meouchy, P.; Haroun, E.; Nehme, R.; Bou Zerdan, M.; Fahed, G.; Petrosino, M.; Dutta, D.; Graziano, S. The Lung Microbiota and Lung Cancer: A Growing Relationship. Cancers 2022, 14, 4813. [Google Scholar] [CrossRef]
- Natalini, J.G.; Singh, S.; Segal, L.N. The Dynamic Lung Microbiome in Health and Disease. Nat. Rev. Microbiol. 2022, 21, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Huffnagle, G.B.; Dickson, R.P.; Lukacs, N.W. The Respiratory Tract Microbiome and Lung Inflammation: A Two-Way Street. Mucosal Immunol. 2017, 10, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Stricker, S.; Hain, T.; Chao, C.M.; Rudloff, S. Respiratory and Intestinal Microbiota in Pediatric Lung Diseases—Current Evidence of the Gut–Lung Axis. Int. J. Mol. Sci. 2022, 23, 6791. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Cheng, L.; Song, Y.; Sun, Y.; Gui, W.; Deng, Y.; Xie, C.; Liu, M. Altered Lung Microbiome and Metabolome Profile in Children With Pulmonary Arterial Hypertension Associated With Congenital Heart Disease. Front. Med. 2022, 9, 940784. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.D.N.; Viscogliosi, E.; Delhaes, L. The Lung Mycobiome: An Emerging Field of the Human Respiratory Microbiome. Front. Microbiol. 2015, 6, 89. [Google Scholar] [CrossRef]
- Li, Z.; Lu, G.; Meng, G. Pathogenic Fungal Infection in the Lung. Front. Immunol. 2019, 10, 01524. [Google Scholar] [CrossRef]
- McDermott, A. Drug-Resistant Fungi on the Rise. Proc. Natl. Acad. Sci. USA 2022, 119, e2217948119. [Google Scholar] [CrossRef]
- Lee, Y.; Robbins, N.; Cowen, L.E. Molecular Mechanisms Governing Antifungal Drug Resistance. NPJ Antimicrob. Resist. 2023, 1, 5. [Google Scholar] [CrossRef]
- Hammond, E.E.; McDonald, C.S.; Vestbo, J.; Denning, D.W. The Global Impact of Aspergillus Infection on COPD. BMC Pulm. Med. 2020, 20, 241. [Google Scholar] [CrossRef]
- Otu, A.; Kosmidis, C.; Mathioudakis, A.G.; Ibe, C.; Denning, D.W. The Clinical Spectrum of Aspergillosis in Chronic Obstructive Pulmonary Disease. Infection 2023, 51, 813–829. [Google Scholar] [CrossRef]
- Shao, W.; Zhang, Z.; Feng, H.; Liang, C.; Liu, D. Pulmonary Mucormycosis: A Case of Pulmonary Arterial Hypertension, Westermark Sign, and Bronchopleural Fistula. J. Int. Med. Res. 2020, 48, 0300060520971450. [Google Scholar] [CrossRef]
- Park, M.; Ho, D.Y.; Wakelee, H.A.; Neal, J.W. Opportunistic Invasive Fungal Infections Mimicking Progression of Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2021, 22, e193–e200. [Google Scholar] [CrossRef] [PubMed]
- Gazzoni, F.F.; Severo, L.C.; Marchiori, E.; Irion, K.L.; Guimarães, M.D.; Godoy, M.C.; Sartori, A.P.G.; Hochhegger, B. Fungal Diseases Mimicking Primary Lung Cancer: Radiologic-Pathologic Correlation. Mycoses 2014, 57, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N.; Akazawa, N.; Murakami, H.; Ishibana, Y.; Takahashi, Y.; Hosoda, W.; Yaguchi, T.; Kamei, K. A Schizophyllum Commune Fungus Ball in a Lung Cancer Cavity: A Case Report. BMC Infect. Dis. 2021, 21, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Kesson, A.M. Respiratory Virus Infections. Paediatr. Respir. Rev. 2007, 8, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Hodinka, R.L. Respiratory RNA Viruses. Diagn. Microbiol. Immunocompromised Host 2016, 4, 233–271. [Google Scholar] [CrossRef]
- Audi, A.; AlIbrahim, M.; Kaddoura, M.; Hijazi, G.; Yassine, H.M.; Zaraket, H. Seasonality of Respiratory Viral Infections: Will COVID-19 Follow Suit? Front. Public Health 2020, 8, 567. [Google Scholar] [CrossRef]
- Britto, C.J.; Brady, V.; Lee, S.; Dela Cruz, C.S. Respiratory Viral Infections in Chronic Lung Diseases. Clin. Chest Med. 2017, 38, 87–96. [Google Scholar] [CrossRef]
- Cool, C.D.; Voelkel, N.F.; Bull, T. Viral Infection and Pulmonary Hypertension: Is There an Association? Expert Rev. Respir. Med. 2011, 5, 207–216. [Google Scholar] [CrossRef]
- Aramini, B.; Masciale, V.; Samarelli, A.V.; Tonelli, R.; Cerri, S.; Clini, E.; Stella, F.; Dominici, M. Biological Effects of COVID-19 on Lung Cancer: Can We Drive Our Decisions. Front. Oncol. 2022, 12, 1029830. [Google Scholar] [CrossRef]
- Hu, Y.; Ren, S.; He, Y.; Wang, L.; Chen, C.; Tang, J.; Liu, W.; Yu, F. Possible Oncogenic Viruses Associated with Lung Cancer. OncoTargets Ther. 2020, 13, 10651–10666. [Google Scholar] [CrossRef] [PubMed]
- Budisan, L.; Zanoaga, O.; Braicu, C.; Pirlog, R.; Covaliu, B.; Esanu, V.; Korban, S.S.; Berindan-Neagoe, I. Links between Infections, Lung Cancer, and the Immune System. Int. J. Mol. Sci. 2021, 22, 9394. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, T. Human Microbiota: A Crucial Gatekeeper in Lung Cancer Initiation, Progression, and Treatment. Med. Microecol. 2022, 13, 100055. [Google Scholar] [CrossRef]
- Campbell, C.; Kandalgaonkar, M.R.; Golonka, R.M.; Yeoh, B.S.; Vijay-Kumar, M.; Saha, P. Crosstalk between Gut Microbiota and Host Immunity: Impact on Inflammation and Immunotherapy. Biomedicines 2023, 11, 294. [Google Scholar] [CrossRef] [PubMed]
- Hufnagl, K.; Pali-Schöll, I.; Roth-Walter, F.; Jensen-Jarolim, E. Dysbiosis of the Gut and Lung Microbiome Has a Role in Asthma. Semin. Immunopathol. 2020, 42, 75–93. [Google Scholar] [CrossRef]
- Enaud, R.; Prevel, R.; Ciarlo, E.; Beaufils, F.; Wieërs, G.; Guery, B.; Delhaes, L. The Gut-Lung Axis in Health and Respiratory Diseases: A Place for Inter-Organ and Inter-Kingdom Crosstalks. Front. Cell. Infect. Microbiol. 2020, 10, 9. [Google Scholar] [CrossRef]
- Krüger, W.; Vielreicher, S.; Kapitan, M.; Jacobsen, I.D.; Niemiec, M.J. Fungal-Bacterial Interactions in Health and Disease. Pathogens 2019, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Diaz, P.I.; Xie, Z.; Sobue, T.; Thompson, A.; Biyikoglu, B.; Ricker, A.; Ikonomou, L.; Dongari-Bagtzoglou, A. Synergistic Interaction between Candida Albicans and Commensal Oral Streptococci in a Novel in Vitro Mucosal Model. Infect. Immun. 2012, 80, 620–632. [Google Scholar] [CrossRef]
- Scott, J.; Sueiro-Olivares, M.; Ahmed, W.; Heddergott, C.; Zhao, C.; Thomas, R.; Bromley, M.; Latgé, J.P.; Krappmann, S.; Fowler, S.; et al. Pseudomonas Aeruginosa-Derived Volatile Sulfur Compounds Promote Distal Aspergillus fumigatus Growth and a Synergistic Pathogen-Pathogen Interaction That Increases Pathogenicity in Co-Infection. Front. Microbiol. 2019, 10, 02311. [Google Scholar] [CrossRef]
- Siavoshi, F.; Heydari, S.; Shafiee, M.; Ahmadi, S.; Saniee, P.; Sarrafnejad, A.; Kolahdoozan, S. Sequestration inside the Yeast Vacuole May Enhance Helicobacter Pylori Survival against Stressful Condition. Infect. Genet. Evol. 2019, 69, 127–133. [Google Scholar] [CrossRef]
- Sánchez-Alonzo, K.; Parra-Sepúlveda, C.; Vega, S.; Bernasconi, H.; Campos, V.L.; Smith, C.T.; Sáez, K.; García-Cancino, A. In Vitro Incorporation of Helicobacter Pylori into Candida Albicans Caused by Acidic PH Stress. Pathogens 2020, 9, 489. [Google Scholar] [CrossRef]
- Erickson, A.K.; Jesudhasan, P.R.; Mayer, M.J.; Narbad, A.; Winter, S.E.; Pfeiffer, J.K. Bacteria Facilitate Enteric Virus Co-Infection of Mammalian Cells and Promote Genetic Recombination. Cell Host Microbe 2018, 23, 77–88.e5. [Google Scholar] [CrossRef]
- Rowe, H.M.; Meliopoulos, V.A.; Iverson, A.; Bomme, P.; Schultz-Cherry, S.; Rosch, J.W. Direct Interactions with Influenza Promote Bacterial Adherence during Respiratory Infections. Nat. Microbiol. 2019, 4, 1328–1336. [Google Scholar] [CrossRef]
- Deinhardt-Emmer, S.; Haupt, K.F.; Garcia-Moreno, M.; Geraci, J.; Forstner, C.; Pletz, M.; Ehrhardt, C.; Löffer, B. Staphylococcus Aureus Pneumonia: Preceding Influenza Infection Paves Theway for Low-Virulent Strains. Toxins 2019, 11, 734. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.N.; Jiao, N.; Tan, J.C.; Wang, Z.; Wu, D.; Wang, A.J.; Chen, J.; Tao, L.; Zhou, C.; Fang, W.; et al. Multi-Kingdom Microbiota Analyses Identify Bacterial–Fungal Interactions and Biomarkers of Colorectal Cancer across Cohorts. Nat. Microbiol. 2022, 7, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Di Spirito, F.; Toti, P.; Pilone, V.; Carinci, F.; Lauritano, D.; Sbordone, L. The Association between Periodontitis and Human Colorectal Cancer: Genetic and Pathogenic Linkage. Life 2020, 10, 211. [Google Scholar] [CrossRef]
- Du, B.; Fu, Y.; Han, Y.; Sun, Q.; Xu, J. The Lung-Gut Crosstalk in Respiratory and Inflammatory Bowel Disease. Front. Cell. Infect. Microbiol. 2023, 13, 1218565. [Google Scholar] [CrossRef]
- Zhou, H.; Suo, J.; Zhu, J. Therapeutic Relevance of Human Microbiota and Lung Cancer. Zhongguo Fei Ai Za Zhi 2019, 22, 464–469. [Google Scholar] [CrossRef]
- Carbone, C.; Piro, G.; Di Noia, V.; D’Argento, E.; Vita, E.; Ferrara, M.G.; Pilotto, S.; Milella, M.; Cammarota, G.; Gasbarrini, A.; et al. Lung and Gut Microbiota as Potential Hidden Driver of Immunotherapy Efficacy in Lung Cancer. Mediators Inflamm. 2019, 2019, 7652014. [Google Scholar] [CrossRef]
- Lamont, R.J.; Koo, H.; Hajishengallis, G. The Oral Microbiota: Dynamic Communities and Host Interactions. Nat. Rev. Microbiol. 2018, 16, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Sudhakara, P.; Gupta, A.; Bhardwaj, A.; Wilson, A. Oral Dysbiotic Communities and Their Implications in Systemic Diseases. Dent. J. 2018, 6, 10. [Google Scholar] [CrossRef]
- Nazir, M.; Al-Ansari, A.; Al-Khalifa, K.; Alhareky, M.; Gaffar, B.; Almas, K. Global Prevalence of Periodontal Disease and Lack of Its Surveillance. Sci. World J. 2020, 2020, 2146160. [Google Scholar] [CrossRef]
- Pardo Romero, F.F.; Hernández, L.J. Enfermedad Periodontal: Enfoques Epidemiológicos Para Su Análisis Como Problema de Salud Pública. Rev. Salud Pública 2018, 20, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Borisy, G.G.; Valm, A.M. Spatial Scale in Analysis of the Dental Plaque Microbiome. Periodontology 2000 2021, 86, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Nocini, R.; Lippi, G.; Mattiuzzi, C. Periodontal Disease: The Portrait of an Epidemic. J. Public Health Emerg. 2020, 4, 2–7. [Google Scholar] [CrossRef]
- Kim, J.; Amar, S. Periodontal Disease and Systemic Conditions: A Bidirectional Relationship. Odontology 2006, 94, 10–21. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Magrin, G.L.; Strauss, F.J.; Benfatti, C.A.M.; Maia, L.C.; Gruber, R. Effects of Short-Chain Fatty Acids on Human Oral Epithelial Cells and the Potential Impact on Periodontal Disease: A Systematic Review of in Vitro Studies. Int. J. Mol. Sci. 2020, 21, 4895. [Google Scholar] [CrossRef]
- Bullman, S.; Pedamallu, C.S.; Sicinska, E.; Clancy, T.E.; Zhang, X.; Cai, D.; Neuberg, D.; Huang, K.; Guevara, F.; Nelson, T.; et al. Analysis of Fusobacterium Persistence and Antibiotic Response in Colorectal Cancer. Science 2017, 358, 1443–1448. [Google Scholar] [CrossRef]
- Li, Y.J.; Chen, X.; Kwan, T.K.; Loh, Y.W.; Singer, J.; Liu, Y.; Ma, J.; Tan, J.; Macia, L.; Mackay, C.R.; et al. Dietary Fiber Protects against Diabetic Nephropathy through Short-Chain Fatty Acid-Mediated Activation of G Protein-Coupled Receptors GPR43 and GPR109A. J. Am. Soc. Nephrol. 2020, 31, 1267–1281. [Google Scholar] [CrossRef]
- Pattayil, L.; Balakrishnan-Saraswathi, H.-T. In Vitro Evaluation of Apoptotic Induction of Butyric Acid Derivatives in Colorectal Carcinoma Cells. Anticancer Res. 2019, 39, 3795–3801. [Google Scholar] [CrossRef] [PubMed]
- Woelber, J.P.; Gärtner, M.; Breuninger, L.; Anderson, A.; König, D.; Hellwig, E.; Al-Ahmad, A.; Vach, K.; Dötsch, A.; Ratka-Krüger, P.; et al. The Influence of an Anti-Inflammatory Diet on Gingivitis. A Randomized Controlled Trial. J. Clin. Periodontol. 2019, 46, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Koliarakis, I.; Messaritakis, I.; Nikolouzakis, T.K.; Hamilos, G.; Souglakos, J.; Tsiaoussis, J. Oral Bacteria and Intestinal Dysbiosis in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 4146. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, K.; Xia, L. The Barrier and beyond: Roles of Intestinal Mucus and Mucin-Type O-Glycosylation in Resistance and Tolerance Defense Strategies Guiding Host-Microbe Symbiosis. Gut Microbes 2022, 14, 2052699. [Google Scholar] [CrossRef]
- Nouri, Z.; Choi, S.W.; Choi, I.J.; Ryu, K.W.; Woo, S.M.; Park, S.-J.; Lee, W.J.; Choi, W.; Jung, Y.-S.; Myung, S.-K.; et al. Exploring Connections between Oral Microbiota, Short-Chain Fatty Acids, and Specific Cancer Types: A Study of Oral Cancer, Head and Neck Cancer, Pancreatic Cancer, and Gastric Cancer. Cancers 2023, 15, 2898. [Google Scholar] [CrossRef]
- Bakhti, S.Z.; Latifi-Navid, S. Oral Microbiota and Helicobacter Pylori in Gastric Carcinogenesis: What Do We Know and Where Next? BMC Microbiol. 2021, 21, 71. [Google Scholar] [CrossRef] [PubMed]
- Krzyżek, P.; Gościniak, G. Oral Helicobacter Pylori: Interactions with Host and Microbial Flora of the Oral Cavity. Dent. Med. Probl. 2018, 55, 75–82. [Google Scholar] [CrossRef]
- Krzyżek, P.; Gościniak, G. A Proposed Role for Diffusible Signal Factors in the Biofilm Formation and Morphological Transformation of Helicobacter Pylori. Turkish J. Gastroenterol. Off. J. Turkish Soc. Gastroenterol. 2018, 29, 7–13. [Google Scholar] [CrossRef]
- Zhao, Y.; Gao, X.; Guo, J.; Yu, D.; Xiao, Y.; Wang, H.; Li, Y. Helicobacter Pylori Infection Alters Gastric and Tongue Coating Microbial Communities. Helicobacter 2019, 24, e12567. [Google Scholar] [CrossRef]
- Huang, K.; Gao, X.; Wu, L.; Yan, B.; Wang, Z.; Zhang, X.; Peng, L.; Yu, J.; Sun, G.; Yang, Y. Salivary Microbiota for Gastric Cancer Prediction: An Exploratory Study. Front. Cell. Infect. Microbiol. 2021, 11, 640309. [Google Scholar] [CrossRef]
- Kesharani, P.; Kansara, P.; Kansara, T.; Kini, A.; Bhat, R.; Shetty, P.; Penugonda, B. Is Periodontitis a Risk Factor for Lung Cancer? A Meta-Analysis and Detailed Review of Mechanisms of Association. Contemp. Clin. Dent. 2022, 13, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Pine, S.R.; Mechanic, L.E.; Enewold, L.; Chaturvedi, A.K.; Katki, H.A.; Zheng, Y.-L.; Bowman, E.D.; Engels, E.A.; Caporaso, N.E.; Harris, C.C. Increased Levels of Circulating Interleukin 6, Interleukin 8, C-Reactive Protein, and Risk of Lung Cancer. J. Natl. Cancer Inst. 2011, 103, 1112–1122. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Reese, R.M.; Harrison, M.M.; Alarid, E.T. Grainyhead-like Protein 2: The Emerging Role in Hormone-Dependent Cancers and Epigenetics. Endocrinology 2019, 160, 1275–1288. [Google Scholar] [CrossRef]
- Bander, Z.A.; Nitert, M.D.; Mousa, A.; Naderpoor, N. The Gut Microbiota and Inflammation: An Overview. Int. J. Environ. Res. Public Health 2020, 17, 7618. [Google Scholar] [CrossRef]
- Ruff, W.E.; Greiling, T.M.; Kriegel, M.A. Host–Microbiota Interactions in Immune-Mediated Diseases. Nat. Rev. Microbiol. 2020, 18, 521–538. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between Microbiota and Immunity in Health and Disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Spor, A.; Koren, O.; Ley, R. Unravelling the Effects of the Environment and Host Genotype on the Gut Microbiome. Nat. Rev. Microbiol. 2011, 9, 279–290. [Google Scholar] [CrossRef]
- Medhasi, S.; Chantratita, N. Human Leukocyte Antigen (HLA) System: Genetics and Association with Bacterial and Viral Infections. J. Immunol. Res. 2022, 2022, 9710376. [Google Scholar] [CrossRef]
- Filler, S.G.; Sheppard, D.C. Fungal Invasion of Normally Non-Phagocytic Host Cells. PLoS Pathog. 2006, 2, 1099–1105. [Google Scholar] [CrossRef]
- Casadevall, A.; Fang, F.C. The Intracellular Pathogen Concept. Mol. Microbiol. 2020, 113, 541–545. [Google Scholar] [CrossRef]
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front. Immunol. 2021, 12, 636568. [Google Scholar] [CrossRef]
- Silva, M.T. Classical Labeling of Bacterial Pathogens According to Their Lifestyle in the Host: Inconsistencies and Alternatives. Front. Microbiol. 2012, 3, 71. [Google Scholar] [CrossRef]
- Manczinger, M.; Boross, G.; Kemény, L.; Müller, V.; Lenz, T.L.; Papp, B.; Pál, C. Pathogen Diversity Drives the Evolution of Generalist MHC-II Alleles in Human Populations. PLoS Biol. 2019, 17, e3000131. [Google Scholar] [CrossRef]
- Wosen, J.E.; Mukhopadhyay, D.; MacAubas, C.; Mellins, E.D. Epithelial MHC Class II Expression and Its Role in Antigen Presentation in the Gastrointestinal and Respiratory Tracts. Front. Immunol. 2018, 9, 02144. [Google Scholar] [CrossRef] [PubMed]
- Heuberger, C.; Pott, J.; Maloy, K.J. Why Do Intestinal Epithelial Cells Express MHC Class II? Immunology 2021, 162, 357–367. [Google Scholar] [CrossRef]
- Corthay, A.; Lorvik, K.B.; Bogen, B. Is Secretion of Tumour-Specific Antigen Important for Cancer Eradication by CD4+ T Cells?—Implications for Cancer Immunotherapy by Adoptive T Cell Transfer. Scand. J. Immunol. 2011, 73, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Das, C.K.; Singh, S.K. Immune Checkpoint Inhibitors in Cancer Therapy: A Ray of Hope. In Biomedical Translational Research from Disease Diagnosis to Treatment; Springer: Singapore, 2022; pp. 393–411. [Google Scholar] [CrossRef]
- Michelakos, T.; Kontos, F.; Kurokawa, T.; Cai, L.; Sadagopan, A.; Krijgsman, D.; Weichert, W.; Durrant, L.G.; Kuppen, P.J.K.; R Ferrone, C.; et al. Differential Role of HLA-A and HLA-B, C Expression Levels as Prognostic Markers in Colon and Rectal Cancer. J. Immunother. Cancer 2022, 10, e004115. [Google Scholar] [CrossRef]
- Anderson, P.; Aptsiauri, N.; Ruiz-Cabello, F.; Garrido, F. HLA Class I Loss in Colorectal Cancer: Implications for Immune Escape and Immunotherapy. Cell. Mol. Immunol. 2021, 18, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Dunne, M.R.; Phelan, J.J.; Michielsen, A.J.; Maguire, A.A.; Dunne, C.; Martin, P.; Noonan, S.; Tosetto, M.; Geraghty, R.; Fennelly, D.; et al. Characterising the Prognostic Potential of HLA-DR during Colorectal Cancer Development. Cancer Immunol. Immunother. 2020, 69, 1577–1588. [Google Scholar] [CrossRef]
- Najafimehr, H.; Hajizadeh, N.; Nazemalhosseini-Mojarad, E.; Pourhoseingholi, M.A.; Abdollahpour-Alitappeh, M.; Ashtari, S.; Zali, M.R. The Role of Human Leukocyte Antigen Class I on Patient Survival in Gastrointestinal Cancers: A Systematic Review and Meta- Analysis. Sci. Rep. 2020, 10, 728. [Google Scholar] [CrossRef]
- Iwasaki, A.; Shinozaki-Ushiku, A.; Kunita, A.; Yamazawa, S.; Sato, Y.; Yamashita, H.; Fukayama, M.; Seto, Y.; Ushiku, T. Human Leukocyte Antigen Class I Deficiency in Gastric Carcinoma: An Adaptive Immune Evasion Strategy Most Common in Microsatellite Instable Tumors. Am. J. Surg. Pathol. 2021, 45, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.R.R.; Kopetz, S.; Patel, S.P.; Welling, T.; Morelli, M.P.; Borad, M.J.; Molina, J.R.; Kirtane, K.; Lin, Y.; Fan-Port, M.; et al. Next Generation Sequencing (NGS) to Identify Relapsed Gastrointestinal (GI) Solid Tumor Patients with Human Leukocyte Antigen (HLA) Loss of Heterozygosity (LOH) for Future Logic-Gated CAR T Therapy to Reduce on Target off Tumor Toxicity. J. Clin. Oncol. 2022, 40, 190. [Google Scholar] [CrossRef]
- Bilici, M.; Okcu, N.; Cayır, K.; Pirim, I.; Tekin, S.B.; Gundogdu, C. Distribution of HLA Tissue Groups in Patients with Gastric Cancer. Eurasian J. Med. 2010, 42, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Mejía-Guarnizo, L.V.; Monroy-Camacho, P.S.; Rincón-Rodríguez, D.E.; Rincón-Riveros, A.; Martinez-Vargas, D.A.; Huertas-Caro, C.A.; Oliveros-Wilches, R.; Sanchez-Pedraza, R.; Nuñez-Lemus, M.; Cristancho-Lievano, C.F.; et al. Soluble HLA-G (SHLA-G) Measurement Might Be Useful as an Early Diagnostic Biomarker and Screening Test for Gastric Cancer. Sci. Rep. 2023, 13, 13119. [Google Scholar] [CrossRef]
- Cuppens, K.; Baas, P.; Geerdens, E.; Cruys, B.; Froyen, G.; Decoster, L.; Thomeer, M.; Maes, B. HLA-I Diversity and Tumor Mutational Burden by Comprehensive next-Generation Sequencing as Predictive Biomarkers for the Treatment of Non-Small Cell Lung Cancer with PD-(L)1 Inhibitors. Lung Cancer 2022, 170, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Abed, A.; Calapre, L.; Lo, J.; Correia, S.; Bowyer, S.; Chopra, A.; Watson, M.; Khattak, M.A.; Millward, M.; Gray, E.S. Prognostic Value of HLA-I Homozygosity in Patients with Non-Small Cell Lung Cancer Treated with Single Agent Immunotherapy. J. Immunother. Cancer 2020, 8, e001620. [Google Scholar] [CrossRef]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e11. [Google Scholar] [CrossRef]
- Hazini, A.; Fisher, K.; Seymour, L. Deregulation of HLA-I in Cancer and Its Central Importance for Immunotherapy. J. Immunother. Cancer 2021, 9, e002899. [Google Scholar] [CrossRef]
- Maróstica, A.S.; Nunes, K.; Castelli, E.C.; Silva, N.S.B.; Weir, B.S.; Goudet, J.; Meyer, D. How HLA Diversity Is Apportioned: Influence of Selection and Relevance to Transplantation. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2022, 377, 20200420. [Google Scholar] [CrossRef]
- Davies, C.S.; Worsley, S.F.; Maher, K.H.; Komdeur, J.; Burke, T.; Dugdale, H.L.; Richardson, D.S. Immunogenetic Variation Shapes the Gut Microbiome in a Natural Vertebrate Population. Microbiome 2022, 10, 41. [Google Scholar] [CrossRef]
- Mayer, R.L.; Verbeke, R.; Asselman, C.; Aernout, I.; Gul, A.; Eggermont, D.; Boucher, K.; Thery, F.; Maia, T.M.; Demol, H.; et al. Immunopeptidomics-Based Design of MRNA Vaccine Formulations against Listeria monocytogenes. Nat. Commun. 2022, 13, 6075. [Google Scholar] [CrossRef]
- Pedraza, L.; Camargo, M.; Moreno-Pérez, D.A.; Sánchez, R.; Del Río-Ospina, L.; Báez-Murcia, I.M.; Patarroyo, M.E.; Patarroyo, M.A. Identifying HLA DRB1-DQB1 Alleles Associated with Chlamydia trachomatis Infection and in Silico Prediction of Potentially-Related Peptides. Sci. Rep. 2021, 11, 12837. [Google Scholar] [CrossRef]
- Scholzen, A.; Richard, G.; Moise, L.; Hartman, E.; Bleeker-Rovers, C.P.; Reeves, P.M.; Paul, S.R.; Martin, W.D.; De Groot, A.S.; Poznansky, M.C.; et al. Coxiella Burnetii Epitope-Specific T-Cell Responses in Patients with Chronic Q Fever. Infect. Immun. 2019, 87, e00213-19. [Google Scholar] [CrossRef]
- Kim, S.J.; Karamooz, E. MR1- and HLA-E-Dependent Antigen Presentation of Mycobacterium Tuberculosis. Int. J. Mol. Sci. 2022, 23, 14412. [Google Scholar] [CrossRef] [PubMed]
- Odera, S.; Mureithi, M.; Aballa, A.; Onyango, N.; Kazungu, S.; Ogolla, S.; Kaiyare, G.; Anzala, O.; Oyugi, J. Association between Human Leukocyte Antigen Class II (HLA-DRB and-DQB) Alleles and Outcome of Exposure to Mycobacterium Tuberculosis: A Cross-Sectional Study in Nairobi, Kenya. Pan Afr. Med. J. 2022, 41, 149. [Google Scholar] [CrossRef] [PubMed]
- Salerno-Gonçalves, R.; Fernandez-Viña, M.; Lewinsohn, D.M.; Sztein, M.B. Identification of a Human HLA-E-Restricted CD8+ T Cell Subset in Volunteers Immunized with Salmonella Enterica Serovar Typhi Strain Ty21a Typhoid Vaccine. J. Immunol. 2004, 173, 5852–5862. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.N.; Lenart, I.; Kriston-Vizi, J.; Iwawaki, T.; Turmaine, M.; McHugh, K.; Ali, S.; Blake, N.; Bowness, P.; Bajaj-Elliott, M.; et al. Salmonella Exploits HLA-B27 and Host Unfolded Protein Responses to Promote Intracellular Replication. Ann. Rheum. Dis. 2019, 78, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Giarola, L.; Dos Santos, R.; Bedendo, J.; Da Silva Júnior, W.; Borelli, S. HLA Molecules and Nasal Carriage of Staphylococcus Aureus Isolated from Dialysis and Kidney Transplant Patients at a Hospital in Southern Brazil. BMC Res. Notes 2012, 5, 90. [Google Scholar] [CrossRef]
- DeLorenze, G.N.; Nelson, C.L.; Scott, W.K.; Allen, A.S.; Ray, G.T.; Tsai, A.-L.; Quesenberry, C.P., Jr.; Fowler, V.G., Jr. Polymorphisms in HLA Class II Genes Are Associated With Susceptibility to Staphylococcus Aureus Infection in a White Population. J. Infect. Dis. 2016, 213, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Gogos, A.; Federle, M.J. Modeling Streptococcus Pyogenespharyngeal Colonization in the Mouse. Front. Cell. Infect. Microbiol. 2019, 9, 137. [Google Scholar] [CrossRef]
- Kasper, K.J.; Zeppa, J.J.; Wakabayashi, A.T.; Xu, S.X.; Mazzuca, D.M.; Welch, I.; Baroja, M.L.; Kotb, M.; Cairns, E.; Cleary, P.P.; et al. Bacterial Superantigens Promote Acute Nasopharyngeal Infection by Streptococcus Pyogenes in a Human MHC Class II-Dependent Manner. PLoS Pathog. 2014, 10, e1004155. [Google Scholar] [CrossRef]
- Meiring, H.D.; Kuipers, B.A.J.; van Gaans-van den Brink, J.A.M.; Poelen, M.C.M.; de Jong, A.P.J.M.; Heck, A.J.R.; van Els, C.A.C.M. Non-Linear in Cis-Spliced Bacterial MHC Class II Epitopes Are Formed in Human Monocyte-Derived Dendritic Cells and Are Recognized by Human CD4+ T Cells. Mol. Immunol. 2022, 150, 24. [Google Scholar] [CrossRef]
- Littmann, M.; Albiger, B.; Frentzen, A.; Normark, S.; Henriques-Normark, B.; Plant, L. Streptococcus Pneumoniae Evades Human Dendritic Cell Surveillance by Pneumolysin Expression. EMBO Mol. Med. 2009, 1, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Weight, C.M.; Venturini, C.; Pojar, S.; Jochems, S.P.; Reiné, J.; Nikolaou, E.; Solórzano, C.; Noursadeghi, M.; Brown, J.S.; Ferreira, D.M.; et al. Microinvasion by Streptococcus Pneumoniae Induces Epithelial Innate Immunity during Colonisation at the Human Mucosal Surface. Nat. Commun. 2019, 10, 3060. [Google Scholar] [CrossRef]
- Mashraqi, M.M.; Alzamami, A.; Alturki, N.A.; Alshamrani, S.; Alshahrani, M.M.; Almasoudi, H.H.; Basharat, Z. Molecular Mimicry Mapping in Streptococcus Pneumoniae: Cues for Autoimmune Disorders and Implications for Immune Defense Activation. Pathogens 2023, 12, 857. [Google Scholar] [CrossRef]
- Armstrong, D.A.; Lee, M.K.; Hazlett, H.F.; Dessaint, J.A.; Mellinger, D.L.; Aridgides, D.S.; Hendricks, G.M.; Abdalla, M.A.K.; Christensen, B.C.; Ashare, A. Extracellular Vesicles from Pseudomonas Aeruginosa Suppress MHC-Related Molecules in Human Lung Macrophages. ImmunoHorizons 2020, 4, 508–519. [Google Scholar] [CrossRef]
- Kulkarni, H.S.; Tsui, K.; Sunder, S.; Ganninger, A.; Tague, L.K.; Witt, C.A.; Byers, D.E.; Trulock, E.P.; Nava, R.; Puri, V.; et al. Pseudomonas Aeruginosa and Acute Rejection Independently Increase the Risk of Donor-Specific Antibodies after Lung Transplantation. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2020, 20, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Kaya, E.; Grassi, L.; Benedetti, A.; Maisetta, G.; Pileggi, C.; Di Luca, M.; Batoni, G.; Esin, S. In Vitro Interaction of Pseudomonas Aeruginosa Biofilms With Human Peripheral Blood Mononuclear Cells. Front. Cell. Infect. Microbiol. 2020, 10, 187. [Google Scholar] [CrossRef] [PubMed]
- Nelson, B.N.; Daugherty, C.S.; Sharp, R.R.; Booth, J.L.; Patel, V.I.; Metcalf, J.P.; Jones, K.L.; Wozniak, K.L. Protective Interaction of Human Phagocytic APC Subsets with Cryptococcus Neoformans Induces Genes Associated with Metabolism and Antigen Presentation. Front. Immunol. 2022, 13, 1054477. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, D.; Zhang, C.; Li, Y.; Cao, B.; Ning, T.; Zhao, Y.; You, W.; Ke, Y. HLA Polymorphisms Are Associated with Helicobacter Pylori Infected Gastric Cancer in a High Risk Population, China. Immunogenetics 2005, 56, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Saribas, S.; Demiryas, S.; Yilmaz, E.; Uysal, O.; Kepil, N.; Demirci, M.; Caliskan, R.; Dinc, H.O.; Akkus, S.; Gareayaghi, N.; et al. Association between Human Leukocyte Antigen Gene Polymorphisms and Multiple EPIYA-C Repeats in Gastrointestinal Disorders. World J. Gastroenterol. 2020, 26, 4817–4832. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.; Le Bihan, C.; Portales, P.; Bourgeois, N.; Vincent, T.; Lachaud, L.; Chanques, G.; Conseil, M.; Corne, P.; Massanet, P.; et al. Monocyte Human Leukocyte Antigen-DR but Not β-d-Glucan May Help Early Diagnosing Invasive Candida Infection in Critically Ill Patients. Ann. Intensive Care 2021, 11, 129. [Google Scholar] [CrossRef]
- Razavi, T.; Falahati, M.; Teimourian, S.; Farahyar, S.; Babaei, V.; Majdabadi, N. HLA-DRB1 Alleles as Predisposing and Resisting Factor in Women Suffering from Vulvovaginal Candidiasis. J. Med. Mycol. 2021, 31, 101200. [Google Scholar] [CrossRef]
- Permata, A.T.; Prasetyo, A.A.; Dharmawan, R. Molecular and Immunological Characteristics of Candida Secreted Aspartyl Proteinase 5. J. Phys. Conf. Ser. 2019, 1153, 012141. [Google Scholar] [CrossRef]
- Liu, J.; Wei, H.; Liu, J.; Peng, L.; Li, G.; Li, M.; Yang, L.; Jiang, Y.; Peng, F. Analysis of the Association of HLA Subtypes with Cryptococcal Meningitis in HIV-Negative Immunocompetent Patients. Future Microbiol. 2022, 17, 1231–1240. [Google Scholar] [CrossRef]
- Sun, Z.; Zhu, P.; Li, L.; Wan, Z.; Zhao, Z.; Li, R. Adoptive Immunity Mediated by HLA-A*0201 Restricted Asp F16 Peptides-Specific CD8+ T Cells against Aspergillus fumigatus Infection. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 3089–3096. [Google Scholar] [CrossRef]
- Schwarz, C.; Eschenhagen, P.; Schmidt, H.; Hohnstein, T.; Iwert, C.; Grehn, C.; Roehmel, J.; Steinke, E.; Stahl, M.; Lozza, L.; et al. Antigen Specificity and Cross-Reactivity Drive Functionally Diverse Anti–Aspergillus fumigatus T Cell Responses in Cystic Fibrosis. J. Clin. Investig. 2023, 133, 1–17. [Google Scholar] [CrossRef]
- Fortes, M.R.P.; Miot, H.A.; Kurokawa, C.S.; Marques, M.E.A.; Marques, S.A. Immunology of Paracoccidioidomycosis. An. Bras. Dermatol. 2011, 86, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Kischkel, B.; Boniche-Alfaro, C.; de Menezes, I.G.; Rossi, S.A.; Angeli, C.B.; de Almeida, S.R.; Palmisano, G.; Lopes-Bezerra, L.; Nosanchuk, J.D.; Taborda, C.P. Immunoproteomic and Immunopeptidomic Analyses of Histoplasma Capsulatum Reveal Promiscuous and Conserved Epitopes Among Fungi With Vaccine Potential. Front. Immunol. 2021, 12, 764501. [Google Scholar] [CrossRef]
- Rubio-Carrasquilla, M.; Ochoa, R.; Santa, C.; Guimarães, A.J.; Cano, L.E.; Moreno, E. Identifying Molecularly Defined Antigens for a Histoplasma Capsulatum-Specific Interferon Gamma Release Assay. Rev. Iberoam. Micol. 2019, 36, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Wiersinga, W.J.; van der Poll, T. Immunopathophysiology of Human Sepsis. eBioMedicine 2022, 86, 104363. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, M. Pattern Recognition Receptors in Health and Diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Gack, M.U. RIG-I-like Receptors: Their Regulation and Roles in RNA Sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Fan, X.; Jin, T. Structures of RIG-I-Like Receptors and Insights into Viral RNA Sensing. Adv. Exp. Med. Biol. 2019, 1172, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tian, Y.; Yin, Q. AIM2 Inflammasome Assembly and Signaling. Adv. Exp. Med. Biol. 2019, 1172, 143–155. [Google Scholar] [CrossRef]
- Willment, J.A. Fc-Conjugated C-Type Lectin Receptors: Tools for Understanding Host–Pathogen Interactions. Mol. Microbiol. 2022, 117, 632–660. [Google Scholar] [CrossRef]
- Mnich, M.E.; van Dalen, R.; van Sorge, N.M. C-Type Lectin Receptors in Host Defense Against Bacterial Pathogens. Front. Cell. Infect. Microbiol. 2020, 10, 309. [Google Scholar] [CrossRef]
- Fischer, S.; Stegmann, F.; Gnanapragassam, V.S.; Lepenies, B. From Structure to Function—Ligand Recognition by Myeloid C-Type Lectin Receptors. Comput. Struct. Biotechnol. J. 2022, 20, 5790–5812. [Google Scholar] [CrossRef]
- Cao, M.; Wu, Z.; Lou, Q.; Lu, W.; Zhang, J.; Li, Q.; Zhang, Y.; Yao, Y.; Zhao, Q.; Li, M.; et al. Dectin-1-Induced RIPK1 and RIPK3 Activation Protects Host against Candida Albicans Infection. Cell Death Differ. 2019, 26, 2622–2636. [Google Scholar] [CrossRef]
- Ambati, S.; Ferarro, A.R.; Kang, S.E.; Lin, J.; Lin, X.; Momany, M.; Lewis, Z.A.; Meagher, R.B. Dectin-1-Targeted Antifungal Liposomes Exhibit Enhanced Efficacy. mSphere 2019, 4, e00025-19. [Google Scholar] [CrossRef]
- Ambati, S.; Ellis, E.C.; Lin, J.; Lin, X.; Lewis, Z.A.; Meagher, R.B. Dectin-2-Targeted Antifungal Liposomes Exhibit Enhanced Efficacy. mSphere 2019, 4, e00715-19. [Google Scholar] [CrossRef] [PubMed]
- Geijtenbeek, T.B.H.; Gringhuis, S.I. Signalling through C-Type Lectin Receptors: Shaping Immune Responses. Nat. Rev. Immunol. 2009, 9, 465–479. [Google Scholar] [CrossRef]
- Gringhuis, S.I.; den Dunnen, J.; Litjens, M.; van het Hof, B.; van Kooyk, Y.; Geijtenbeek, T.B.B.H. C-Type Lectin DC-SIGN Modulates Toll-like Receptor Signaling via Raf-1 Kinase-Dependent Acetylation of Transcription Factor NF-ΚB. Immunity 2007, 26, 605–616. [Google Scholar] [CrossRef]
- Ganguly, K.; Kishore, U.; Madan, T. Role of C-Type Lectins in the Tumor Microenvironment BT—Handbook of Cancer and Immunology. In Handbook of Cancer and Immunology; Rezaei, N., Ed.; Springer International Publishing: Cham, Switzerland, 2023; pp. 1–23. ISBN 978-3-030-80962-1. [Google Scholar]
- Velloso, F.J.; Trombetta-Lima, M.; Anschau, V.; Sogayar, M.C.; Correa, R.G. NOD-like Receptors: Major Players (and Targets) in the Interface between Innate Immunity and Cancer. Biosci. Rep. 2019, 29, BSR20181709. [Google Scholar] [CrossRef]
- Mukherjee, T.; Hovingh, E.S.; Foerster, E.G.; Abdel-Nour, M.; Philpott, D.J.; Girardin, S.E. NOD1 and NOD2 in Inflammation, Immunity and Disease. Arch. Biochem. Biophys. 2019, 670, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Godkowicz, M.; Druszczyńska, M. NOD1, NOD2, and NLRC5 Receptors in Antiviral and Antimycobacterial Immunity. Vaccines 2022, 10, 1487. [Google Scholar] [CrossRef] [PubMed]
- Gocher, A.M.; Workman, C.J.; Vignali, D.A.A. Interferon-γ: Teammate or Opponent in the Tumour Microenvironment? Nat. Rev. Immunol. 2022, 22, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Delneste, Y.; Beauvillain, C.; Jeannin, P. Immunité Naturelle. Médecine/Sciences 2007, 23, 67–74. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef]
- Qu, F.; She, Q.; Li, J.; Zeng, X.; Li, Y.; Liu, X.; Ren, L.; Liu, Z.; Gao, C.; Lu, X.; et al. Molecular Characterization of MyD88 in Anodonta Woodiana and Its Involvement in the Innate Immune Response to Bacterial Infection. Front. Immunol. 2022, 13, 925168. [Google Scholar] [CrossRef]
- Lee, S.M.-Y.; Yip, T.-F.; Yan, S.; Jin, D.-Y.; Wei, H.-L.; Guo, R.-T.; Peiris, J.S.M. Recognition of Double-Stranded RNA and Regulation of Interferon Pathway by Toll-Like Receptor 10. Front. Immunol. 2018, 9, 516. [Google Scholar] [CrossRef]
- Sameer, A.S.; Nissar, S. Toll-Like Receptors (TLRs): Structure, Functions, Signaling, and Role of Their Polymorphisms in Colorectal Cancer Susceptibility. Biomed Res. Int. 2021, 2021, 1157023. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Wang, X.; Zhang, D. TLRs as a Promise Target Along With Immune Checkpoint Against Gastric Cancer. Front. Cell Dev. Biol. 2021, 8, 611444. [Google Scholar] [CrossRef] [PubMed]
- Hoden, B.; DeRubeis, D.; Martinez-Moczygemba, M.; Ramos, K.S.; Zhang, D. Understanding the Role of Toll-like Receptors in Lung Cancer Immunity and Immunotherapy. Front. Immunol. 2022, 13, 1033483. [Google Scholar] [CrossRef] [PubMed]
- Urban-Wojciuk, Z.; Khan, M.M.; Oyler, B.L.; Fåhraeus, R.; Marek-Trzonkowska, N.; Nita-Lazar, A.; Hupp, T.R.; Goodlett, D.R. The Role of Tlrs in Anti-Cancer Immunity and Tumor Rejection. Front. Immunol. 2019, 10, 02388. [Google Scholar] [CrossRef]
- Marietta, E.; Rishi, A.; Taneja, V. Immunogenetic Control of the Intestinal Microbiota. Immunology 2015, 145, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Nichols, R.G.; Davenport, E.R. The Relationship between the Gut Microbiome and Host Gene Expression: A Review. Hum. Genet. 2021, 140, 747–760. [Google Scholar] [CrossRef]
- Maudet, C.; Mano, M.; Eulalio, A. MicroRNAs in the Interaction between Host and Bacterial Pathogens. FEBS Lett. 2014, 588, 4140–4147. [Google Scholar] [CrossRef]
- Shock, T.; Badang, L.; Ferguson, B.; Martinez-Guryn, K. The Interplay between Diet, Gut Microbes, and Host Epigenetics in Health and Disease. J. Nutr. Biochem. 2021, 95, 108631. [Google Scholar] [CrossRef]
- Krautkramer, K.A.; Kreznar, J.H.; Romano, K.A.; Vivas, E.I.; Barrett-Wilt, G.A.; Rabaglia, M.E.; Keller, M.P.; Attie, A.D.; Rey, F.E.; Denu, J.M. Diet-Microbiota Interactions Mediate Global Epigenetic Programming in Multiple Host Tissues. Mol. Cell 2016, 64, 982–992. [Google Scholar] [CrossRef]
- Richards, A.L.; Muehlbauer, A.L.; Alazizi, A.; Burns, M.B.; Findley, A.; Messina, F.; Gould, T.J.; Cascardo, C.; Pique-Regi, R.; Blekhman, R.; et al. Gut Microbiota Has a Widespread and Modifiable Effect on Host Gene Regulation. mSystems 2019, 4, e00323-18. [Google Scholar] [CrossRef] [PubMed]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The Crucial Role of Protein Phosphorylation in Cell Signaling and Its Use as Targeted Therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef]
- Ibarra, I.L.; Hollmann, N.M.; Klaus, B.; Augsten, S.; Velten, B.; Hennig, J.; Zaugg, J.B. Mechanistic Insights into Transcription Factor Cooperativity and Its Impact on Protein-Phenotype Interactions. Nat. Commun. 2020, 11, 124. [Google Scholar] [CrossRef] [PubMed]
- Simon, I.; Barnett, J.; Hannett, N.; Harbison, C.T.; Rinaldi, N.J.; Volkert, T.L.; Wyrick, J.J.; Zeitlinger, J.; Gifford, D.K.; Jaakkola, T.S.; et al. Serial Regulation of Transcriptional Regulators in the Yeast Cell Cycle. Cell 2001, 106, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Hochedlinger, K.; Plath, K. Epigenetic Reprogramming and Induced Pluripotency. Development 2009, 136, 509–523. [Google Scholar] [CrossRef] [PubMed]
- van den Elsen, P.J. Expression Regulation of Major Histocompatibility Complex Class I and Class II Encoding Genes. Front. Immunol. 2011, 2, 48. [Google Scholar] [CrossRef]
- Lambert, M.; Jambon, S.; Depauw, S.; David-Cordonnier, M.H. Targeting Transcription Factors for Cancer Treatment. Molecules 2018, 23, 1479. [Google Scholar] [CrossRef]




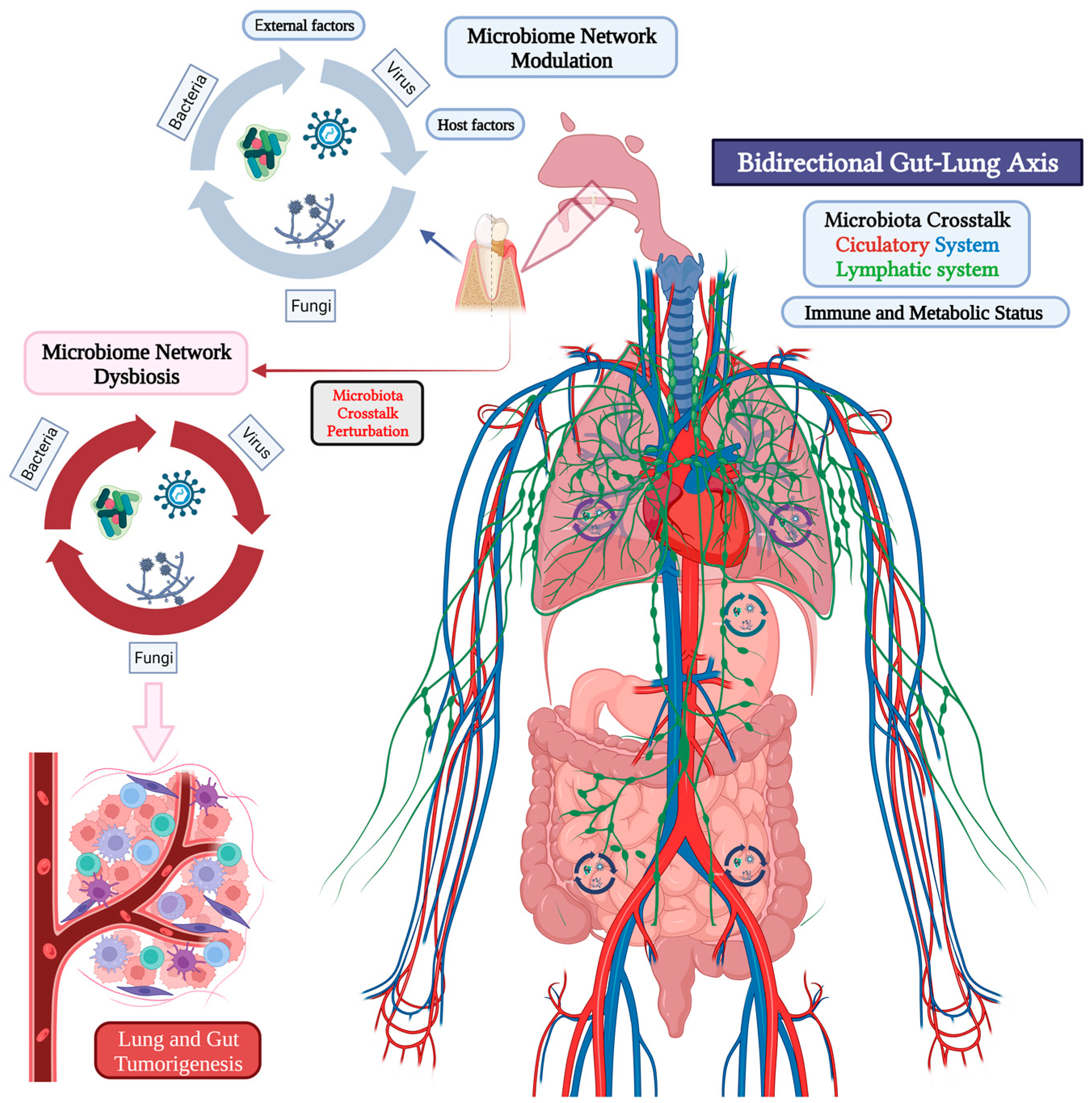{kind=link}
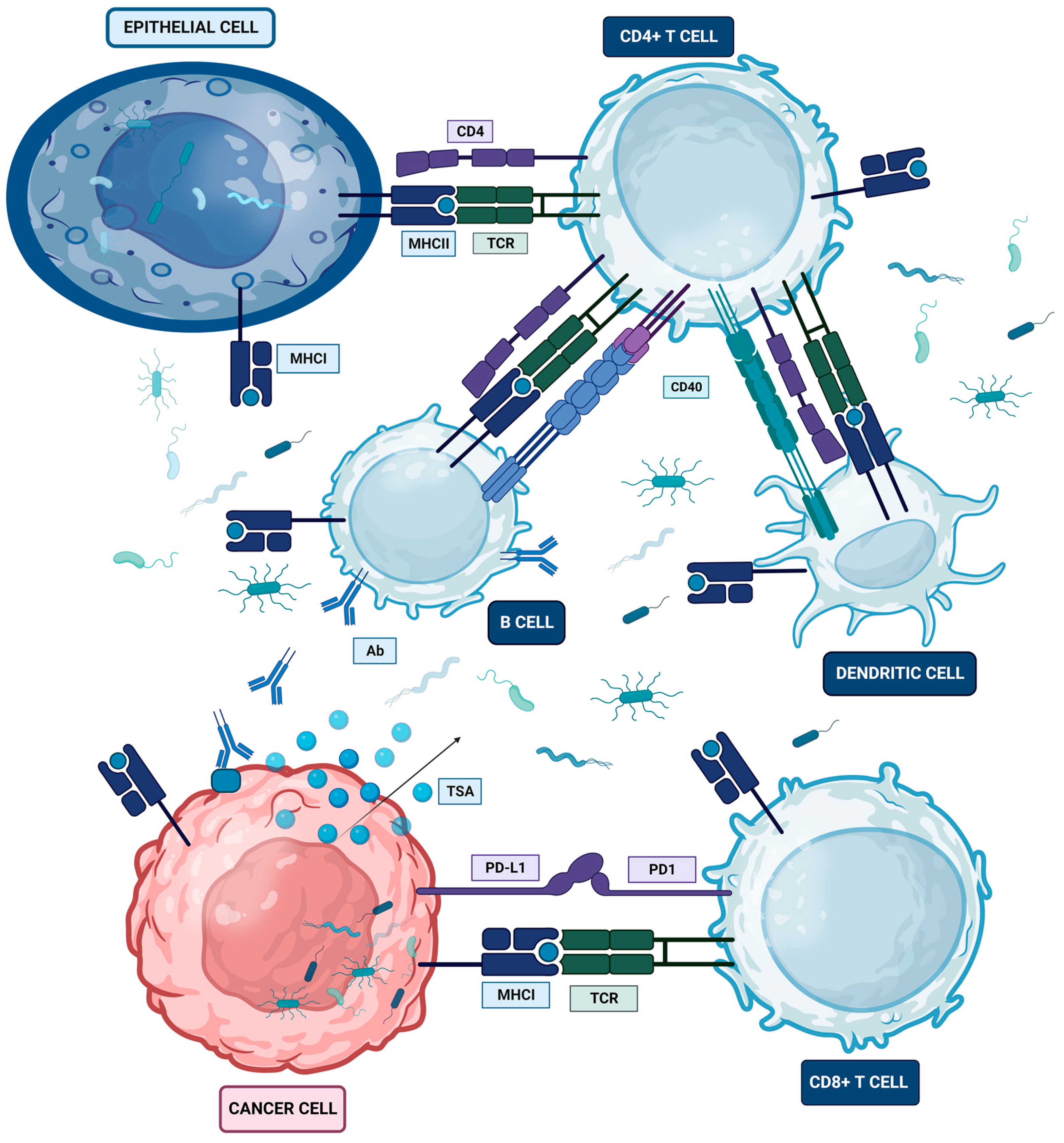{kind=link}
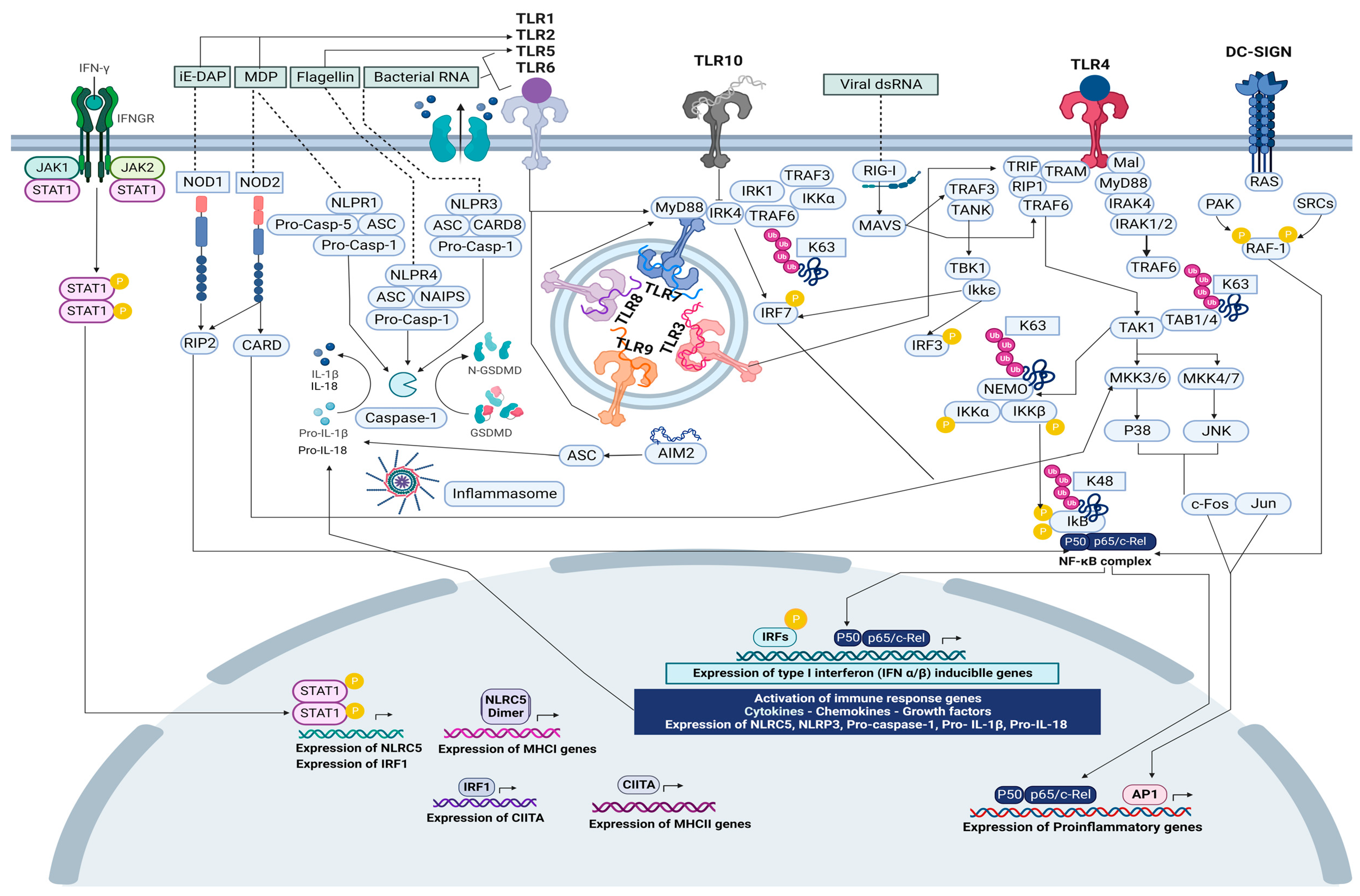{kind=link}
| Microorganism | Immune Receptor (IR) | References |
|---|---|---|
| Listeria monocytogenes | HLA-C_HLA-DR | [213] |
| Chlamydia trachomatis | HLA-A2_HLA-DRB1_HLA-DQB1 | [214] |
| Coxiella burnetii | HLA-A_HLA-B_HLA-DRB1 | [215] |
| Mycobacterium tuberculosis | HLA-E_HLA-DRB5*2_HLA-DRB1*14 | [216,217] |
| Salmonella enterica | HLA-E_HLA-B27 | [218,219] |
| Staphylococcus aureus | HLA-A_HLA-B_HLA-DRB1_HLA-DRA | [220,221] |
| Streptococcus pyogenes | HLA-B6_HLA-DR4_HLA-DQA1_HLA-DQ8 | [222,223] |
| Neisseria meningitidis | HLA-DRB | [224] |
| Streptococcus pneumoniae | HLA-A_HLA-B_HLA-DR_HLA-DRB4 | [225,226,227] |
| Pseudomonas aeruginosa | HLA-DRA_HLA-DRB3_HLA-DQ | [228,229,230] |
| Cryptococcus neoformans | HLA-A_HLA-B_HLA-C_HLA-DQB5 | [231] |
| Helicobacter pylori | HLA-CW*03_HLA-DRB1*01_HLA-DQA1_HLA-DQB1_HLA-A*2 | [232,233] |
| Candida albicans | HLA-DR_HLA-DRB1_HLA-DPA1 | [234,235,236] |
| Cryptococcus neoformans | HLA-A_HLA-DRA_HLA-DRB5_HLA-DQB1 | [231,237] |
| Aspergillus fumigatus | HLA-A2 | [238,239] |
| Paracoccidioides brasiliensis | HLA-DR | [240] |
| Histoplasma capsulatum | HLA-A_HLA-B_HLA-DR_HLA-DQ_HLA-DP | [241,242] |
| Immune Receptor (IR) | Microorganism–Ligand | Localization |
|---|---|---|
| TLR1 | Bacteria–Triacyl lipopeptide, lipoteichoic acid, and peptidoglycans Gram(+) bacteria–Lipopolysaccharide Fungi–Zymosan | Lung carcinoma Metastatic colorectal cancer Gastric cancer |
| TLR2 | Bacteria–Triacyl lipopeptide, lipoteichoic acid, peptidoglycans, and diacylated lipopeptides Viruses surface GP Gram(+) bacteria–LP and PG Gram(−) bacteria–Porin and PG Fungi–Zymosan, β-glycan, and mannan Protozoa GPI anchors | Colorectal cancer Gastric cancer Lewis lung carcinoma |
| TLR3 | dsDNA Viruses–dsRNA | Colorectal cancer Gastric cancer Lung tumoral exosomes NSCLC |
| TLR4 | Viruses surface GP Gram(−) bacteria–Lipopolysaccharide Fungi–Mannan Protozoa–GPI anchors | Colorectal cancer Gastric cancer Lung cancer NSCLC |
| TLR5 | Gram(−) bacteria–Flagellin | Colorectal cancer Gastric cancer NSCLC |
| TLR6 | Gram(+) bacteria–Lipopolysaccharide | Colorectal cancer Lung cancer |
| TLR7 | Viruses–ssRNA | Colorectal cancer Gastric cancer NSCLC |
| TLR8 | Viruses–ssRNA | Primary lung tumors |
| TLR9 | Viruses Gram(+)/(−) bacteria Bacterial unmethylated CpG DNA Fungi–dsDNA Protozoa–dsDNA | Colorectal cancer Gastric cancer Lung carcinoma Lung cancer PBMCs |
| TLR10 | dsDNA | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Otálora-Otálora, B.A.; López-Rivera, J.J.; Aristizábal-Guzmán, C.; Isaza-Ruget, M.A.; Álvarez-Moreno, C.A. Host Transcriptional Regulatory Genes and Microbiome Networks Crosstalk through Immune Receptors Establishing Normal and Tumor Multiomics Metafirm of the Oral-Gut-Lung Axis. Int. J. Mol. Sci. 2023, 24, 16638. https://doi.org/10.3390/ijms242316638
Otálora-Otálora BA, López-Rivera JJ, Aristizábal-Guzmán C, Isaza-Ruget MA, Álvarez-Moreno CA. Host Transcriptional Regulatory Genes and Microbiome Networks Crosstalk through Immune Receptors Establishing Normal and Tumor Multiomics Metafirm of the Oral-Gut-Lung Axis. International Journal of Molecular Sciences. 2023; 24(23):16638. https://doi.org/10.3390/ijms242316638
Chicago/Turabian StyleOtálora-Otálora, Beatriz Andrea, Juan Javier López-Rivera, Claudia Aristizábal-Guzmán, Mario Arturo Isaza-Ruget, and Carlos Arturo Álvarez-Moreno. 2023. "Host Transcriptional Regulatory Genes and Microbiome Networks Crosstalk through Immune Receptors Establishing Normal and Tumor Multiomics Metafirm of the Oral-Gut-Lung Axis" International Journal of Molecular Sciences 24, no. 23: 16638. https://doi.org/10.3390/ijms242316638
APA StyleOtálora-Otálora, B. A., López-Rivera, J. J., Aristizábal-Guzmán, C., Isaza-Ruget, M. A., & Álvarez-Moreno, C. A. (2023). Host Transcriptional Regulatory Genes and Microbiome Networks Crosstalk through Immune Receptors Establishing Normal and Tumor Multiomics Metafirm of the Oral-Gut-Lung Axis. International Journal of Molecular Sciences, 24(23), 16638. https://doi.org/10.3390/ijms242316638