
Alpha-Lipoic Acid Reduces Cell Growth, Inhibits Autophagy, and Counteracts Prostate Cancer Cell Migration and Invasion: Evidence from In Vitro Studies


 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
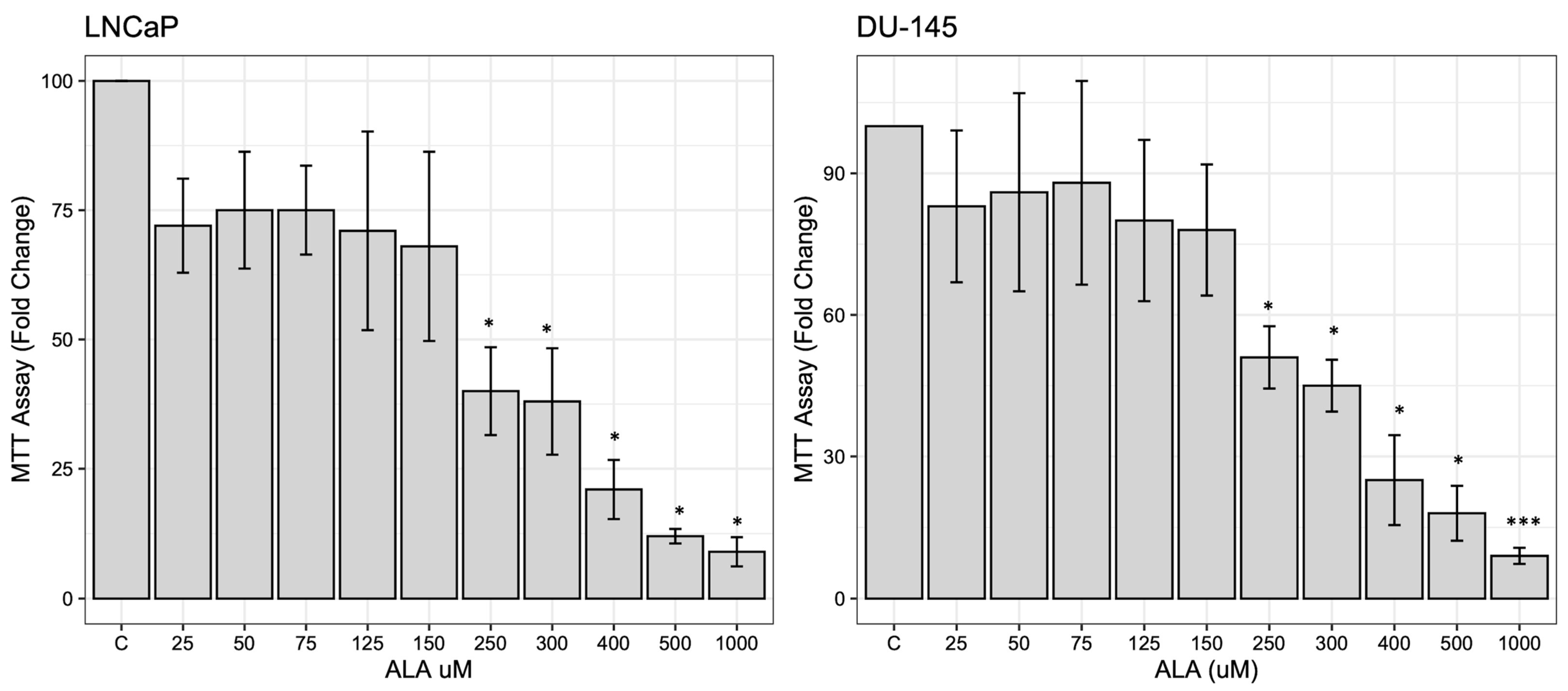
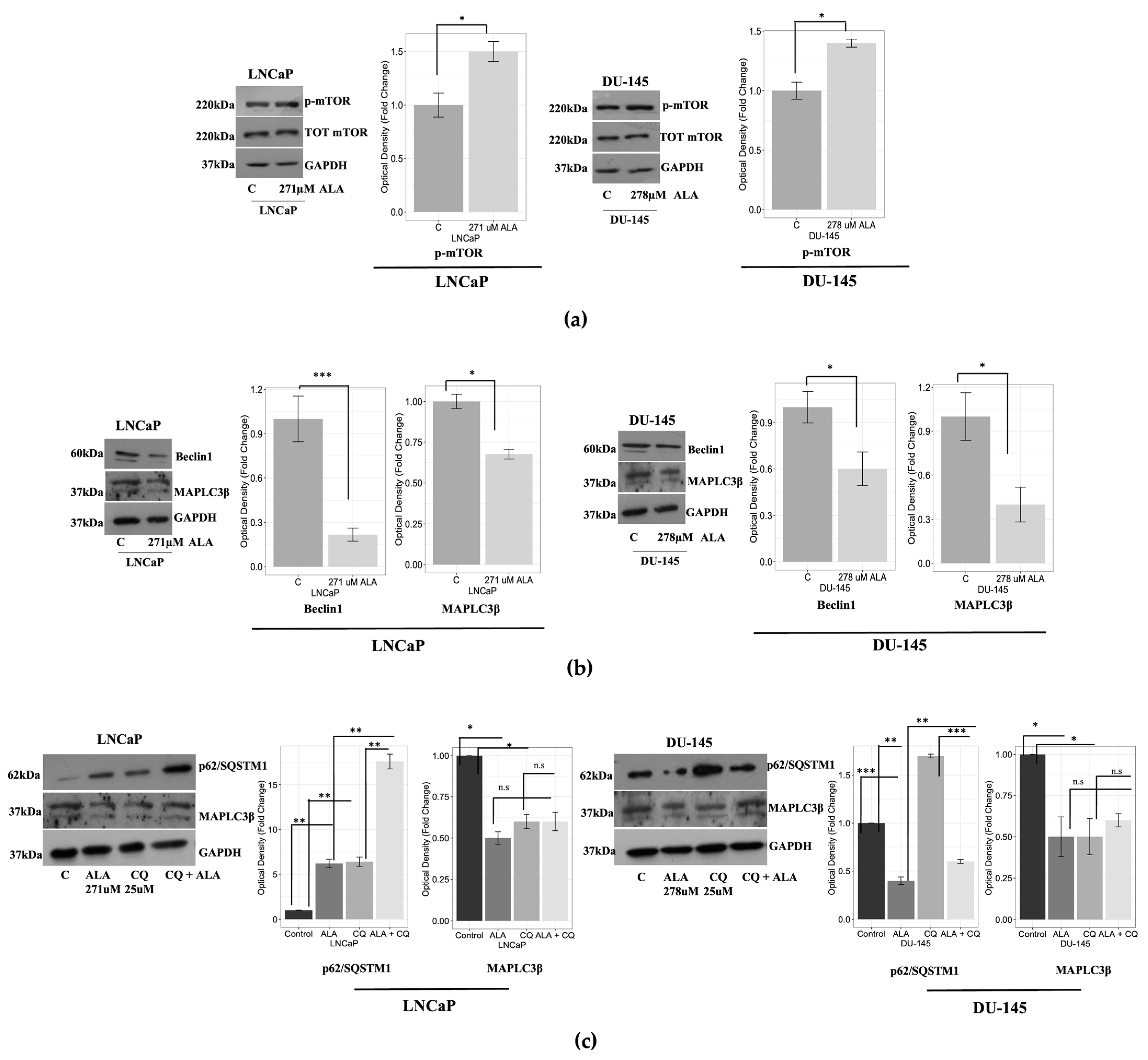
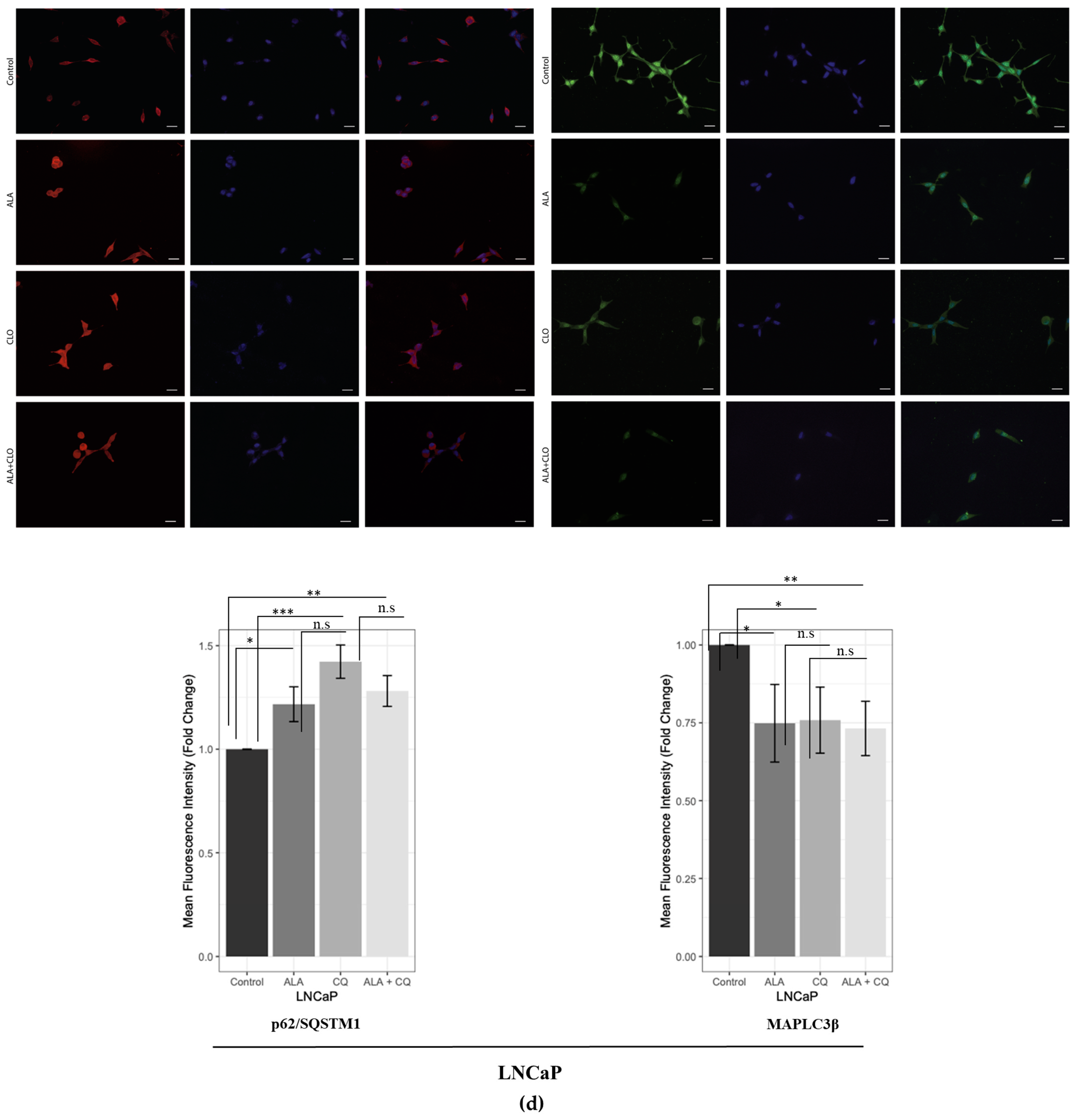
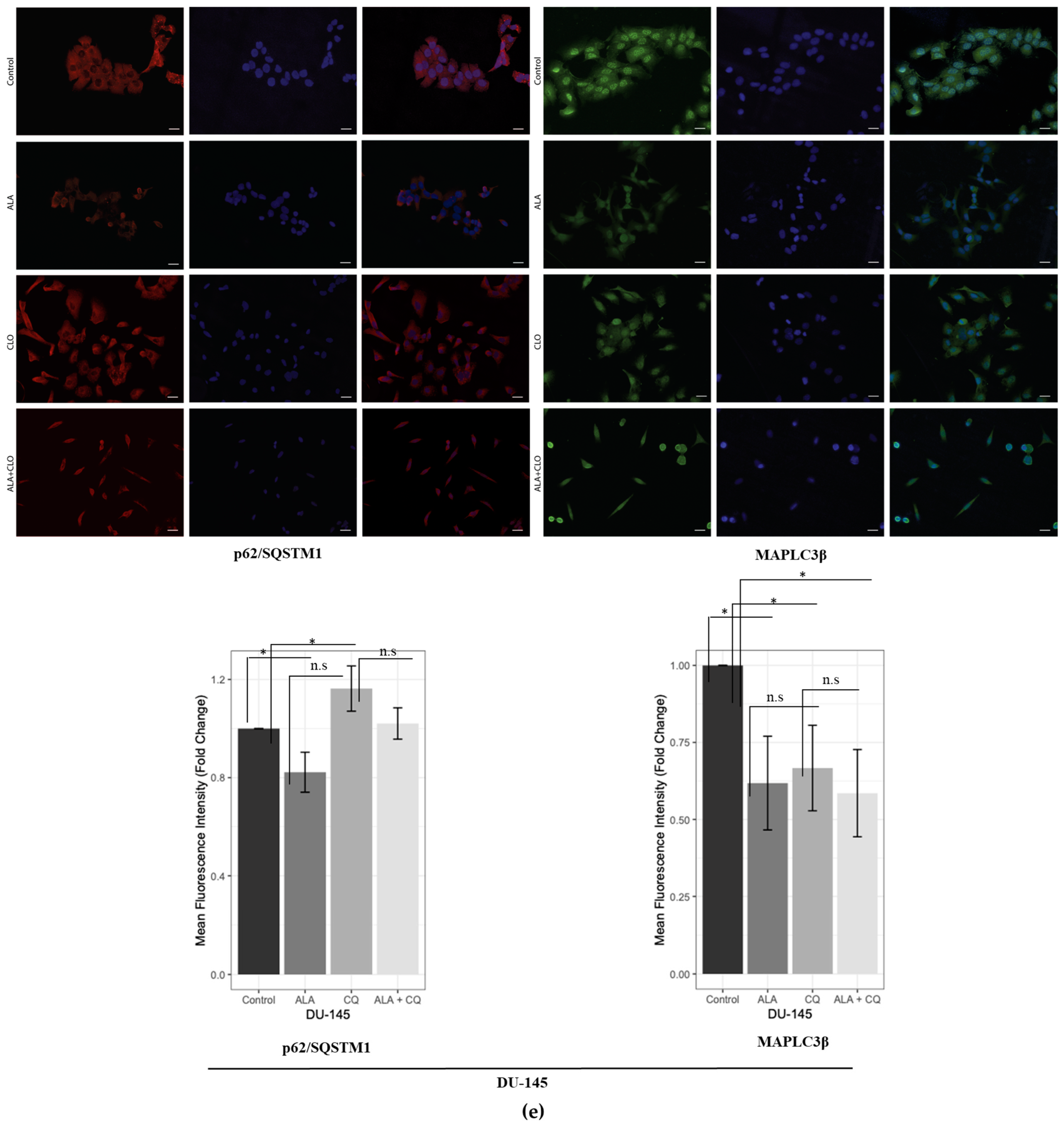
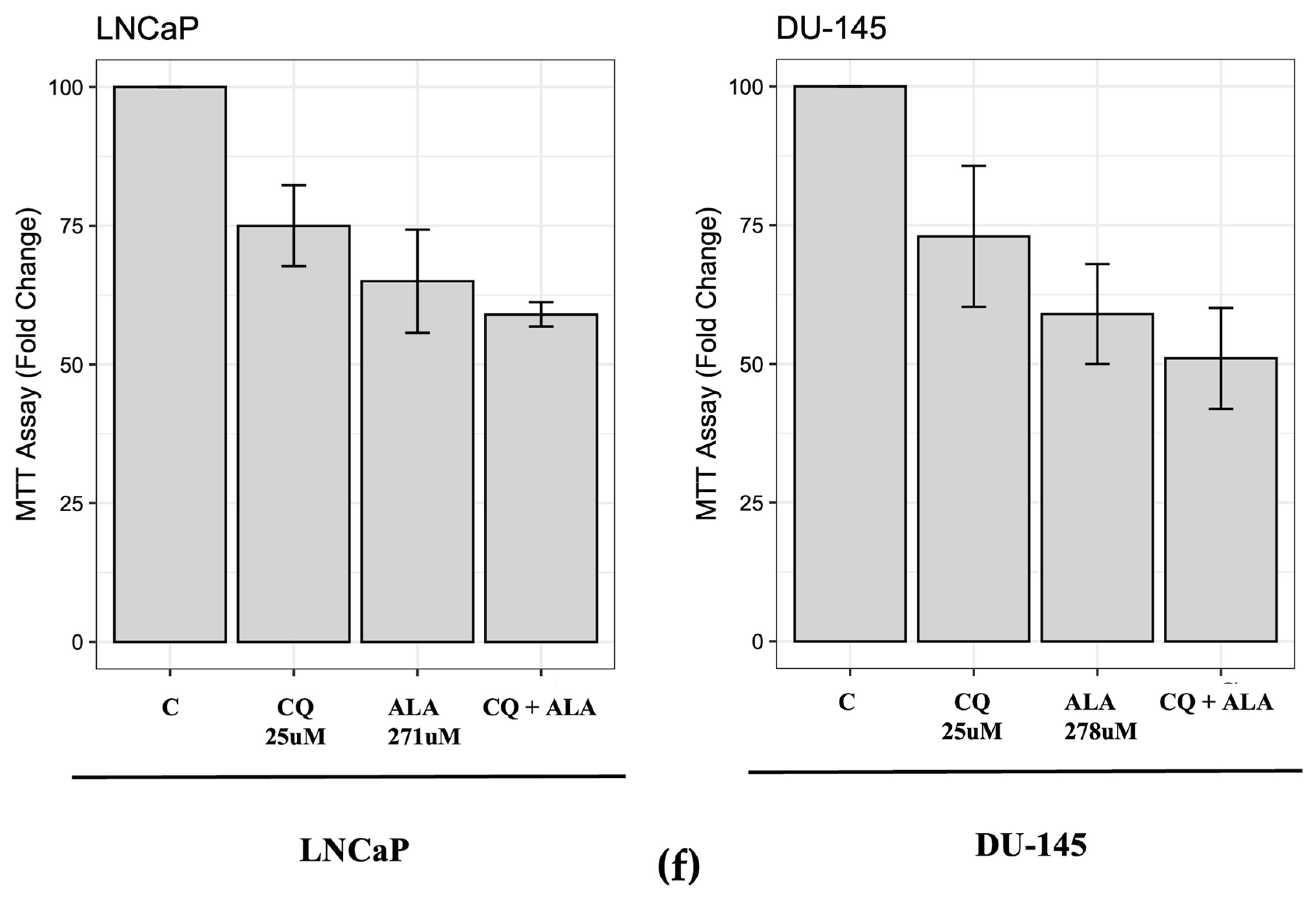
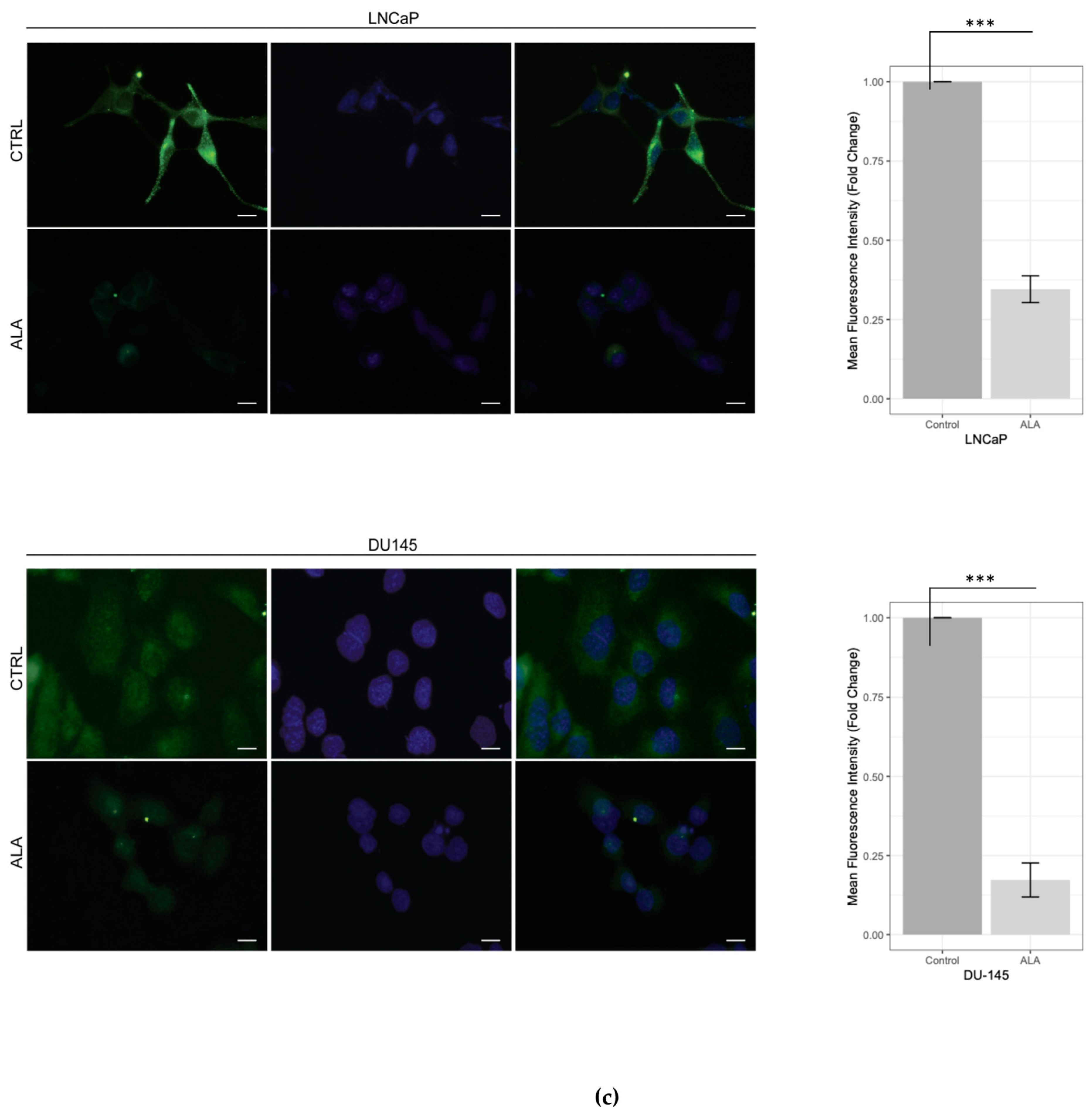
2.1. ALA Reduces Prostate Cancer Cells’ Viability by Inhibiting Autophagy
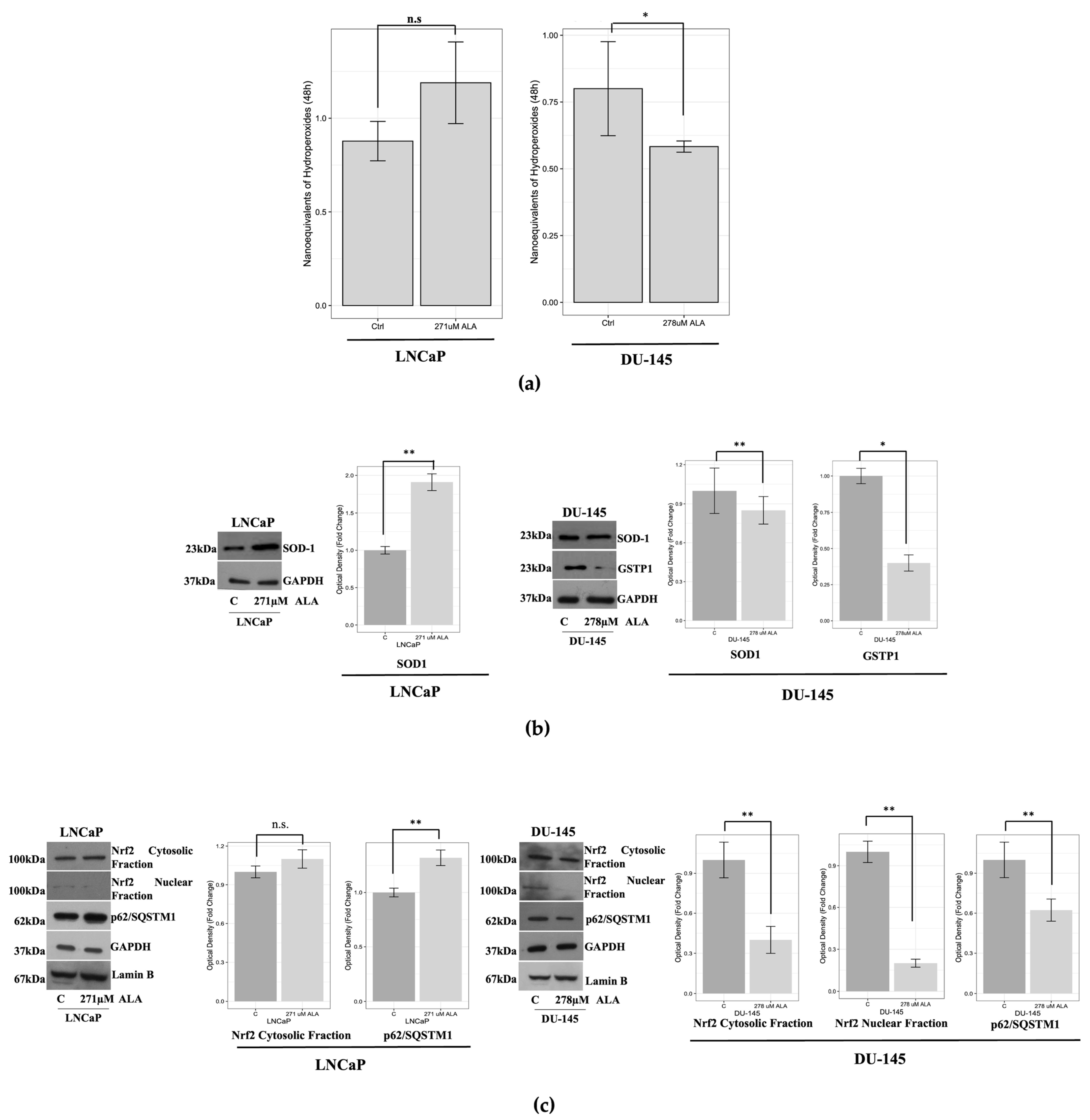
2.2. ALA Affects the Oxidative/Redox System Balance of Prostate Cancer Cells
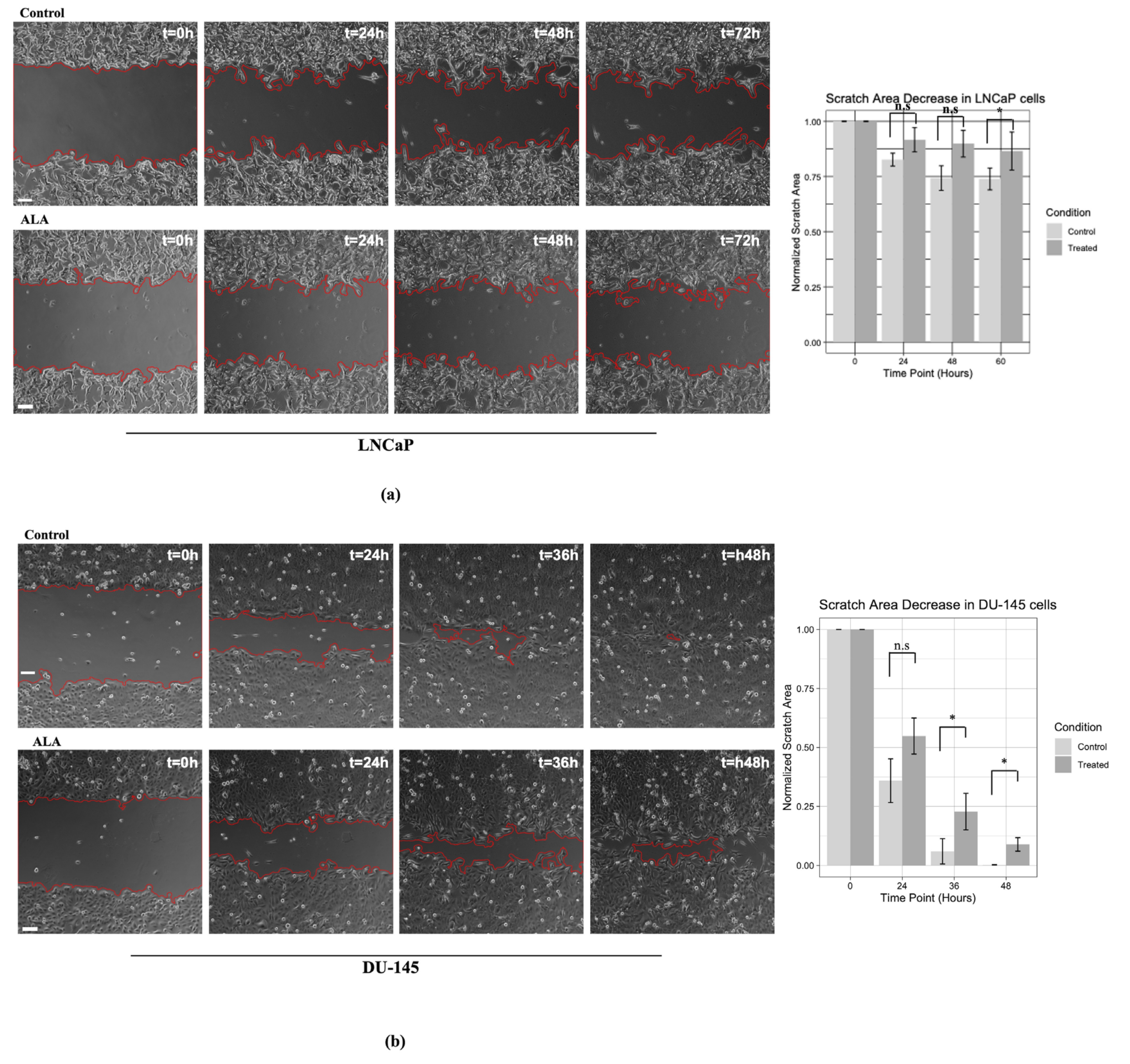
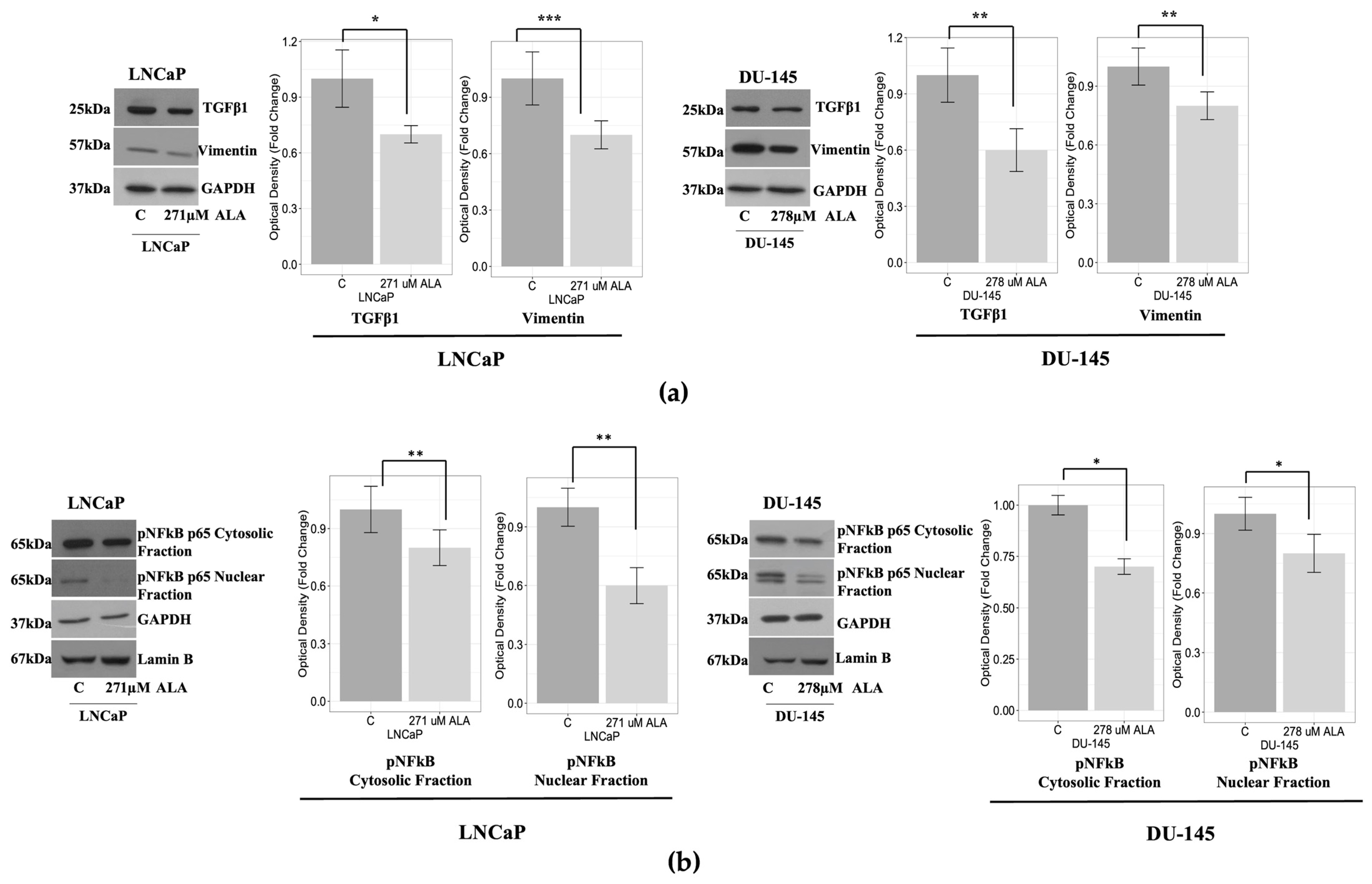
2.3. ALA Counteracts Prostate Cancer Cell Motility by Reducing Epithelial to Mesenchymal Transition (EMT)
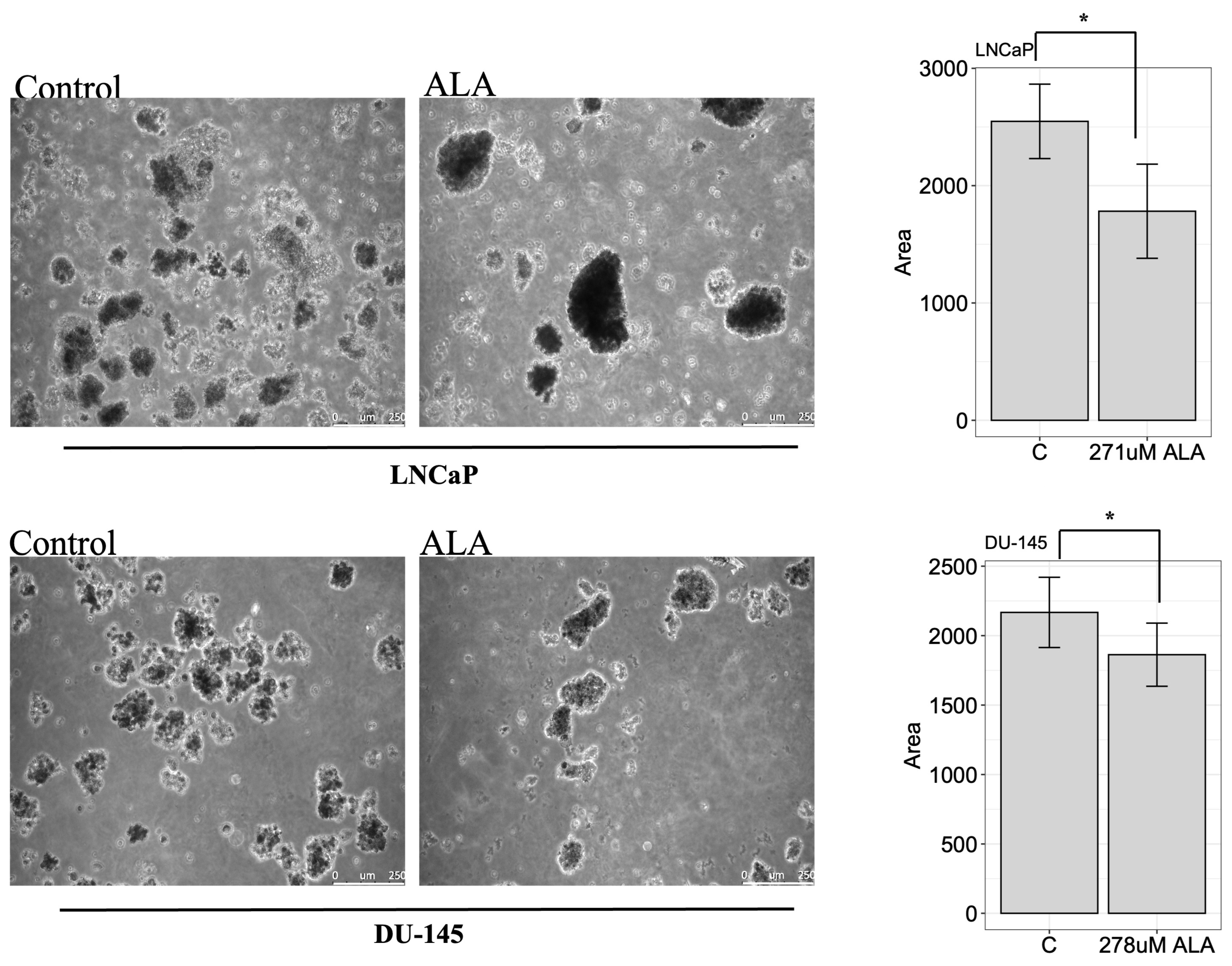
2.4. ALA Inhibits the Colony-Forming Ability of Prostate Cancer Cell Lines
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. Cell Viability Assay
4.3. Protein Extraction and Western Blotting Analysis
4.4. Wound-Healing Assay
4.5. Lipocell Assay
4.6. Immunofluorescence Assay
4.7. Soft Agar Assay
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Jemal, A.; Wender, R.C.; Gansler, T.; Ma, J.; Brawley, O.W. An Assessment of Progress in Cancer Control. CA Cancer J. Clin. 2018, 68, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate Cancer. Nat. Rev. Dis. Primers 2021, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; Shende, P. Dual Role of Autophagy for Advancements from Conventional to New Delivery Systems in Cancer. Biochim. Biophys. Acta Gen. Subj. 2023, 1867, 130430. [Google Scholar] [CrossRef] [PubMed]
- Ashrafizadeh, M.; Paskeh, M.D.A.; Mirzaei, S.; Gholami, M.H.; Zarrabi, A.; Hashemi, F.; Hushmandi, K.; Hashemi, M.; Nabavi, N.; Crea, F.; et al. Targeting Autophagy in Prostate Cancer: Preclinical and Clinical Evidence for Therapeutic Response. J. Exp. Clin. Cancer Res. 2022, 41, 105. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.H.; Abraham, R.T. The Autophagy Conundrum in Cancer: Influence of Tumorigenic Metabolic Reprogramming. Oncogene 2011, 30, 4687–4696. [Google Scholar] [CrossRef] [PubMed]
- Loizzo, D.; Pandolfo, S.D.; Rogers, D.; Cerrato, C.; di Meo, N.A.; Autorino, R.; Mirone, V.; Ferro, M.; Porta, C.; Stella, A.; et al. Novel Insights into Autophagy and Prostate Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2022, 23, 3826. [Google Scholar] [CrossRef] [PubMed]
- Chueh, K.-S.; Lu, J.-H.; Juan, T.-J.; Chuang, S.-M.; Juan, Y.-S. The Molecular Mechanism and Therapeutic Application of Autophagy for Urological Disease. Int. J. Mol. Sci. 2023, 24, 14887. [Google Scholar] [CrossRef]
- Ahmad, A.; Ginnebaugh, K.R.; Li, Y.; Padhye, S.B.; Sarkar, F.H. Molecular Targets of Naturopathy in Cancer Research: Bridge to Modern Medicine. Nutrients 2015, 7, 321–334. [Google Scholar] [CrossRef]
- Chopra, H.; Bibi, S.; Goyal, R.; Gautam, R.K.; Trivedi, R.; Upadhyay, T.K.; Mujahid, M.H.; Shah, M.A.; Haris, M.; Khot, K.B.; et al. Chemopreventive Potential of Dietary Nanonutraceuticals for Prostate Cancer: An Extensive Review. Front. Oncol. 2022, 12, 925379. [Google Scholar] [CrossRef]
- Dozio, E.; Ruscica, M.; Passafaro, L.; Dogliotti, G.; Steffani, L.; Marthyn, P.; Pagani, A.; Demartini, G.; Esposti, D.; Fraschini, F.; et al. The Natural Antioxidant Alpha-Lipoic Acid Induces P27(Kip1)-Dependent Cell Cycle Arrest and Apoptosis in MCF-7 Human Breast Cancer Cells. Eur. J. Pharmacol. 2010, 641, 29–34. [Google Scholar] [CrossRef]
- Tripathy, J.; Tripathy, A.; Thangaraju, M.; Suar, M.; Elangovan, S. α-Lipoic Acid Inhibits the Migration and Invasion of Breast Cancer Cells through Inhibition of TGFβ Signaling. Life Sci. 2018, 207, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Damnjanovic, I.; Kocic, G.; Najman, S.; Stojanovic, S.; Stojanovic, D.; Veljkovic, A.; Conic, I.; Langerholc, T.; Pesic, S. Chemopreventive Potential of Alpha Lipoic Acid in the Treatment of Colon and Cervix Cancer Cell Lines. Bratisl. Lek. Listy 2014, 115, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.-Y.; Liu, H.-L.; Stern, J.S.; Yu, P.-Z.; Liu, S.-L. Alpha-Lipoic Acid Induces Apoptosis in Hepatoma Cells via the PTEN/Akt Pathway. FEBS Lett. 2008, 582, 1667–1671. [Google Scholar] [CrossRef] [PubMed]
- Celik, A.; Bakar-Ates, F. Alpha-Lipoic Acid Induced Apoptosis of PC3 Prostate Cancer Cells through an Alteration on Mitochondrial Membrane Depolarization and MMP-9 mRNA Expression. Med. Oncol. 2023, 40, 244. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, U.A. Augmentation of Fluvastatin Cytotoxicity Against Prostate Carcinoma PC3 Cell Line Utilizing Alpha Lipoic-Ellagic Acid Nanostructured Lipid Carrier Formula. AAPS PharmSciTech 2018, 19, 3454–3461. [Google Scholar] [CrossRef] [PubMed]
- Alhakamy, N.A.; Al-Rabia, M.W.; Asfour, H.Z.; Alshehri, S.; Alharbi, W.S.; Halawani, A.; Alamoudi, A.J.; Noor, A.O.; Bannan, D.F.; Fahmy, U.A.; et al. 2-Methoxy-Estradiol Loaded Alpha Lipoic Acid Nanoparticles Augment Cytotoxicity in MCF-7 Breast Cancer Cells. Dose Response 2021, 19, 15593258211055023. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, B.; Rajput, S.; Raza, S.; Rajak, S.; Tewari, A.; Gupta, P.; Upadhyay, A.; Chattopadhyay, N.; Sinha, R.A. Lipoic Acid Blocks Autophagic Flux and Impairs Cellular Bioenergetics in Breast Cancer and Reduces Stemness. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166455. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Kim, J.H.; Jang, S.J.; Yun, J.W.; Kang, K.M.; Jeong, H.; Ha, I.B.; Jeong, B.K. Synergistic Tumoricidal Effects of Alpha-Lipoic Acid and Radiotherapy on Human Breast Cancer Cells via HMGB1. Cancer Res. Treat. 2021, 53, 685–694. [Google Scholar] [CrossRef]
- Farhat, D.; Ghayad, S.E.; Icard, P.; Le Romancer, M.; Hussein, N.; Lincet, H. Lipoic Acid-Induced Oxidative Stress Abrogates IGF-1R Maturation by Inhibiting the CREB/Furin Axis in Breast Cancer Cell Lines. Oncogene 2020, 39, 3604–3610. [Google Scholar] [CrossRef]
- Izadi, A.; Soukhtanloo, M.; Mirzavi, F.; Jalili-Nik, M.; Sadeghi, A. Alpha-Lipoic Acid, Auraptene, and Particularly Their Combination Prevent the Metastasis of U87 Human Glioblastoma Cells. Evid. Based Complement. Alternat. Med. 2023, 2023, 8618575. [Google Scholar] [CrossRef]
- Jeon, M.J.; Kim, W.G.; Lim, S.; Choi, H.-J.; Sim, S.; Kim, T.Y.; Shong, Y.K.; Kim, W.B. Alpha Lipoic Acid Inhibits Proliferation and Epithelial Mesenchymal Transition of Thyroid Cancer Cells. Mol. Cell. Endocrinol. 2016, 419, 113–123. [Google Scholar] [CrossRef]
- Kafara, P.; Icard, P.; Guillamin, M.; Schwartz, L.; Lincet, H. Lipoic Acid Decreases Mcl-1, Bcl-xL and up Regulates Bim on Ovarian Carcinoma Cells Leading to Cell Death. J. Ovarian Res. 2015, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Kuban-Jankowska, A.; Gorska-Ponikowska, M.; Wozniak, M. Lipoic Acid Decreases the Viability of Breast Cancer Cells and Activity of PTP1B and SHP2. Anticancer Res. 2017, 37, 2893–2898. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, U.; Nickel, A.; Daniel, H. Alpha-Lipoic Acid Induces Apoptosis in Human Colon Cancer Cells by Increasing Mitochondrial Respiration with a Concomitant O2-*-Generation. Apoptosis 2005, 10, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Yoo, T.-H.; Lee, J.-H.; Chun, H.-S.; Chi, S.-G. α-Lipoic Acid Prevents P53 Degradation in Colon Cancer Cells by Blocking NF-κB Induction of RPS6KA4. Anticancer Drugs 2013, 24, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Ren, Y.; Yue, Q.; Ding, Z.; Wang, K.; Zheng, T.; Chen, G.; Chen, X.; Li, M.; Fan, L. α-Lipoic Acid Targeting PDK1/NRF2 Axis Contributes to the Apoptosis Effect of Lung Cancer Cells. Oxid. Med. Cell Longev. 2021, 2021, 6633419. [Google Scholar] [CrossRef]
- Peng, P.; Zhang, X.; Qi, T.; Cheng, H.; Kong, Q.; Liu, L.; Cao, X.; Ding, Z. Alpha-Lipoic Acid Inhibits Lung Cancer Growth via mTOR-Mediated Autophagy Inhibition. FEBS Open Bio 2020, 10, 607–618. [Google Scholar] [CrossRef]
- Dörsam, B.; Fahrer, J. The Disulfide Compound α-Lipoic Acid and Its Derivatives: A Novel Class of Anticancer Agents Targeting Mitochondria. Cancer Lett 2016, 371, 12–19. [Google Scholar] [CrossRef]
- Lin, X.; Tascilar, M.; Lee, W.H.; Vles, W.J.; Lee, B.H.; Veeraswamy, R.; Asgari, K.; Freije, D.; van Rees, B.; Gage, W.R.; et al. GSTP1 CpG Island Hypermethylation Is Responsible for the Absence of GSTP1 Expression in Human Prostate Cancer Cells. Am. J. Pathol. 2001, 159, 1815–1826. [Google Scholar] [CrossRef]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. P62 Links Autophagy and Nrf2 Signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef]
- Deng, Z.; Lim, J.; Wang, Q.; Purtell, K.; Wu, S.; Palomo, G.M.; Tan, H.; Manfredi, G.; Zhao, Y.; Peng, J.; et al. ALS-FTLD-Linked Mutations of SQSTM1/P62 Disrupt Selective Autophagy and NFE2L2/NRF2 Anti-Oxidative Stress Pathway. Autophagy 2020, 16, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Arnedo, A.; Torres Figueroa, F.; Clavijo, C.; Arbeláez, P.; Cruz, J.C.; Muñoz-Camargo, C. An Image J Plugin for the High Throughput Image Analysis of in Vitro Scratch Wound Healing Assays. PLoS ONE 2020, 15, e0232565. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Blando, J.; Perez, C.J.; Wang, H.; Benavides, F.J.; Kazanietz, M.G. Activation of Nuclear Factor κB (NF-κB) in Prostate Cancer Is Mediated by Protein Kinase C Epsilon (PKCepsilon). J. Biol. Chem. 2012, 287, 37570–37582. [Google Scholar] [CrossRef] [PubMed]
- Staal, J.; Beyaert, R. Inflammation and NF-κB Signaling in Prostate Cancer: Mechanisms and Clinical Implications. Cells 2018, 7, 122. [Google Scholar] [CrossRef]
- Smith, A.R.; Shenvi, S.V.; Widlansky, M.; Suh, J.H.; Hagen, T.M. Lipoic Acid as a Potential Therapy for Chronic Diseases Associated with Oxidative Stress. Curr. Med. Chem. 2004, 11, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Kamt, S.F.; Liu, J.; Yan, L.-J. Renal-Protective Roles of Lipoic Acid in Kidney Disease. Nutrients 2023, 15, 1732. [Google Scholar] [CrossRef]
- Venigalla, M.; Sonego, S.; Gyengesi, E.; Sharman, M.J.; Münch, G. Novel Promising Therapeutics against Chronic Neuroinflammation and Neurodegeneration in Alzheimer’s Disease. Neurochem. Int. 2016, 95, 63–74. [Google Scholar] [CrossRef]
- Moungjaroen, J.; Nimmannit, U.; Callery, P.S.; Wang, L.; Azad, N.; Lipipun, V.; Chanvorachote, P.; Rojanasakul, Y. Reactive Oxygen Species Mediate Caspase Activation and Apoptosis Induced by Lipoic Acid in Human Lung Epithelial Cancer Cells through Bcl-2 down-Regulation. J. Pharmacol. Exp. Ther. 2006, 319, 1062–1069. [Google Scholar] [CrossRef]
- Attia, M.; Essa, E.A.; Zaki, R.M.; Elkordy, A.A. An Overview of the Antioxidant Effects of Ascorbic Acid and Alpha Lipoic Acid (in Liposomal Forms) as Adjuvant in Cancer Treatment. Antioxidants 2020, 9, 359. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Sivridis, E.; Mendrinos, S.; Koutsopoulos, A.V.; Koukourakis, M.I. Autophagy Proteins in Prostate Cancer: Relation with Anaerobic Metabolism and Gleason Score. Urol. Oncol. 2014, 32, 39.e11–39.e18. [Google Scholar] [CrossRef]
- Farrow, J.M.; Yang, J.C.; Evans, C.P. Autophagy as a Modulator and Target in Prostate Cancer. Nat. Rev. Urol. 2014, 11, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The Role of Autophagy in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Moldogazieva, N.T.; Lutsenko, S.V.; Terentiev, A.A. Reactive Oxygen and Nitrogen Species-Induced Protein Modifications: Implication in Carcinogenesis and Anticancer Therapy. Cancer Res. 2018, 78, 6040–6047. [Google Scholar] [CrossRef] [PubMed]
- Mondal, D.; Narwani, D.; Notta, S.; Ghaffar, D.; Mardhekar, N.; Quadri, S.S.A. Oxidative Stress and Redox Signaling in CRPC Progression: Therapeutic Potential of Clinically-Tested Nrf2-Activators. Cancer Drug Resist. 2021, 4, 96–124. [Google Scholar] [CrossRef] [PubMed]
- Wible, D.J.; Bratton, S.B. Reciprocity in ROS and Autophagic Signaling. Curr. Opin. Toxicol. 2018, 7, 28–36. [Google Scholar] [CrossRef]
- Leinonen, H.M.; Kansanen, E.; Pölönen, P.; Heinäniemi, M.; Levonen, A.-L. Role of the Keap1-Nrf2 Pathway in Cancer. Adv. Cancer Res. 2014, 122, 281–320. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Hsu, W.-L.; Wang, C.-M.; Yao, C.-L.; Chen, S.-C.; Nien, C.-Y.; Sun, Y.-H.; Tseng, T.-Y.; Luo, Y.-H. Blockage of Nrf2 and Autophagy by L-Selenocystine Induces Selective Death in Nrf2-Addicted Colorectal Cancer Cells through P62-Keap-1-Nrf2 Axis. Cell Death Dis. 2022, 13, 1060. [Google Scholar] [CrossRef]
- Tossetta, G.; Fantone, S.; Marzioni, D.; Mazzucchelli, R. Role of Natural and Synthetic Compounds in Modulating NRF2/KEAP1 Signaling Pathway in Prostate Cancer. Cancers 2023, 15, 3037. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Lee, H.S.; Na, M.H.; Kim, W.K. Alpha-Lipoic Acid Reduces Matrix Metalloproteinase Activity in MDA-MB-231 Human Breast Cancer Cells. Nutr. Res. 2010, 30, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.K.; Ray, A.K.; Mishra, S.K.; Bishen, S.M.; Mishra, H.; Khurana, A. Molecular and Therapeutic Insights of Alpha-Lipoic Acid as a Potential Molecule for Disease Prevention. Rev. Bras. Farmacogn. 2023, 33, 272–287. [Google Scholar] [CrossRef] [PubMed]
- Lupinacci, S.; Perri, A.; Toteda, G.; Vizza, D.; Lofaro, D.; Pontrelli, P.; Stallone, G.; Divella, C.; Tessari, G.; La Russa, A.; et al. Rapamycin Promotes Autophagy Cell Death of Kaposi’s Sarcoma Cells through P75NTR Activation. Exp. Dermatol. 2022, 31, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Bossio, S.; Perri, A.; Malivindi, R.; Giordano, F.; Rago, V.; Mirabelli, M.; Salatino, A.; Brunetti, A.; Greco, E.A.; Aversa, A. Oleuropein Counteracts Both the Proliferation and Migration of Intra- and Extragonadal Seminoma Cells. Nutrients 2022, 14, 2323. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bossio, S.; Perri, A.; Gallo, R.; De Bartolo, A.; Rago, V.; La Russa, D.; Di Dio, M.; La Vignera, S.; Calogero, A.E.; Vitale, G.; et al. Alpha-Lipoic Acid Reduces Cell Growth, Inhibits Autophagy, and Counteracts Prostate Cancer Cell Migration and Invasion: Evidence from In Vitro Studies. Int. J. Mol. Sci. 2023, 24, 17111. https://doi.org/10.3390/ijms242317111
Bossio S, Perri A, Gallo R, De Bartolo A, Rago V, La Russa D, Di Dio M, La Vignera S, Calogero AE, Vitale G, et al. Alpha-Lipoic Acid Reduces Cell Growth, Inhibits Autophagy, and Counteracts Prostate Cancer Cell Migration and Invasion: Evidence from In Vitro Studies. International Journal of Molecular Sciences. 2023; 24(23):17111. https://doi.org/10.3390/ijms242317111
Chicago/Turabian StyleBossio, Sabrina, Anna Perri, Raffaella Gallo, Anna De Bartolo, Vittoria Rago, Daniele La Russa, Michele Di Dio, Sandro La Vignera, Aldo E. Calogero, Giovanni Vitale, and et al. 2023. "Alpha-Lipoic Acid Reduces Cell Growth, Inhibits Autophagy, and Counteracts Prostate Cancer Cell Migration and Invasion: Evidence from In Vitro Studies" International Journal of Molecular Sciences 24, no. 23: 17111. https://doi.org/10.3390/ijms242317111
APA StyleBossio, S., Perri, A., Gallo, R., De Bartolo, A., Rago, V., La Russa, D., Di Dio, M., La Vignera, S., Calogero, A. E., Vitale, G., & Aversa, A. (2023). Alpha-Lipoic Acid Reduces Cell Growth, Inhibits Autophagy, and Counteracts Prostate Cancer Cell Migration and Invasion: Evidence from In Vitro Studies. International Journal of Molecular Sciences, 24(23), 17111. https://doi.org/10.3390/ijms242317111