
Calcium’s Role and Signaling in Aging Muscle, Cellular Senescence, and Mineral Interactions

Abstract
:1. Introduction
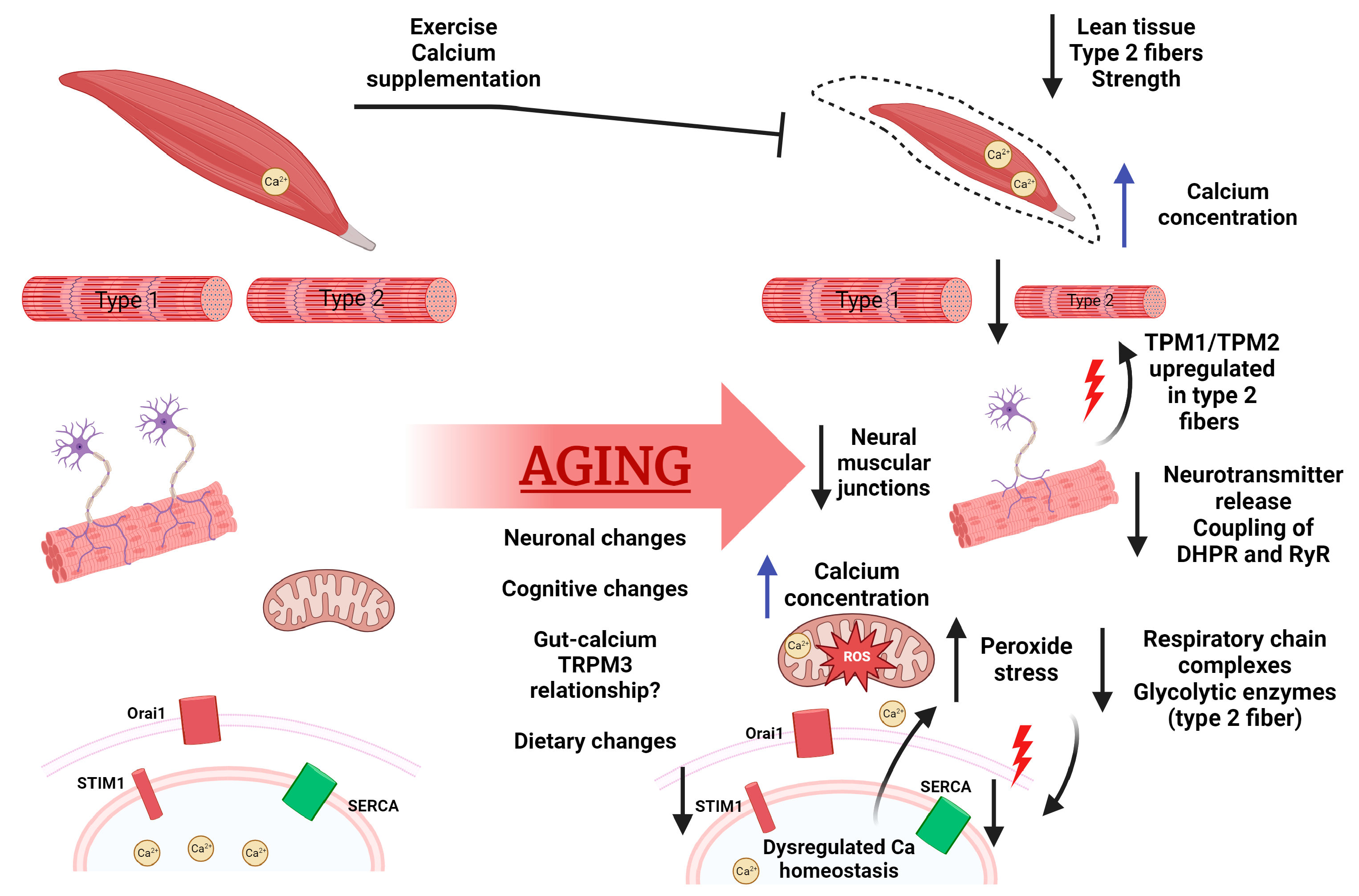
2. Calcium Signaling and Aging Muscle
2.1. Role of Calcium in Muscle
2.2. Aging’s Effect on Muscle
2.3. Aging’s Effect on Calcium
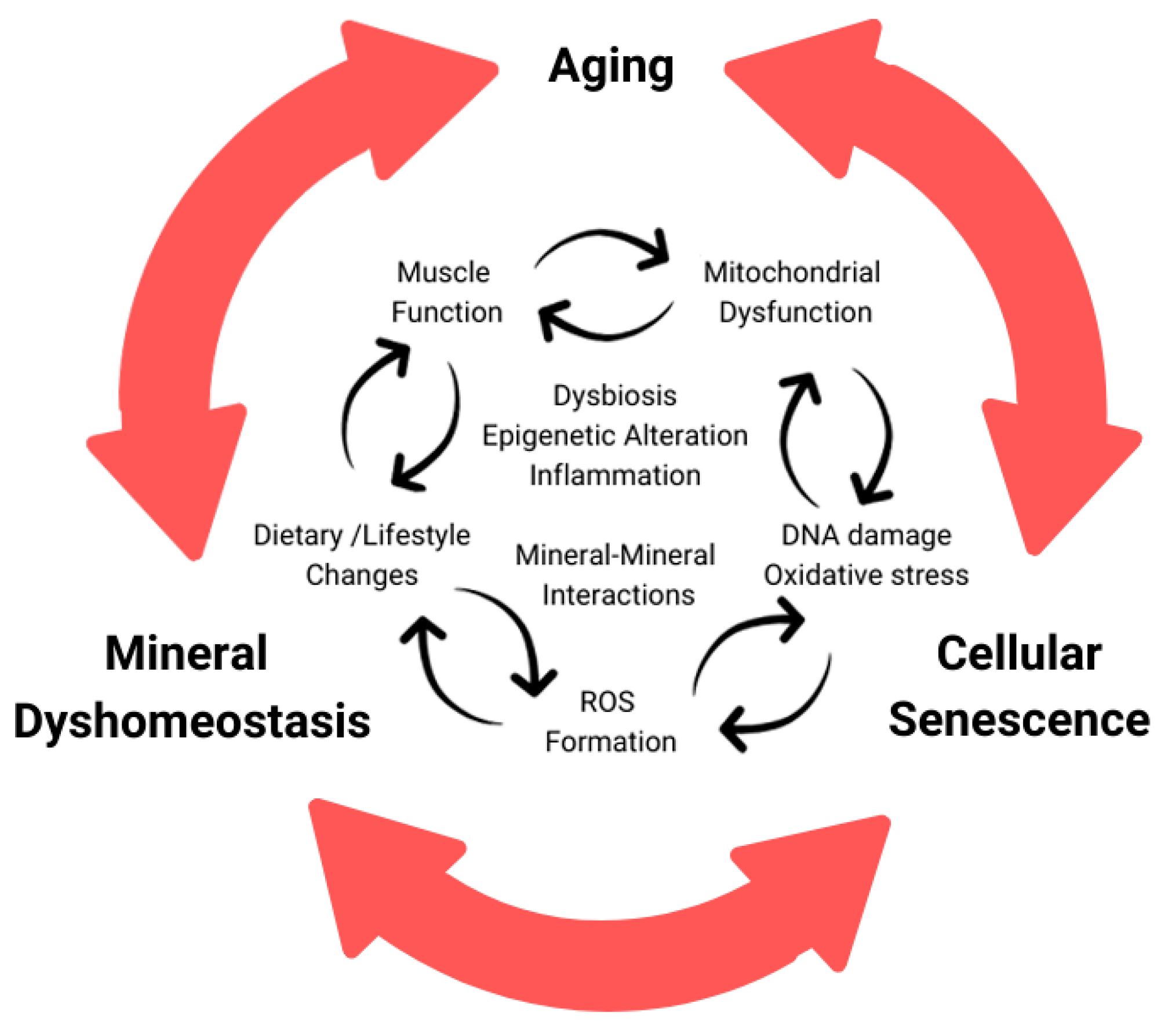
3. Cellular Senescence
3.1. Aging Muscle and Senescence
3.2. Mitochondria Dysfunction and Senescence
4. Crosstalk between Minerals
4.1. Zinc
4.1.1. Interaction with Calcium Channels and Calcium-Binding/Dependent Proteins
4.1.2. Zinc Transporters and Calcium
4.2. Iron
4.2.1. Iron’s Effect on Calcium Signaling and Transport
4.2.2. Iron and Calcium ROS Generation and Ferroptosis
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviation | Full name |
| ATAD3A | ATPase family AAA domain containing 3A |
| CaM | calmodulin |
| CaMK | calmodulin kinase |
| CaMKK | calmodulin kinase kinase |
| CDKN | cyclin-dependent kinase inhibitor |
| CRD | cysteine-rich domain isoforms of the neuregulin-1 |
| DHPR | dihydropyridine receptor |
| DMT | divalent metal transporter |
| ERK | extracellular signal-regulated kinase |
| FAP | fibro/adipogenic progenitor |
| FGF | fibroblast growth factor |
| GCV | ganciclovir |
| GLUT | glucose transporter type 4 |
| HMGBI | high-mobility group box 1 |
| HRC | histidine-rich Ca2+-binding protein |
| IGF1 | insulin-growth like factor 1 |
| IL-6 | interleukin-6 |
| IP3 | inositol-1,4,5-triphosphate |
| IP3R | inositol-1,4,5-triphosphate receptor |
| IR | insulin receptor |
| IRS | insulin receptor substrate |
| ITPRKO | inositol 1,4,5-trisphosphate receptorknock out |
| KO | knock out |
| LETM1 | leucine zipper-EF-hand-containing transmembrane protein 1 |
| LTCC | L-type calcium channel |
| MAPK | mitogen-activated protein kinase |
| MCAT | mitochondrial targeted catalase |
| MCU | mitochondrial calcium uniporter |
| MFI | multidimensional fatigue inventory |
| MG | mitsugumin 29 |
| mPTP | mitochondrial permeability transition pore |
| mTOR | mammalian target of rapamycin |
| NCS | neuronal calcium sensor |
| NF-κB | nuclear factor kappa B |
| NFAT | nuclear factor of activated T cells |
| NOX1 | NADPH oxidase |
| Nrf2 | nuclear factor-erythroid factor 2-related factor 2 |
| PI3K | phosphoinositide 3-kinase |
| PIP3 | phosphatidylinositol-3,4,5-triphosphate |
| PKC | protein kinase C |
| PLC | phospholipase C |
| PRDX3 | Peroxiredoxin 3 |
| ROS | reactive oxygen species |
| RyR | ryanodine receptor |
| SA-β-gal | senescence-associated beta-galactosidase |
| SASP | senescence-associated secretory phenotype |
| SERCA | sarcoplasmic/endoplasmic reticulum Ca2+-ATPase |
| SkM | skeletal muscle |
| SLC | solute carrier |
| SOCE | store-operated calcium entry |
| SOD | superoxide dismutase |
| SR | sarcoplasmic reticulum |
| STIM1 | stromal Interaction Molecule 1 |
| TAF | telomere-associated foci |
| TfR | transferrin receptor |
| TNF-a | tumor necrosis factor-alpha |
| TNiC | C-terminal one third of troponin I |
| TRPC | transient receptor potential canonical |
| TRPM | transient receptor potential melastin |
| TTCC | T-type calcium channels |
| VGCC | voltage-gated calcium channel |
| WT | wild type |
| ZIP | Zrt-/Irt-like protein |
| ZnT | zinc transporter |
| γ-H2AX | gamma-H2A histone family member X |
References
- Arango-Lopera, V.E.; Arroyo, P.; Gutierrez-Robledo, L.M.; Perez-Zepeda, M.U.; Cesari, M. Mortality as An Adverse Outcome of Sarcopenia. J. Nutr. Health Aging 2013, 17, 259–262. [Google Scholar] [CrossRef]
- Moiseeva, V.; Cisneros, A.; Sica, V.; Deryagin, O.; Lai, Y.; Jung, S.; Ortet, L.; Lukesova, V.; Volpe, G. Senescence atlas reveals an aged-like inflamed niche that blunts muscle regeneration. Nature 2023, 613, 169. [Google Scholar] [CrossRef] [PubMed]
- Zembron-Lacny, A.; Dziubek, W.; Wolny-Rokicka, E.; Dabrowska, G.; Wozniewski, M. The Relation of Inflammaging with Skeletal Muscle Properties in Elderly Men. Am. J. Mens. Health 2019, 13, 1557988319841934. [Google Scholar] [CrossRef] [PubMed]
- Rosa, S.C.S.; Nayak, N.; Caymo, A.M.; Gordon, J.W. Mechanisms of muscle insulin resistance and the cross-talk with liver and adipose tissue. Physiol. Rep. 2020, 8, e14607. [Google Scholar]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Fraser, D.G.; Onken, J.L.; Johnson, K.O.; Verzosa, G.C.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246. [Google Scholar] [CrossRef]
- Mijares, A.; Allen, P.D.; Lopez, J.R. Senescence Is Associated With Elevated Intracellular Resting [Ca2+] in Mice Skeletal Muscle Fibers. An in vivo Study. Front. Physiol. 2021, 11, 601189. [Google Scholar] [CrossRef]
- Martin, N.; Zhu, K.; Cxarnecka-Herok, J.; Vernier, M.; Bernard, D. Regulation and Role of Calcium in Cellular Senescence. Cell Calcium 2023, 110, 102701. [Google Scholar] [CrossRef]
- Seturo, E.; Fumiko, E.; Ayako, K. Troponin as the Ca++-receptive protein in the contractile system. J. Biochem. 1967, 62, 137–138. [Google Scholar]
- Kamiński, M.; Kręgielska-Narożna, M.; Bogdański, P. Determination of the Popularity of Dietary Supplements Using Google Search Rankings. Nutrients 2020, 12, 908. [Google Scholar] [CrossRef]
- Lim, K.H.; Riddell, L.J.; Nowson, C.A.; Booth, A.O.; Szymlek-Gay, E.A. Iron and Zinc Nutrition in the Economically-Developed World: A Review. Nutrients 2013, 5, 3184–3211. [Google Scholar] [CrossRef]
- Morel, J.; Sauzéat, L.; Goeminne, L.J.; Jha, P.; Williams, E.; Houtkooper, R.H.; Aebersold, R.; Auwerx, J.; Balter, V. The mouse metallomic landscape of aging and metabolism. Nat. Commun. 2022, 13, 607. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Barone, V.; Giacomello, E.; Cusimano, V.; Sorrentino, V. Sarcoplasmic Reticulum: An Organized Patchwork of Specialized Domains. Traffic 2008, 9, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Xu, D.; Wu, A.Z.; Kranias, E.; Lin, S.; Chen, P.; Chen, Z. Phospholamban regulates nuclear Ca2+ stores and inositol 1,4,5-trisphosphate mediated nuclear Ca2+ cycling in cardiomyocytes. J. Mol. Cell. Cardiol. 2018, 123, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Carafoli, E.; Krebs, J. Why Calcium? How Calcium Became the Best Communicator. J. Biol. Chem. 2016, 291, 20849–20857. [Google Scholar] [CrossRef] [PubMed]
- Lanner, J.T.; Georgiou, D.K.; Joshi, A.D.; Hamilton, S.L. Ryanodine Receptors: Structure, Expression, Molecular Details, and Function in Calcium Release. Cold Spring Harb. Perspect. Biol. 2010, 2, a003996. [Google Scholar] [CrossRef] [PubMed]
- Foskett, J.K.; White, C.; Cheung, K.; Mak, D.D. Inositol Trisphosphate Receptor Ca2+ Release Channels. Physiol. Rev. 2007, 87, 593–658. [Google Scholar] [CrossRef] [PubMed]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 Proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef]
- Johnson, C.K.; Harms, G.S. Tracking and localization of calmodulin in live cells. Biochim. Biophys. Acta 2016, 1863, 2017–2026. [Google Scholar] [CrossRef]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca2+ homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef]
- Brunello, E.; Marcucci, L.; Irving, M.; Fusi, L. Activation of skeletal muscle is controlled by a dual-filament mechano-sensing mechanism. Proc. Natl. Acad. Sci. USA 2023, 120, e2302837120. [Google Scholar] [CrossRef]
- Yamada, Y.; Namba, K.; Fujii, T. Cardiac muscle thin filament structures reveal calcium regulatory mechanism. Nat. Commun. 2020, 11, 153. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Craig, R.; Tobacman, L.; Horowitz, R.; Lehman, W. Tropomyosin positions in regulated thin filaments revealed by cryoelectron microscopy. Biophys. J. 1999, 77, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Pierantozzi, E.; Szentesi, P.; Paolini, C.; Dienes, B.; Fodor, J.; Oláh, T.; Colombini, B.; Rassier, D.E.; Rubino, E.M.; Lange, S.; et al. Impaired Intracellular Ca2+ Dynamics, M-Band and Sarcomere Fragility in Skeletal Muscles of Obscurin KO Mice. Int. J. Mol. Sci. 2022, 23, 1319. [Google Scholar] [CrossRef] [PubMed]
- Randazzo, D.; Blaauw, B.; Paolini, C.; Pierantozzi, E.; Spinozzi, S.; Lange, S.; Chen, J.; Protasi, F.; Reggiani, C.; Sorrentino, V. Exercise-induced alterations and loss of sarcomeric M-line organization in the diaphragm muscle of obscurin knockout mice. Am. J. Physiol.-Cell Physiol. 2017, 312, C16. [Google Scholar] [CrossRef]
- Bravo-Sagua, R.; Parra, V.; Muñoz-Cordova, F.; Sanchez-Aguilera, P.; Garrido, V.; Contreras-Ferrat, A.; Chiong, M.; Lavandero, S. Sarcoplasmic reticulum and calcium signaling in muscle cells: Homeostasis and disease. Int. Rev. Cell Mol. Biol. 2020, 350, 197–264. [Google Scholar]
- Yoshida, T.; Delafontaine, P. Mechanisms of IGF-1-Mediated Regulation of Skeletal Muscle Hypertrophy and Atrophy. Cells 2020, 9, 1970. [Google Scholar] [CrossRef]
- Tu, M.K.; Levin, J.B.; Hamilton, A.M.; Borodinsky, L.N. Calcium signaling in skeletal muscle development, maintenance and regeneration. Cell Calcium 2016, 59, 91–97. [Google Scholar] [CrossRef]
- Valdés, J.A.; Flores, S.; Fuentes, E.N.; Osorio-Fuentealba, C.; Jaimovich, E.; Molina, A. IGF-1 induces IP3-dependent calcium signal involved in the regulation of myostatin gene expression mediated by NFAT during myoblast differentiation. J. Cell. Physiol. 2013, 228, 1452–1463. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Venturelli, M.; Morgan, G.R.; Donato, A.J.; Reese, V.; Bottura, R.; Tarperi, C.; Milanese, C.; Schena, F.; Reggiani, C.; Naro, F.; et al. Cellular aging of skeletal muscle: Telomeric and free radical evidence that physical inactivity is responsible and not age. Clin. Sci. 2014, 127, 415–421. [Google Scholar] [CrossRef]
- Bruce, A.; Alexander, J.; Julian, L.; Martin, R.; Keith, R. Walter, Peter, Genesis, Modulation, and Regeneration of Skeletal Muscle, Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2022. [Google Scholar]
- Coletta, G.; Phillips, S.M. An elusive consensus definition of sarcopenia impedes research and clinical treatment: A narrative review. Ageing Res. Rev. 2023, 86, 101883. [Google Scholar] [CrossRef] [PubMed]
- Goodpaster, B.H.; Park, S.W.; Harris, T.B.; Kritchevsky, S.B.; Nevitt, M.; Schwartz, A.V.; Simonsick, E.M.; Tylacsky, F.A.; Visser, M.; Newman, A.B. The Loss of Skeletal Muscle Strength, Mass, and Quality in Older Adults: The Health, Aging and Body Composition Study. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2006, 61, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Murgia, M.; Toniolo, L.; Nagaraj, N.; Ciciliot, S.; Vindigni, V.; Schiaffino, S. Single Muscle Fiber Proteomics Reveals Fiber-Type-Specific Features of Human Muscle Aging. Cell Rep. 2017, 19, 2396–2409. [Google Scholar] [CrossRef] [PubMed]
- Walton, R.G.; Dungan, C.M.; Long, D.E.; Tuggle, S.C.; Kosmac, K.; Peck, B.D.; Bush, H.M.; Villasante Tezanos, A.G.; McGwin, G.; Windham, S.T.; et al. Metformin blunts muscle hypertrophy in response to progressive resistance exercise training in older adults: A randomized, double-blind, placebo-controlled, multicenter trial: The MASTERS trial. Aging Cell 2019, 18, e13039. [Google Scholar] [CrossRef] [PubMed]
- Therakomen, V.; Petchlorlian, A.; Lakananurak, N. Prevalence and risk factors of primary sarcopenia in community-dwelling outpatient elderly: A cross-sectional study. Sci. Rep. 2020, 10, 19551. [Google Scholar] [CrossRef] [PubMed]
- Hester, G.M.; VanDusseldorp, T.A.; Ha, P.L.; Kiani, K.; Olmos, A.A.; Jabbari, M.; Kalladanthyil, S.; An, S.; Bailly, A.R.; Dalton, B.E.; et al. Microbiopsy Sampling for Examining Age-Related Differences in Skeletal Muscle Fiber Morphology and Composition. Front. Physiol. 2022, 12, 756626. [Google Scholar] [CrossRef]
- Bres, E.; Bouvier, J.; Courtay, A.; Delaire, L.; Humblot, J.; Cuerq, C.; Tripoz-Dit-Masson, S.; Fauvernier, M.; Gilbert, T.; Bonnefoy, M. FGF19 and muscle architecture in older patients. Exp. Gerontol. 2023, 174, 112120. [Google Scholar] [CrossRef]
- Lang, F.; Khaghani, S.; Turk, C.; Wiederstein, J.L.; Holper, S.; Piller, T.; Nogara, L.; Blaauw, B.; Gunther, S.; Muller, S.; et al. Single Muscle Fiber Proteomics Reveals Distinct Protein Changes in Slow and Fast Fibers during Muscle Atrophy. J. Proteome Res. 2018, 17, 3333–3347. [Google Scholar] [CrossRef]
- Lukjanenko, L.; Karaz, S.; Stuelsatz, P.; Gurriaran-Rodriguez, U.; Michaud, J.; Dammone, G.; Sizzano, F.; Mashinchian, O.; Ancel, S.; Miglivacca, E.; et al. Aging Disrupts Muscle Stem Cell Function by Impairing Matricellular WISP1 Secretion from Fibro-Adipogenic Progenitors. Cell Stem Cell 2019, 24, 433–446.e7. [Google Scholar] [CrossRef]
- Xu, H.; Ranjit, R.; Richardson, A.; Van Remmen, H. Muscle mitochondrial catalase expression prevents neuromuscular junction disruption, atrophy, and weakness in a mouse model of accelerated sarcopenia. J. Cachexia Sarcopenia Muscle 2021, 12, 1582–1596. [Google Scholar] [CrossRef]
- Kim, K.H.; Chung, Y.; Huh, J.; Park, D.J.; Cho, Y.; Oh, Y.; Jeong, H.; Yoon, J.; Kang, J.; Shin, H.; et al. Gut microbiota of the young ameliorates physical fitness of the aged in mice. Microbiome 2022, 10, 238. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.; Rehm, C.; Rogers, G.; Ruan, M.; Wang, D.; Hu, F.; Mozaffarian, D.; Zhang, F.F. Bhupathiraju, Shilpa. Trends in Dietary Carbohydrate, Protein, and Fat Intake and Diet Quality Among US Adults, 1999–2016. JAMA 2019, 322, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Freire, M.; de Cabo, R.; Studenski, S.A.; Ferrucci, L. The Neuromuscular Junction: Aging at the Crossroad between Nerves and Muscle. Front. Aging Neurosci. 2014, 6, 208. [Google Scholar] [CrossRef] [PubMed]
- Paintin, J.; Cooper, C.; Dennison, E. Osteosarcopenia. Br. J. Hosp. Med. 2018, 79, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Kemmler, W.; Kohl, M.; Fröhlich, M.; Jakob, F.; Engelke, K.; Stengel, S.; Schoene, D. Effects of High-Intensity Resistance Training on Osteopenia and Sarcopenia Parameters in Older Men with Osteosarcopenia—One-Year Results of the Randomized Controlled Franconian Osteopenia and Sarcopenia Trial (FrOST). J. Bone Min. Res. 2020, 35, 1634. [Google Scholar] [CrossRef] [PubMed]
- Hill, T.R.; Verlaan, S.; Biesheuvel, E.; Eastell, R.; Bauer, J.M.; Bautmans, I.; Brandt, K.; Donini, L.M.; Maggio, M.; Mets, T.; et al. A Vitamin D, Calcium and Leucine-Enriched Whey Protein Nutritional Supplement Improves Measures of Bone Health in Sarcopenic Non-Malnourished Older Adults: The PROVIDE Study. Calcif. Tissue Int. 2019, 105, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Wiedmer, P.; Jung, T.; Castro, J.P.; Pomatto, L.C.; Sun, P.Y.; Davies, K.J.; Tilman, G. Sarcopenia—Molecular mechanisms and open questions. Ageing Res. Rev. 2021, 65, 101200. [Google Scholar] [CrossRef] [PubMed]
- Dayal, A.; Schrötter, K.; Pan, Y.; Föhr, K.; Melzer, W.; Grabner, M. The Ca2+ influx through the mammalian skeletal muscle dihydropyridine receptor is irrelevant for muscle performance. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Liu, Y.; Sugiura, Y.; Chen, F.; Lee, K.; Ye, Q.; Lin, W. Blocking skeletal muscle DHPRs/Ryr1 prevents neuromuscular synapse loss in mutant mice deficient in type III Neuregulin 1 (CRD-Nrg1). PLoS Genet. 2019, 15, e1007857. [Google Scholar] [CrossRef]
- Edwards, J.N.; Blackmore, D.G.; Gilbert, D.F.; Murphy, R.M.; Launikonis, B.S. Store-operated calcium entry remains fully functional in aged mouse skeletal muscle despite a decline in STIM1 protein expression. Aging Cell 2011, 10, 675–685. [Google Scholar] [CrossRef]
- Weisleder, N.; Brotto, M.; Komazaki, S.; Pan, Z.; Zhao, X.; Nosek, T.; Parness, J.; Takeshima, H.; Ma, J. Muscle Aging Is Associated with Compromised Ca2+ Spark Signaling and Segregated Intracellular Ca2+ Release. J. Cell Biol. 2006, 174, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Grimes, D.; Johnson, R.; Pashos, M.; Cummings, C.; Kang, C.; Sampedro, G.R.; Tycksen, E.; McBride, H.J.; Sah, R.; Lowell, C.A.; et al. ORAI1 and ORAI2 modulate murine neutrophil calcium signaling, cellular activation, and host defense. Proc. Natl. Acad. Sci. USA 2020, 117, 24403–24414. [Google Scholar] [CrossRef] [PubMed]
- Lacruz, R.S.; Feske, S. Diseases caused by mutations in ORAI1 and STIM1. Ann. N. Y. Acad. Sci. 2015, 1356, 45–79. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Zhou, X.; Gumpper, K.; Zhu, H. MG29 Interacts with Bin-1 to Maintain T-Tubule Structure in Skeletal Muscle Physiology and Regeneration. FASEB J. 2019, 33, 868-24. [Google Scholar] [CrossRef]
- Fraysse, B.; Desaphy, J.; Rolland, J.; Pierno, S.; Liantonio, A.; Giannuzzi, V.; Camerino, C.; Didonna, M.P.; Cocchi, D.; Luca, A.D.; et al. Fiber type-related changes in rat skeletal muscle calcium homeostasis during aging and restoration by growth hormone. Neurobiol. Dis. 2006, 21, 372–380. [Google Scholar] [CrossRef]
- Kim, Y.; Hong, K.; Han, K.; Park, Y.C.; Park, J.; Kim, K.; Kim, B.T. Longitudinal Observation of Muscle Mass over 10 Years According to Serum Calcium Levels and Calcium Intake among Korean Adults Aged 50 and Older: The Korean Genome and Epidemiology Study. Nutrients 2020, 12, 2856. [Google Scholar] [CrossRef]
- Asghar, M.Y.; Törnquist, K. Transient Receptor Potential Canonical (TRPC) Channels as Modulators of Migration and Invasion. Int. J. Mol. Sci. 2020, 21, 1739. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Zhang, X.; Habiballa, L.; Aversa, Z.; Ng, Y.E.; Sakamoto, A.E.; Englund, D.A.; Pearsall, V.M.; White, T.A.; Robinson, M.M.; Rivas, D.A. Characterization of cellular senescence in aging skeletal muscle. Nat. Aging 2022, 2, 601. [Google Scholar] [CrossRef]
- Tsuyoshi, K.; David, A.B.; Richard, W.; Tomas, P.A. Influences of aging and caloric restriction on the transcriptional profile of transcriptional profile of skeletal muscle from rhesus monkeys. Proc. Natl. Acad. Sci. USA 2001, 98, 5093–5098. [Google Scholar]
- Perez, K.; Ciotlos, S.; McGirr, J.; Limbad, C.; Doi, R.; Nederveen, J.P.; Nilsson, M.I.; Winer, D.A.; Evans, W.; Tarnopolsky, M. Single nuclei profiling identifies cell specific markers of skeletal muscle aging, frailty, and senescence. Aging 2022, 14, 9393–9422. [Google Scholar] [CrossRef] [PubMed]
- Dungan, C.M.; Peck, B.D.; Walton, R.G.; Huang, Z.; Bamman, M.M.; Kern, P.A.; Peterson, C.A. In vivo analysis of γH2AX+ cells in skeletal muscle from aged and obese humans. FASEB J. 2020, 34, 7018. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Ohtani, N.; Youssef, S.; Rodier, F.; Toussaint, W.; Mitchell, J.; Laberge, R.M.; Vijg, J.; Steeg, H.V.; Dolle, M.E.T.; et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Prieto, L.; Graves, S.; Baker, D. Insights from In Vivo Studies of Cellular Senescence. Cells 2020, 9, 954. [Google Scholar] [CrossRef] [PubMed]
- Raynard, C.; Tessier, N.; Huna, A.; Warnier, M.; Flaman, J.; Van Coppenolle, F.; Ducreux, S.; Martin, N.; Bernard, D. Expression of the Calcium-Binding Protein CALB1 Is Induced and Controls Intracellular Ca2+ Levels in Senescent Cells. Int. J. Mol. Sci. 2022, 23, 9376. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, D.V.; Vindrieux, D.; Goehrig, D.; Jaber, S.; Collin, G.; Griveau, A.; Wiel, C.; Bendridi, N.; Djebali, S.; Farfariello, V.; et al. Calcium channel ITPR2 and mitochondria–ER contacts promote cellular senescence and aging. Nat. Commun. 2021, 12, 720. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Dhakal, K.; Yi, J. Mitochondrial Ca2+ uptake in skeletal muscle health and disease. Sci. China Life Sci. 2016, 59, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Harrington, J.S.; Ryter, S.W.; Plataki, M.; Price, D.R.; Choi, A.M.K. Mitochondria in Health, Disease, and Aging. Physiol. Rev. 2023, 103, 2349–2422. [Google Scholar] [CrossRef]
- Abrisch, R.G.; Gumbin, S.C.; Wisniewski, B.T.; Lackner, L.L.; Voeltz, G.K. Fission and fusion machineries converge at ER contact sites to regulate mitochondrial morphology. J. Cell Biol. 2020, 219, 1. [Google Scholar] [CrossRef]
- Miwa, S.; Kashyap, S.; Chini, E.; Von Zglinicki, T. Mitochondrial dysfunction in cell senescence and aging. J. Clin. Investig. 2022, 132, e158447. [Google Scholar] [CrossRef]
- Li, Y.; Tran, Q.; Shrestha, R.; Piao, L.; Park, S.; Park, J.; Park, J. LETM1 is required for mitochondrial homeostasis and cellular viability. Mol. Med. Rep. 2019, 19, 3367–3375. [Google Scholar] [CrossRef] [PubMed]
- Samanta, K.; Mirams, G.R.; Parekh, A.B. Sequential forward and reverse transport of the Na+ Ca2+ exchanger generates Ca2+ oscillations within mitochondria. Nat. Commun. 2018, 9, 156. [Google Scholar] [CrossRef] [PubMed]
- Harrington, J.L.; Murphy, E. The mitochondrial calcium uniporter: Mice can live and die without it. J. Mol. Cell. Cardiol. 2015, 78, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Young, M.P.; Schug, Z.T.; Booth, D.M.; Yule, D.I.; Mikoshiba, K.; Hajnoczky, G.; Joseph, S.K. Metabolic adaptation to the chronic loss of Ca2+ signaling induced by KO of IP3 receptors or the mitochondrial Ca2+ uniporter. J. Biol. Chem. 2022, 298, 101436. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.; Chandel, N. Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto-Imoto, H.; Minami, S.; Shioda, T.; Yamashita, Y.; Sakai, S.; Maeda, S.; Yamamoto, T.; Oki, S.; Takashima, M.; Yamamuro, T.; et al. Age-associated decline of MondoA drives cellular senescence through impaired autophagy and mitochondrial homeostasis. Cell Rep. 2022, 38, 110444. [Google Scholar] [CrossRef] [PubMed]
- Debattisti, V.; Horn, A.; Singh, R.; Seifert, E.L.; Hogarth, M.W.; Mazala, D.A.; Huang, K.T.; Horvath, R.; Jaiswal, J.K.; Hajnoczky, G. Dysregulation of Mitochondrial Ca2+ Uptake and Sarcolemma Repair Underlie Muscle Weakness and Wasting in Patients and Mice Lacking MICU1. Cell Rep. 2019, 29, 1274–1286.e6. [Google Scholar] [CrossRef]
- Logan, C.V.; Szabadkai, G.; Sharpe, J.A.; Parry, D.A.; Torelli, S.; Childs, A.; Kriek, M.; Phadke, R.; Johnson, C.A.; Roberts, N.Y.; et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet. 2014, 46, 188–193. [Google Scholar] [CrossRef]
- Bonaventura, P.; Benedetti, G.; Albarède, F.; Miossec, P. Zinc and its role in immunity and inflammation. Autoimmun. Rev. 2015, 14, 277–285. [Google Scholar] [CrossRef]
- Huber, A.M.; Gershoff, S.N. Effects of dietary zinc on zinc enzymes in the rat. J. Nutr. 1973, 103, 1175–1181. [Google Scholar] [CrossRef]
- Hernández-Camacho, J.D.; Vicente-García, C.; Parsons, D.S.; Navas-Enamorado, I. Zinc at the crossroads of exercise and proteostasis. Redox Biol. 2020, 35, 101529. [Google Scholar] [CrossRef] [PubMed]
- Afzali, A.; Goli, S.; Moravveji, A.; Bagheri, H.; Mirhosseini, S.; Ebrahimi, H. The effect of zinc supplementation on fatigue among elderly community dwellers: A parallel clinical trial. Health Sci. Rep. 2021, 4, e301. [Google Scholar] [CrossRef] [PubMed]
- Killilea, D.W.; Atamna, H.; Liao, C.; Ames, B.N. Iron Accumulation During Cellular Senescence in Human Fibroblasts In Vitro. Antioxid. Redox Signal. 2003, 5, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; Shih, R.S.; Schoene, N.W.; Lei, K.Y. Zinc-induced G2/M blockage is P53 and P21 dependent in normal human bronchial epithelial cells. Am. J. Physiol.-Cell Physiol. 2008, 294, C1342–C1349. [Google Scholar] [CrossRef] [PubMed]
- Salazar, G.; Huang, J.; Feresin, R.G.; Zhao, Y.; Griendling, K.K. Zinc regulates Nox1 expression through a NF-κB and mitochondrial ROS dependent mechanism to induce senescence of vascular smooth muscle cells. Free Radic. Biol. Med. 2017, 108, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Dabravolski, S.A.; Sadykhov, N.K.; Kartuesov, A.G.; Borisov, E.E.; Sukhorukov, V.N.; Orekhov, A.N. Interplay between Zn2+ Homeostasis and Mitochondrial Functions in Cardiovascular Diseases and Heart Ageing. Int. J. Mol. Sci. 2022, 23, 6890. [Google Scholar] [CrossRef]
- Busti, F.; Campostrini, N.; Martinelli, N.; Girelli, D. Iron deficiency in the elderly population, revisited in the hepcidin era. Front. Pharmacol. 2014, 5, 83. [Google Scholar] [CrossRef]
- Alves, F.M.; Ayton, S.; Bush, A.I.; Lynch, G.S.; Koopman, R. Age-Related Changes in Skeletal Muscle Iron Homeostasis. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2023, 78, 16–24. [Google Scholar] [CrossRef]
- Alves, F.M.; Kysenius, K.; Caldow, M.K.; Hardee, J.P.; Crouch, P.J.; Ayton, S.; Bush, A.I.; Lynch, G.S.; Koopman, R. Iron accumulation in skeletal muscles of old mice is associated with impaired regeneration after ischaemia–reperfusion damage. J. Cachexia Sarcopenia Muscle 2021, 12, 476. [Google Scholar] [CrossRef]
- Huang, Y.; Wu, B.; Shen, D.; Chen, J.; Yu, Z.; Chen, C. Ferroptosis in a sarcopenia model of senescence accelerated mouse prone 8 (SAMP8). Int. J. Biol. Sci. 2021, 17, 151–162. [Google Scholar] [CrossRef]
- Nemeth, E.; Ganz, T. Hepcidin and Iron in Health and Disease. Annu. Rev. Med. 2023, 74, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Masaldan, S.; Clatworthy, S.A.; Gamell, C.; Meggyesy, P.M.; Rigopoulos, A.; Haupt, S.; Haupt, Y.; Denoyer, D.; Alard, P.A.; Bush, A.I.; et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2018, 14, 100–115. [Google Scholar] [CrossRef]
- Seo, A.Y.; Xu, J.; Servais, S.; Hofer, T.; Marzetti, E.; Wohlgemuth, S.E.; Knutson, M.D.; Chung, H.Y.; Leeuwenburgh, C. Mitochondrial iron accumulation with age and functional consequences. Aging Cell 2008, 7, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Bird, A.J. Zinc’ing sensibly: Controlling zinc homeostasis at the transcriptional level. Metallomics 2014, 6, 1198–1215. [Google Scholar] [CrossRef]
- Bouron, A.; Oberwinkler, J. Contribution of calcium-conducting channels to the transport of zinc ions. Pflug. Arch. Eur. J. Physiol. 2014, 466, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Kerchner, G.A.; Canzoniero, L.M.; Yu, S.P.; Ling, C.; Choi, D.W. Zn2+ current is mediated by voltage-gated Ca2+ channels and enhanced by extracellular acidity in mouse cortical neurones. J. Physiol. 2000, 528, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Atar, D.; Backx, P.H.; Appel, M.M.; Gao, W.D.; Marban, E. Excitation-Transcription Coupling Mediated by Zinc Influx through Voltage-dependent Calcium Channels (∗). J. Biol. Chem. 1995, 270, 2473–2477. [Google Scholar] [CrossRef]
- Gyulkhandanyan, A.V.; Lee, S.C.; Bikopoulos, G.; Dai, F.; Wheeler, M.B. The Zn2+-transporting pathways in pancreatic beta-cells: A role for the L-type voltage-gated Ca2+ channel. J. Biol. Chem. 2006, 281, 9361–9372. [Google Scholar] [CrossRef]
- Yamasaki, S.; Hasegawa, A.; Hojyo, S.; Ohashi, W.; Fukada, T.; Nishida, K.; Hirano, T. A Novel Role of the L-Type Calcium Channel [alpha].sub.1D Subunit as a Gatekeeper for Intracellular Zinc Signaling: Zinc Wave. PLoS ONE 2012, 7, e39654. [Google Scholar] [CrossRef]
- Monteilh-Zoller, M.K.; Hermosura, M.C.; Nadler, M.J.; Scharenberg, A.M.; Penner, R.; Fleig, A. TRPM7 Provides an Ion Channel Mechanism for Cellular Entry of Trace Metal Ions. J. Gen. Physiol. 2003, 121, 49–60. [Google Scholar] [CrossRef]
- Topala, C.N.; Groenestege, W.T.; Thébault, S.; van den Berg, D.; Nilius, B.; Hoenderop, J.G.; Bindels, R.J. Molecular determinants of permeation through the cation channel TRPM6. Cell Calcium 2007, 41, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Jiang, J.; Yue, L. Functional Characterization of Homo- and Heteromeric Channel Kinases TRPM6 and TRPM7. J. Gen. Physiol. 2006, 127, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Sander, S.; Pick, J.; Gattkowski, E.; Fliegert, R.; Tidow, H. The crystal structure of TRPM2 MHR1/2 domain reveals a conserved Zn2+-binding domain essential for structural integrity and channel activity. Protein Sci. 2022, 31, e4320. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.T.; Munsey, T.S.; Abuarab, N.; Li, F.; Asipu, A.; Howell, G.; Sedo, A.; Yang, W.; Naylor, J.; Beech, D.J.; et al. TRPM2-mediated intracellular Zn2+ release triggers pancreatic β-cell death. Biochem. J. 2015, 466, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.; Moran, A.; Hershfinkel, M.; Sekler, I. Inhibitory Mechanism of Store-operated Ca2+ Channels by Zinc. J. Biol. Chem. 2004, 279, 11106–11111. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Cui, C.; Luo, Y.; Kim, S.; Ko, J.; Huo, X.; Ma, J.; Fu, L.; Souza, R.; Korichneva, I.; et al. Selective inhibitory effects of zinc on cell proliferation in esophageal squamous cell carcinoma through Orai1. FASEB J. 2018, 32, 404–416. [Google Scholar] [CrossRef]
- Carvalho, A.P. Effects of Potentiators of Muscular Contraction on Binding of Cations by Sarcoplasmic Reticulum. J. Gen. Physiol. 1968, 51, 427–442. [Google Scholar] [CrossRef]
- Rossowska, M.J.; Nakamoto, T. Interaction between zinc and calcium in skeletal muscle in young growing rats. Biol. Trace Elem. Res. 1993, 38, 301–309. [Google Scholar] [CrossRef]
- Nasu, T. Zinc ions block the intracellular calcium release induced by caffeine in guinea-pig taenia caeci. Experientia 1995, 51, 113–116. [Google Scholar] [CrossRef]
- Pitt, S.J.; Stewart, A.J. Examining a new role for zinc in regulating calcium release in cardiac muscle. Biochem. Soc. Trans. 2015, 43, 359–363. [Google Scholar] [CrossRef]
- Wang, H.; Wei, Q.Q.; Cheng, X.Y.; Chen, K.Y.; Zhu, P.H. Inhibition of ryanodine binding to sarcoplasmic reticulum vesicles of cardiac muscle by Zn2+ ions. Cell. Physiol. Biochem. 2001, 11, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Woodier, J.; Rainbow, R.D.; Stewart, A.J.; Pitt, S.J. Intracellular Zinc Modulates Cardiac Ryanodine Receptor-mediated Calcium Release. J. Biol. Chem. 2015, 290, 17599–17610. [Google Scholar] [CrossRef] [PubMed]
- Reilly-O’Donnell, B.; Robertson, G.B.; Karumbi, A.; McIntyre, C.; Bal, W.; Nishi, M.; Takeshima, H.; Stewart, A.J.; Pitt, S.J. Dysregulated Zn2+ homeostasis impairs cardiac type-2 ryanodine receptor and mitsugumin 23 functions, leading to sarcoplasmic reticulum Ca2+ leakage. J. Biol. Chem. 2017, 292, 13361–13373. [Google Scholar] [CrossRef]
- Dorward, A.M.; Stewart, A.J.; Pitt, S.J. The role of Zn2+ in shaping intracellular Ca2+ dynamics in the heart. J. Gen. Physiol. 2023, 155, e202213206. [Google Scholar] [CrossRef] [PubMed]
- Heilmann, C.; Spamer, C.; Leberer, E.; Gerok, W.; Michalak, M. Human Liver Calreticulin: Characterization and Zn2+-Dependent Interaction with Phenyl-Sepharose. Biochem. Biophys. Res. Commun. 1993, 193, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Chen, M.; Li, Z.; Mabuchi, K.; Bouvier, M. The calcium- and zinc-responsive regions of calreticulin reside strictly in the N-/C-domain. Biochim. Biophys. Acta 2006, 1760, 745–753. [Google Scholar] [CrossRef]
- Rivera, J.F.; Baral, A.J.; Nadat, F.; Boyd, G.; Smyth, R.; Patel, H.; Burman, E.L.; Alameer, G.; Boxall, S.A.; Jackson, B.R.; et al. Zinc-dependent multimerization of mutant calreticulin is required for MPL binding and MPN pathogenesis. Blood Adv. 2021, 5, 1922–1932. [Google Scholar] [CrossRef]
- Baksh, S.; Spamer, C.; Oikawa, K.; McCubbin, W.D.; Heilmann, C.; Kay, C.M.; Michalak, M. Zn2+ Binding to Cardiac Calsequestrin. Biochem. Biophys. Res. Commun. 1995, 209, 310–315. [Google Scholar] [CrossRef]
- Picello, E.; Damiani, E.; Margreth, A. Low-affinity Ca2+-binding sites versus Zn2+-binding sites in histidine-rich Ca2+-binding protein of skeletal muscle sarcoplasmic reticulum. Biochem. Biophys. Res. Commun. 1992, 186, 659–667. [Google Scholar] [CrossRef]
- Adebayo, O.L.; Khera, A.; Sandhir, R.; Adenuga, G.A. Reduced expressions of calmodulin genes and protein and reduced ability of calmodulin to activate plasma membrane Ca2+-ATPase in the brain of protein undernourished rats: Modulatory roles of selenium and zinc supplementation. Cell Biochem. Funct. 2016, 34, 95–103. [Google Scholar] [CrossRef]
- Baudier, J.; Haglid, K.; Haiech, J.; Gérard, D. Zinc ion binding to human brain calcium binding proteins, Calmodulin and S100b protein. Biochem. Biophys. Res. Commun. 1983, 114, 1138–1146. [Google Scholar] [CrossRef]
- Brewer, G.J.; Aster, J.C.; Knutsen, C.A.; Kruckeberg, W.C. Zinc inhibition of calmodulin: A proposed molecular mechanism of zinc action on cellular functions. Am. J. Hematol. 1979, 7, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.Y.; Lin, W.H.; Tsou, C.T.; Song, Y.M.; Chen, M.D. Effect of Zinc on Cellular Levels of Calmodulin and Cyclic Adenosine Monophosphate in the Adipocyte. Biol. Trace Elem. Res. 2000, 76, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Heng, M.K.; Song, M.K.; Heng, M.C. Reciprocity between tissue calmodulin and cAMP levels: Modulation by excess zinc. Br. J. Dermatol. 1993, 129, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, R.P.; Rostas, J.A. Effect of Zinc on Calmodulin-Stimulated Protein Kinase II and Protein Phosphorylation in Rat Cerebral Cortex. J. Neurochem. 1991, 57, 605–614. [Google Scholar] [CrossRef]
- Baudier, J.; Glasser, N.; Gerard, D. Ions binding to S100 proteins. I. Calcium- and zinc-binding properties of bovine brain S100 alpha alpha, S100a (alpha beta), and S100b (beta beta) protein: Zn2+ regulates Ca2+ binding on S100b protein. J. Biol. Chem. 1986, 261, 8192–8203. [Google Scholar] [CrossRef]
- Baudier, J.; Deloulme, J.C.; Shaw, G.S. The Zn2+ and Ca2+-binding S100B and S100A1 proteins: Beyond the myths. Biol. Rev. Camb. Philos. Soc. 2020, 95, 738–758. [Google Scholar] [CrossRef]
- Brodersen, D.E.; Nyborg, J.; Kjeldgaard, M. Zinc-Binding Site of an S100 Protein Revealed. Two Crystal Structures of Ca2+-Bound Human Psoriasin (S100A7) in the Zn2+-Loaded and Zn2+-Free States. Biochemistry 1999, 38, 1695–1704. [Google Scholar] [CrossRef]
- Moroz, O.V.; Wilson, K.S.; Bronstein, I.B. The role of zinc in the S100 proteins: Insights from the X-ray structures. Amino Acids 2011, 41, 761–772. [Google Scholar] [CrossRef]
- Wang, Q.; Aleshintsev, A.; Jose, A.N.; Aramini, J.M.; Gupta, R. Calcium Regulates S100A12 Zinc Sequestration by Limiting Structural Variations. ChemBioChem A Eur. J. Chem. Biol. 2020, 21, 1372–1382. [Google Scholar] [CrossRef]
- Baksheeva, V.E.; Tsvetkov, P.O.; Zalevsky, A.O.; Vladimirov, V.I.; Gorokhovets, N.V.; Zinchenko, D.V.; Permyakov, S.E.; Devred, F.; Zernii, E.Y. Zinc Modulation of Neuronal Calcium Sensor Proteins: Three Modes of Interaction with Different Structural Outcomes. Biomolecules 2022, 12, 956. [Google Scholar] [CrossRef] [PubMed]
- Baksheeva, V.E.; Baldin, A.V.; Zalevsky, A.O.; Nazipova, A.A.; Kazakov, A.S.; Vladimirov, V.I.; Gorokhovets, N.V.; Devred, F.; Philippov, P.P.; Bazhin, A.V.; et al. Disulfide Dimerization of Neuronal Calcium Sensor-1: Implications for Zinc and Redox Signaling. Int. J. Mol. Sci. 2021, 22, 12602. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.O.; Roman, A.Y.; Baksheeva, V.E.; Nazipova, A.A.; Shevelyova, M.P.; Vladimirov, V.I.; Buyanova, M.F.; Zinchenko, D.V.; Zamyatnin, A.A., Jr. Functional Status of Neuronal Calcium Sensor-1 Is Modulated by Zinc Binding. Front. Mol. Neurosci. 2018, 11, 459. [Google Scholar] [CrossRef] [PubMed]
- Permyakov, S.E.; Cherskaya, A.M.; Wasserman, L.A.; Khokhlova, T.I.; Senin, I.I.; Zargarov, A.A.; Zinchenko, D.V.; Zernii, E.U.; Lipkin, V.M.; Philippov, P.P.; et al. Recoverin Is a Zinc-Binding Protein. J. Proteome Res. 2003, 2, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cheng, X.; Zhao, H.; Yang, Q.; Xu, Z. Downregulation of the zinc transporter SLC39A13 (ZIP13) is responsible for the activation of CaMKII at reperfusion and leads to myocardial ischemia/reperfusion injury in mouse hearts. J. Mol. Cell. Cardiol. 2021, 152, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Segal, D.; Ohana, E.; Besser, L.; Hershfinkel, M.; Moran, A.; Sekler, I. A role for ZnT-1 in regulating cellular cation influx. Biochem. Biophys. Res. Commun. 2004, 323, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.; Beharier, O.; Etzion, Y.; Mor, M.; Buzaglo, L.; Shaltiel, L.; Gheber, L.A.; Kahn, J.; Muslin, A.J.; Katz, A.; et al. Molecular Basis for Zinc Transporter 1 Action as an Endogenous Inhibitor of L-type Calcium Channels. J. Biol. Chem. 2009, 284, 32434–32443. [Google Scholar] [CrossRef]
- Gottesman, N.; Asraf, H.; Bogdanovic, M.; Sekler, I.; Tzounopoulos, T.; Aizenman, E.; Hershfinkel, M. ZnT1 is a neuronal Zn2+/Ca2+ exchanger. Cell Calcium 2022, 101, 102505. [Google Scholar] [CrossRef]
- Lönnerdal, B. Calcium and Iron Absorption—Mechanisms and Public Health Relevance. Int. J. Vitam. Nutr. Res. 2010, 80, 293–299. [Google Scholar] [CrossRef]
- Shawki, A.; Mackenzie, B. Interaction of calcium with the human divalent metal-ion transporter-1. Biochem. Biophys. Res. Commun. 2010, 393, 471–475. [Google Scholar] [CrossRef]
- Hidalgo, C.; Nunez, M.T. Calcium, iron and neuronal function. IUBMB Life 2007, 59, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Yalcintepe, L.; Halis, E. Modulation of iron metabolism by iron chelation regulates intracellular calcium and increases sensitivity to doxorubicin. Bosn. J. Basic. Med. Sci. 2016, 16, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Phoaubon, S.; Lertsuwan, K.; Teerapornpuntakit, J.; Charoenphandhu, N. Hepcidin induces intestinal calcium uptake while suppressing iron uptake in Caco-2 cells. PLoS ONE 2021, 16, e0258433. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.Y.; Trivieri, M.G.; Khaper, N.; Liu, P.P.; Peter, H.B. Role of L-type Ca2+ channels in iron transport and iron-overload cardiomyopathy. J. Mol. Med. 2006, 84, 349–364. [Google Scholar] [CrossRef]
- Tsushima, R.G.; Wickenden, A.D.; Bouchard, R.A.; Oudit, G.Y.; Liu, P.P.; Backx, P.H. Modulation of Iron Uptake in Heart by L-Type Ca2+ Channel Modifiers: Possible Implications in Iron Overload. Circ. Res. 1999, 84, 1302–1309. [Google Scholar] [CrossRef]
- Li, G.; Xu, Y.; He, Y.; Du, B.; Zhang, P.; Zhao, D.; Yu, C.; Qin, C.; Li, K. Effect of hepcidin on intracellular calcium in human osteoblasts. Mol. Cell Biochem. 2012, 366, 169–174. [Google Scholar] [CrossRef]
- Xu, Y.; Li, G.; Du, B.; Zhang, P.; Xiao, L.; Sirois, P.; Li, K. Hepcidin increases intracellular Ca2+ of osteoblast hFOB1.19 through L-type Ca2+ channels. Regul. Pept. 2011, 172, 58–61. [Google Scholar] [CrossRef]
- Kumfu, S.; Chattipakorn, S.; Srichairatanakool, S.; Settakorn, J.; Fucharoen, S.; Chattipakorn, N. T-type calcium channel as a portal of iron uptake into cardiomyocytes of beta-thalassemic mice. Eur. J. Haematol. 2011, 86, 156–166. [Google Scholar] [CrossRef]
- Lopin, K.V.; Gray, I.P.; Obejero-Paz, C.A.; Thévenod, F.; Jones, S.W. Fe2+ block and permeation of CaV3.1 (α1G) T-type calcium channels: Candidate mechanism for non-transferrin-mediated Fe2+ influx. Mol. Pharmacol. 2012, 82, 1194–1204. [Google Scholar] [CrossRef]
- Kim, E.; Giri, S.N.; Pessah, I.N. Iron(II) Is a Modulator of Ryanodine-Sensitive Calcium Channels of Cardiac Muscle Sarcoplasmic Reticulum. Toxicol. Appl. Pharmacol. 1995, 130, 57–66. [Google Scholar] [CrossRef]
- Sanmartín, C.D.; Paula-Lima, A.C.; García, A.; Barattini, P.; Hartel, S.; Núñez, M.T.; Hidalgo, C. Ryanodine receptor-mediated Ca2+ release underlies iron-induced mitochondrial fission and stimulates mitochondrial Ca2+ uptake in primary hippocampal neurons. Front. Mol. Neurosci. 2014, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, P.; Zavala, G.; Castillo, K.; Aguirre, P.; Hidalgo, C.; Núñez, M.T. Effect of iron on the activation of the MAPK/ERK pathway in PC12 neuroblastoma cells. Biol. Res. 2006, 39, 189–190. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.J.; Luo, A.; Park, K.C.; Loonat, A.A.; Lakhal-Littleton, S.; Robbins, P.A.; Swietach, P. Iron-deficiency anemia reduces cardiac contraction by downregulating RyR2 channels and suppressing SERCA pump activity. JCI Insight 2019, 4, e125618. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, C.N.; Ruwe, T.A.; Shawki, A.; Xin, V.; Vieth, K.R.; Valore, E.V.; Qiao, B.; Ganz, T.; Nemeth, E.; Mackenzia, B.; et al. Calcium is an essential cofactor for metal efflux by the ferroportin transporter family. Nat. Commun. 2018, 9, 3075. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Wilbon, A.S.; Zhou, M.; Pan, Y. Mechanism of Ca2+ transport by ferroportin. eLife 2023, 12, e82947. [Google Scholar] [CrossRef] [PubMed]
- Ci, W.; Li, W.; Ke, Y.; Qian, Z.; Shen, X. Intracellular Ca2+ regulates the cellular iron uptake in K562 cells. Cell Calcium 2003, 33, 257–266. [Google Scholar] [CrossRef]
- Sabbir, M.G. CAMKK2-CAMK4 signaling regulates transferrin trafficking, turnover, and iron homeostasis. Cell Commun. Signal. 2020, 18, 80. [Google Scholar] [CrossRef]
- Sabbir, M.G. Loss of Ca2+/Calmodulin Dependent Protein Kinase Kinase 2 Leads to Aberrant Transferrin Phosphorylation and Trafficking: A Potential Biomarker for Alzheimer’s Disease. Front. Mol. Biosci. 2018, 5, 99. [Google Scholar] [CrossRef]
- Sabbir, M.G.; Taylor, C.G.; Zahradka, P. Hypomorphic CAMKK2 in EA.hy926 endothelial cells causes abnormal transferrin trafficking, iron homeostasis and glucose metabolism. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118763. [Google Scholar] [CrossRef]
- Ke, K.; Li, L.; Lu, C.; Zhu, Q.; Wang, Y.; Mou, Y.; Wang, H.; Jin, W. The crosstalk effect between ferrous and other ions metabolism in ferroptosis for therapy of cancer. Front. Oncol. 2022, 12, 916082. [Google Scholar] [CrossRef]
- Núñez, M.T.; Hidalgo, C. Noxious Iron–Calcium Connections in Neurodegeneration. Front. Neurosci. 2019, 13, 48. [Google Scholar] [CrossRef] [PubMed]
- Gleitze, S.; Paula-Lima, A.; Núñez, M.T.; Hidalgo, C. The calcium–iron connection in ferroptosis-mediated neuronal death. Free Radic. Biol. Med. 2021, 175, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Pelizzoni, I.; Macco, R.; Zacchetti, D.; Grohovaz, F.; Codazzi, F. Iron and calcium in the central nervous system: A close relationship in health and sickness. Biochem. Soc. Trans. 2008, 36, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Maher, P.; van Leyen, K.; Dey, P.N.; Honrath, B.; Dolga, A.; Methner, A. The role of Ca2+ in cell death caused by oxidative glutamate toxicity and ferroptosis. Cell Calcium 2018, 70, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Braughler, J.M.; Duncan, L.A.; Chase, R.L. Interaction of Lipid Peroxidation and Calcium in the Pathogenesis of Neuronal Injury. Cent. Nerv. Syst. Trauma 1985, 2, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Siri-Angkul, N.; Song, Z.; Fefelova, N.; Gwathmey, J.K.; Chattipakorn, S.C.; Qu, Z.; Chattipakorn, N.; Xie, L. Activation of TRPC (Transient Receptor Potential Canonical) Channel Currents in Iron Overloaded Cardiac Myocytes. Circ. Arrhythmia Electrophysiol. 2021, 14, e009291. [Google Scholar] [CrossRef] [PubMed]
- Nikolaienko, R.; Bovo, E.; Kahn, D.; Gracia, R.; Jamrozik, T.; Zima, A.V. Cysteines 1078 and 2991 cross-linking plays a critical role in redox regulation of cardiac ryanodine receptor (RyR). Nat. Commun. 2023, 14, 4498. [Google Scholar] [CrossRef]
- Goldberg, J.; Antonio, C.; Gamze, A.; Huang, L.; Maxim, S.; Maher, P.; Schubert, D. Targeting of intracellular Ca2+ stores as a therapeutic strategy against age-related neurotoxicities. npj Aging Mech. Dis. 2020, 6, 10. [Google Scholar] [CrossRef]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gelé, P.; Pétrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef]
- Wang, S.; Yi, X.; Wu, Z.; Guo, S.; Dai, W.; Wang, H.; Shi, Q.; Zeng, K.; Guo, W.; Li, C. CAMKK2 Defines Ferroptosis Sensitivity of Melanoma Cells by Regulating AMPK—NRF2 Pathway. J. Investig. Dermatol. 2022, 142, 189–200.e8. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Author/Year | Species/Age | Finding | Implication | Ref. |
|---|---|---|---|---|
| Human Studies | ||||
| Goodpaster, B.H. et al. (2006) | Three-year changes in old (70–79 years; n = 1880) subjects (female, 51%; male, 48%) | Initially functioning older adults exhibited three-fold greater loss in strength than the loss of muscle mass over the course of 3 years. Maintenance or even gain of lean mass did not necessarily prevent loss of strength. | Loss of strength is more rapid and suggests a decline in the quality of muscle. Losses of strength can increase risks of falls and serious injury. | [33] |
| Venturelli, M. et al. (2014) | Young (25 ± 2 years; n = 12), old-mobile (87 ± 3 years of age; n = 12) and 12 old-immobile (88 ± 4 years; n = 12) sex-matched subjects (female, n = 9; male, n = 3) | Mean skeletal telomere length of thigh decreased with age, but not of arm. Mean free radical increased with age in thigh but not in arm. | Chronological age does not affect the cellular aging of skeletal muscle evenly. Physical inactivity could be mediated by the free radical effect. | [30] |
| Murgia, M. et al. (2017) | Young (22–27 years; n = 4) and old (65–75 years of age, n = 4) non-sarcopenic subjects | Fiber size of fast-twitch (type 2a) but not slow-twitch (type 1) muscles decreased with age. Decreased respiratory chain complexes were found in aging muscles. Changes in protein quality, turnover, and metabolic pathways were changed with age in muscles. | Many glycolytic enzymes were expressed higher in slow fibers of the older cohort, and these same enzymes declined within fast-twitch fibers, showing changes in mitochondria in line with previous studies. These proteomic data support the idea that aging may differentially affect type 2 muscle fibers, protein homeostasis, mitochondria function, and metabolic pathways. | [34] |
| Walton, R.G. et al. (2019) | Randomized, double-blind trials in placebo (n = 55) and Metformin (n = 54) groups of old (over 65 years) subjects | Metformin inhibited progressive resistance training-induced lean mass gain but did not change the effect of weight loss from training. Metformin prevented decreases in type 1 fiber frequency. | Metformin inhibits gains in fat-free mass in response to concurrent aerobic and resistance training in subjects with prediabetes. Metformin may inhibit hypertrophy via mTORC1 inhibition. | [35] |
| Therakomen, V. et al. (2020) | Old (over 60 years, n = 330) male subjects | Development of sarcopenia is positively correlated with age, as is prefrailty and low physical activity. | The study supports previously understood risk factors for primary sarcopenia: age, prefrailty, physical activity, and nutritional status, but not sex. | [36] |
| Hester, G.M. et al. (2021) | Young (n = 15, age = 20.7 ± 2.2 years) and old (n = 15, age = 71.6 ± 3.9 years) male non-sarcopenic subjects | Peak torque was lower in the older group at all velocities compared to the young group. The whole-muscle cross-sectional area was smaller in old muscles. Type 1 fiber was larger and type 2 fiber was smaller in muscles in the older group. | Microbiopsy methods appear to be viable alternatives that are less intrusive but with similar results. The lack of association among neuromuscular junction deterioration, strength, and age-related muscle fiber atrophy may be due to the fact that samples were from a non-sarcopenic healthy elderly population. | [37] |
| Bres, E. et al. (2023) | Sarcopenic (n = 30) and non-sarcopenic (n = 22) old (over 70 years) subjects | Serum fibroblast growth factor (FGF) 19 was correlated with muscle ultrasound parameters of pennatation angle and muscle fiber length. FGF19 levels were not correlated with age, BMI, nutritional parameters, or tissue mass. | The association of FGF19 and the pennetation angle implies that a high-FGF19 environment promotes both the development of fast-twitch muscles as well as a negative association with balance and lower extremity strength, suggesting the role of FGF19 in muscle function and architecture. | [38] |
| Animal studies | ||||
| Lang, F. et al. (2018) | Manual denervation model of sarcopenia in adult C57BL/6J mice | Skeletal muscle exhibits varied protein changes after denervation, with opposing protein changes between type 1 and type 2a muscle fibers of Soleus during muscle atrophy. | Using a manual denervation method to study muscle atrophy, at 7 days post-denervation, this group showed complexities of response between different muscle fibers and tissues. | [39] |
| Lukjanenko, L. et al. (2020) | Young (9–13 weeks) and aged (20–25 months) C57BL/6J mice | Aging impaired fibro/adipogenic progenitor (FAP) functions with a failure to support muscle stem cells. Transcriptome analysis relieved the downregulated WNT1 inducible signaling pathway protein 1 (WISP1) gene comparing aged to young activated FAPs. | Aging damages functions of FAPs and their ability to support myogenesis, the regenerative capacity. WISP1 is a FAP-derived factor controlling muscle stem cell expansion and differentiation, and age-induced loss of WISP occurs due to a lack of FAP population. | [40] |
| Xu, H. et al. (2021) | Whole body CuZnSOD KO mice and muscle-specific mitochondrial targeted catalase (mMCAT) transgenic mice with C57BL/6J background. accelerated sarcopenia model in female C57BL/6J mice (n = 12) | An altered oxygen consumption rate and peroxide generation in CuZnSOD KO mice is reversed through mMCAT expression. Significant muscle loss and function is observed in CuZnSOD KO mice. Muscle fiber composition and diameter in CuZnSOD KO mice were decreased through mMCAT. | In an accelerated sarcopenia model, mMCAT is sufficient to prevent the majority of muscle atrophy and weaknessin CuZnSOD KO mice. The absence of CuZnSOD leads to a reduction in force and disruption of neuromuscular junction with increased mitochondrial ROS. Mitochondrial scavenging capacity is important for the prevention of the loss of innervation, muscle atrophy, and weakness. | [41] |
| Kim, K.H. et al. (2022) | Young (5 weeks) and aged (25 month) C57BL/6J female mice | Fecal microbiota transplantation of young mice improved grip strength, muscle fiber thickness, and other fitness markers. Young donor transplantation increased genes involved in cell differentiation, proliferation, and fatty acid synthesis in muscle. | A group of Bacteroidetes from the young-derived microbiota are discriminative in old mice, increasing gene expression in recipients’ muscles and skin. This supports that the age related changes in the microbiome can play a role with changes in aging muscles. | [42] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terrell, K.; Choi, S.; Choi, S. Calcium’s Role and Signaling in Aging Muscle, Cellular Senescence, and Mineral Interactions. Int. J. Mol. Sci. 2023, 24, 17034. https://doi.org/10.3390/ijms242317034
Terrell K, Choi S, Choi S. Calcium’s Role and Signaling in Aging Muscle, Cellular Senescence, and Mineral Interactions. International Journal of Molecular Sciences. 2023; 24(23):17034. https://doi.org/10.3390/ijms242317034
Chicago/Turabian StyleTerrell, Kristofer, Suyun Choi, and Sangyong Choi. 2023. "Calcium’s Role and Signaling in Aging Muscle, Cellular Senescence, and Mineral Interactions" International Journal of Molecular Sciences 24, no. 23: 17034. https://doi.org/10.3390/ijms242317034
APA StyleTerrell, K., Choi, S., & Choi, S. (2023). Calcium’s Role and Signaling in Aging Muscle, Cellular Senescence, and Mineral Interactions. International Journal of Molecular Sciences, 24(23), 17034. https://doi.org/10.3390/ijms242317034