

Recent Studies of Artificial Intelligence on In Silico Drug Distribution Prediction



Abstract
:1. Introduction

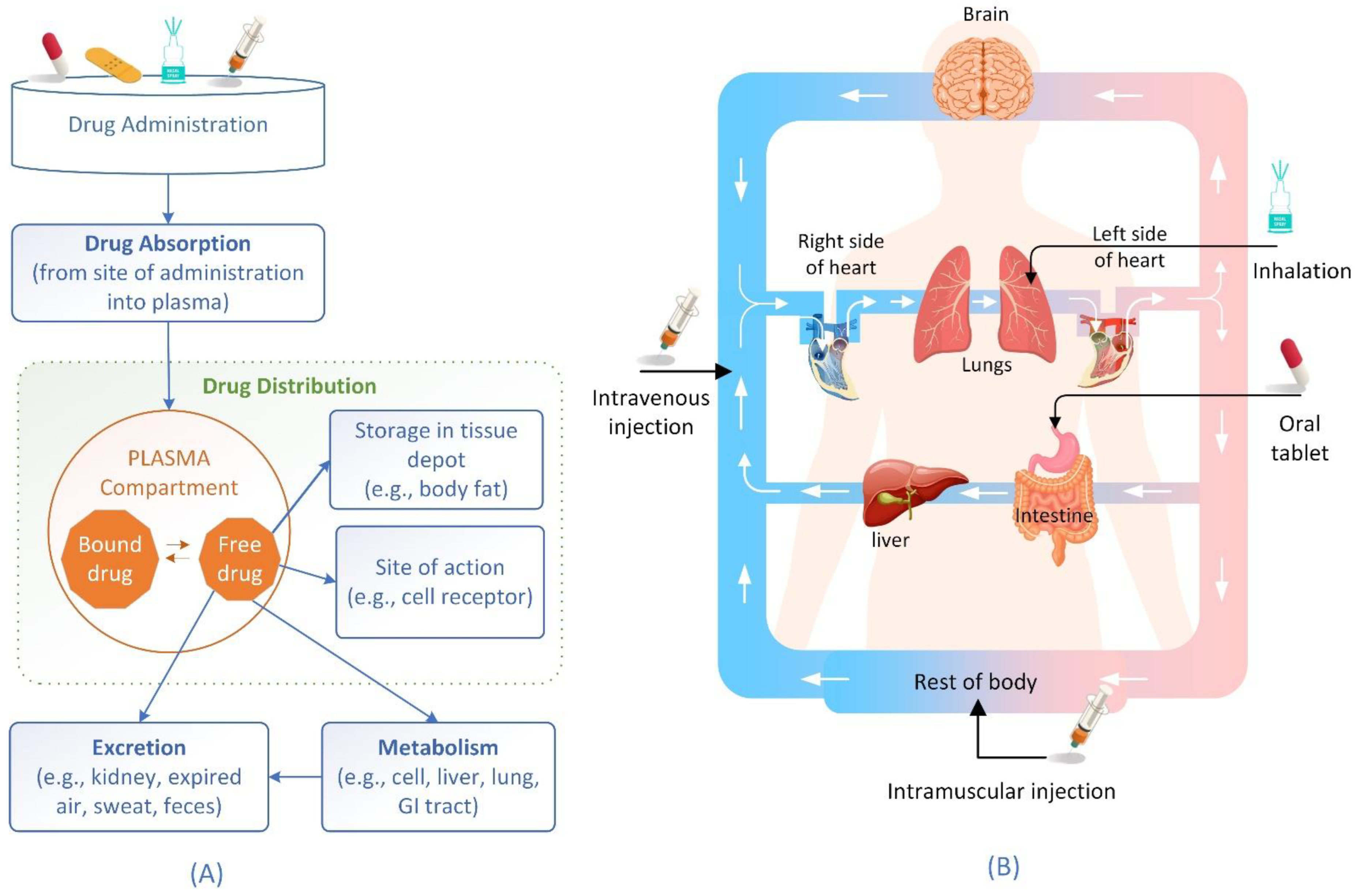
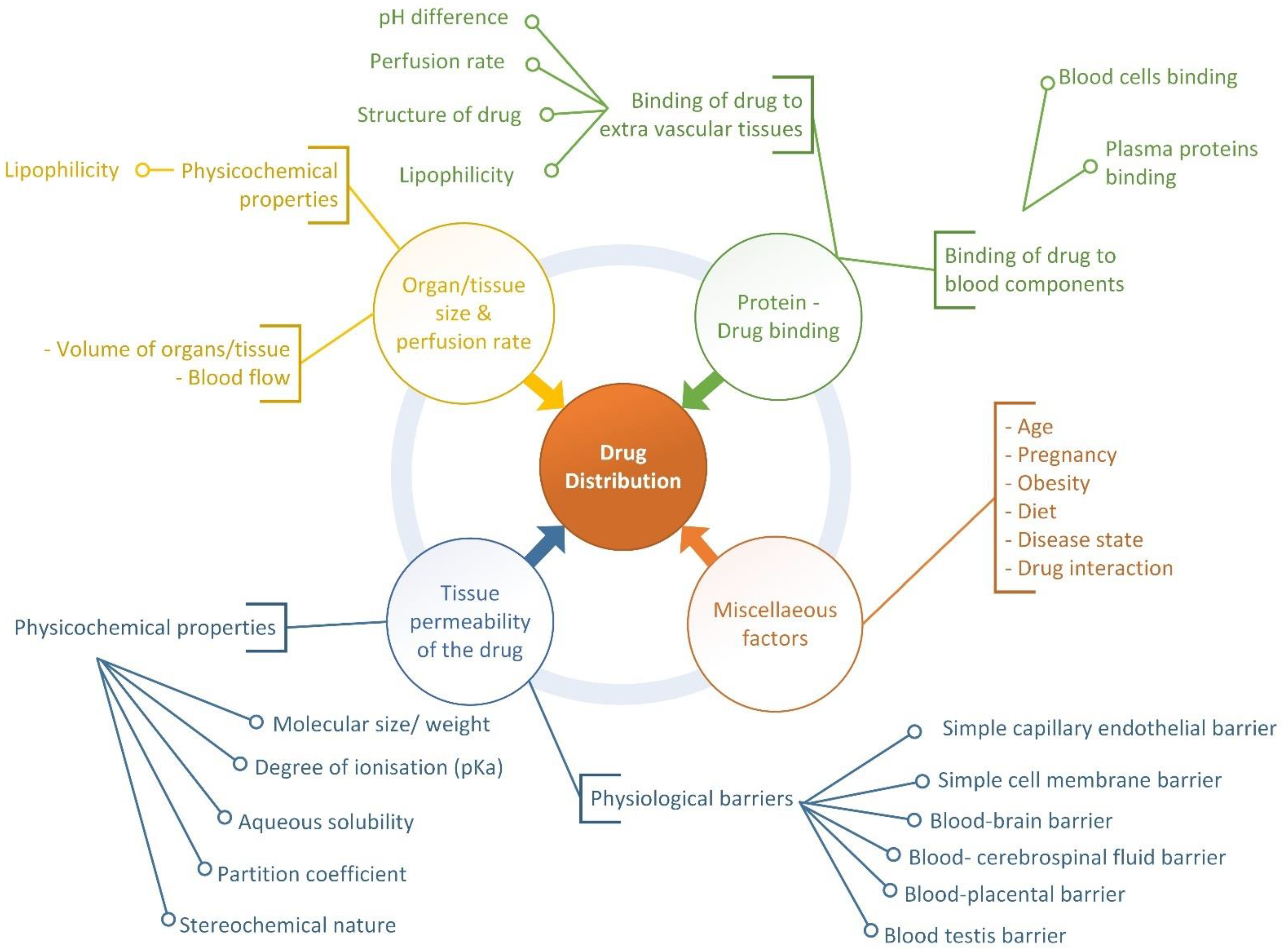
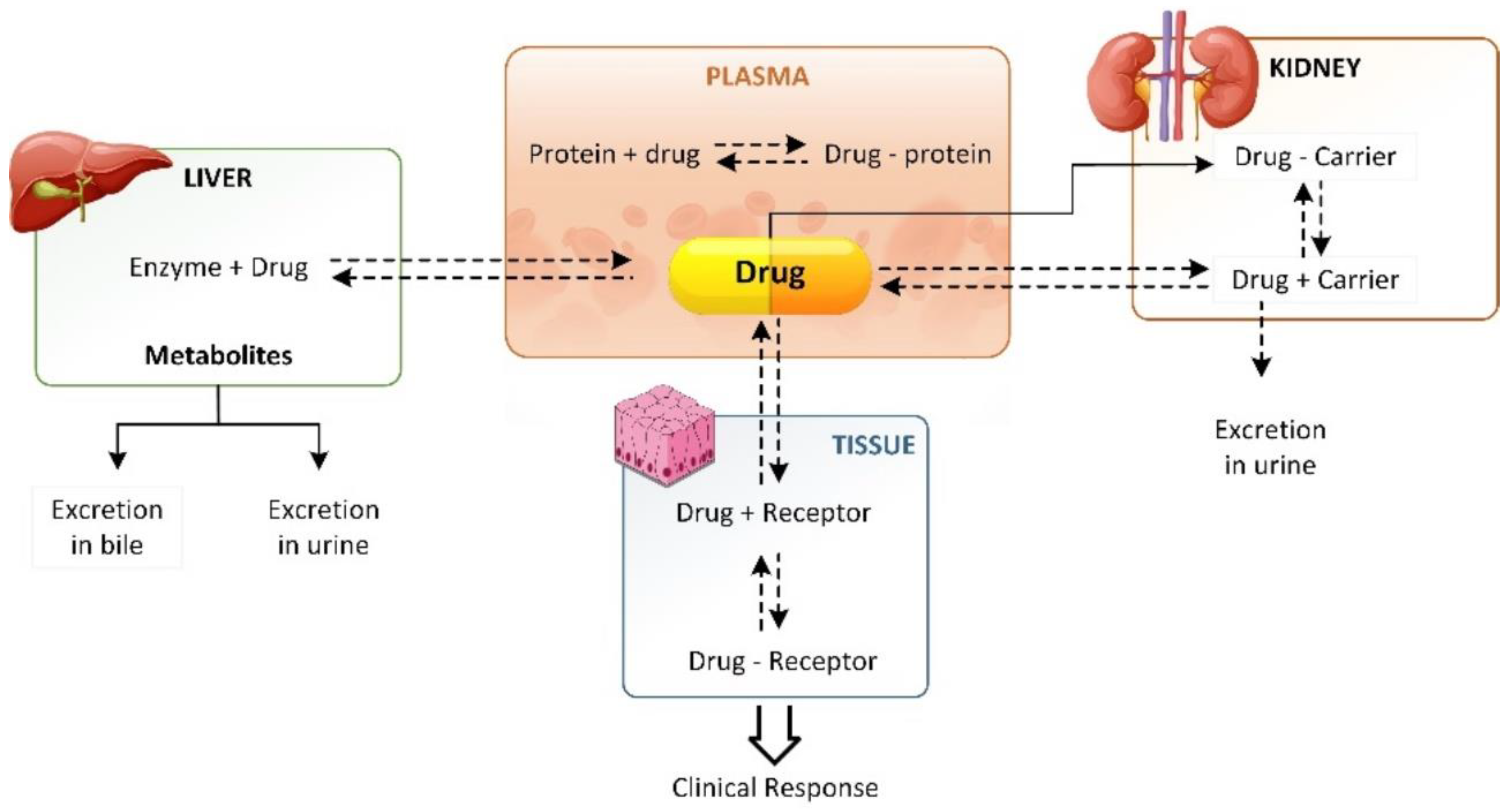
2. Drug Distribution Process and Factors Affecting the Process
3. Performance Metrics to Evaluate and Compare AI-Based Distribution Prediction Methods
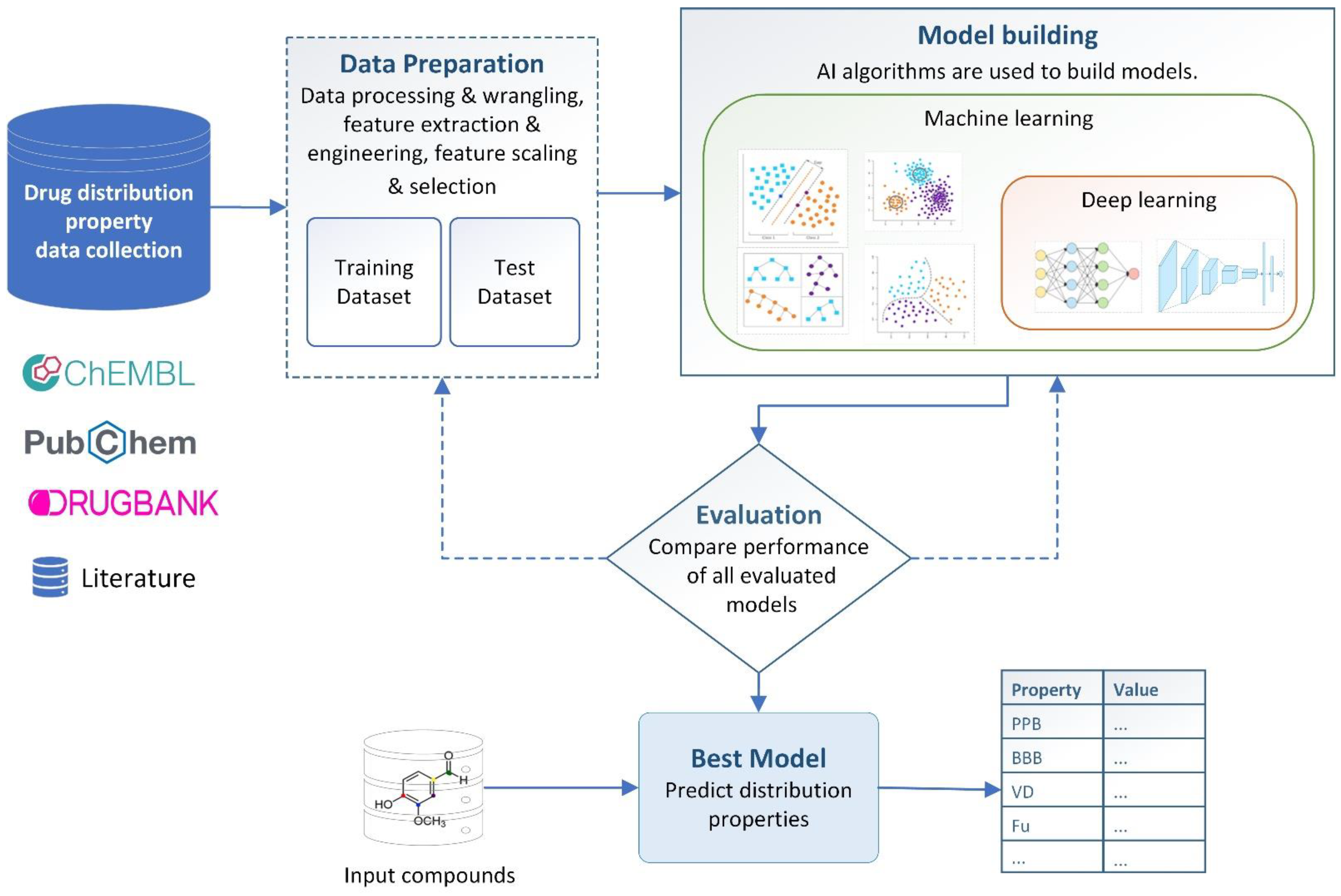
4. AI-Based Distribution Property Prediction
4.1. Blood–Brain Barrier Permeability Prediction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Data Sources | No. of Compounds | Performance | Ref. |
|---|---|---|---|---|
| SVM, RF, XGB | [32,33,34,35] | 1970 | AUC = 0.957, ACC = 0.910 | [15] |
| LightGBM | [17,18,32,36,37,38,39,40] | 7162 | AUC = 0.94, ACC = 0.89 | [16] |
| Mixed DL: Multilayer Perceptron (MLP), CNN | [16] | 7162 | AUC = 0.96, ACC = 0.92 | [20] |
| RF, MLP, Sequential Minimal Optimization | [33,36,41] | 2313 | ACC = 0.88 | [17] |
| Logistic Regression, DT, RF, GB | [42] | 968 | AUC = 0.78, ACC = 0.817 | [18] |
| SVM, k-NN, DT, DNN | SIDER [40,43] | 1000 | ACC = 0.97, AUC = 0.98 | [19] |
| Multichannel Substructure-Graph Gated Recurrent Unit Architecture | [37] | 2053 | AUC = 0.753 | [23] |
| CNN | [37] | 2039 | AUC = 0.694 | [24] |
| CNN | [35,44] | 2254 | ACC = 0.755, AUC = 0.784 | [25] |
| CNN | [15,16,18,36,37,40] | 7224 | ACC = 0.74, AUC = 0.83 | [45] |
| ANN | [46,47] | 300 | RMSE = 0.171 | [26] |
| SVM and GCNN | [48] | 940 | ACC = 0.96, F1 score = 0.95 | [27] |
| Fully Connected Neural Network, CNN | [37,40] | 2264 | AUC = 0.995 | [28] |
| RNN | [32] | 2350 | ACC = 0.965, AUC = 0.98 | [29] |
| DNN | [32] | 2350 | ACC = 0.962, AUC = 0.968 | [30] |
| XGraphBoost | [38,49] | 2039 | AUC = 0.932 | [31] |
4.2. Plasma Protein Binding Prediction
| Method | Data Sources | No. of Compounds | Performance | Ref. |
|---|---|---|---|---|
| RF | [57] | 670 | R2 = 0.74, RMSE = 0.12 | [52] |
| RF | [53,58] | 8103 | ACC = 0.84, AUC = 0.92 | [22] |
| SVM | AstraZeneca in-house | 100,550 | RMSE = 0.444, R2 = 0.721 | [59] |
| k-NN, SVR, RF, BT, and GER | [55,60,61,62,63,64,65,66,67,68,69], CHEMBL and DrugBank | 6741 | MAE = 0.076 | [53] |
| GCNN | [62] | 1209 | R2= 0.668, RMSE = 0.191 | [21] |
| Multitask graph attention framework | ChEMBL, PubChem, OCHEM, Literature | 4712 | R2 = 0.733, RMSE = 0.135 | [50] |
| GNN | [61,62] | 1744 | R2 = 0.747 | [70] |
| MolGIN method | [55] | 1830 | R2 = 0.738 | [54] |
| GCNN, GAT | ChEMBL, PubChem, DrugBank, Literature | 1830 | R2 = 0.563, RMSE = 0.211 | [71] |
| Attentive fingerprint algorithm (GNN) | [56] | 3921 | R2 = 0.841, RMSE = 0.112 | [56] |
4.3. Fraction Unbound in Plasma Prediction
| Method | Data Sources | No. of Compounds | Performance | Ref. |
|---|---|---|---|---|
| SVM, RF, GB, XGB | [76] | 1352 | R2 = 0.82, RMSE = 0.291 | [75] |
| AutoML Framework | ChEMBL v.27 | 5471 | R2 = 0.85, RMSE = 8.44 | [77] |
| QSAR/Partial Least Squares (PLS) model | [69,80] | 599 | Q2 = 0.69 | [81] |
| DNNs | ADMET assays | 9730 | RMSE = 0.086 | [78] |
| PotentialNet GCNNs | ADMET assays | 17,850 | R2 = 0.919 | [79] |
4.4. Volume of Distribution Prediction
| Method | Data Sources | No. of Compounds | Performance | Ref. |
|---|---|---|---|---|
| RF | [89] | 1303 | GMFE = 2.15 % < 2-fold = 54 % < 3-fold = 73 | [86] |
| RF | AstraZeneca in-house | 1440 | RMSE = 0.371, R2 = 0.67 | [59] |
| SVM, RF, GB machine, XGB | [76] | 1352 | R2 = 0.87, RMSE = 0.208 | [75] |
| PLSANN, RF | ChEMBL [76,90] | 1442 | R2 = 0.61, RMSE = 0.41 | [87] |
| PLS regression, SVM, ANN, RF, k-NN, multitask learning feed-forward neural network, DeepPharm | Drugbank | 412 | ACC = 0.63, MAE = 0.174 | [88] |
| PotentialNet GCNNs | ADMET assays | 63,305 | R2 = 0.525 | [79] |
5. Public AI-Based ADMET Prediction Tools
6. Data Sources for Distribution Prediction Research
7. Challenges for AI-Based Distribution Prediction Researcher
8. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated research and development investment needed to bring a new medicine to market, 2009–2018. JAMA 2020, 323, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Peng, J.; Ma, J. Next Decade’s AI-Based Drug Development Features Tight Integration of Data and Computation. Health Data Sci. 2022, 2022, 9816939. [Google Scholar] [CrossRef]
- Hsiao, Y.; Su, B.H.; Tseng, Y.J. Current development of integrated web servers for preclinical safety and pharmacokinetics assessments in drug development. Brief. Bioinform. 2021, 22, bbaa160. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef] [PubMed]
- Ksir, C.J.; Carl, L.; Hart, D. Drugs, Society, and Human Behavior; McGraw-Hill Education: New York, NY, USA, 2017. [Google Scholar]
- Dong, J.; Wang, N.N.; Yao, Z.J.; Zhang, L.; Cheng, Y.; Ouyang, D.; Lu, A.P.; Cao, D.S. ADMETlab: A platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J. Cheminformatics 2018, 10, 29. [Google Scholar] [CrossRef]
- Onetto, A.J.; Sharif, S. Drug Distribution; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Brahmankar, D.M.; Jaiswal, S.B. Distribution of drugs. In Biopharmaceutics and Pharmacokinetics: A Treatise, 3rd ed.; Vallabh Prakashan: Delhi, India, 2019. [Google Scholar]
- Rifaioglu, A.S.; Atas, H.; Martin, M.J.; Cetin-Atalay, R.; Atalay, V.; Doğan, T. Recent applications of deep learning and machine intelligence on in silico drug discovery: Methods, tools and databases. Brief. Bioinform. 2019, 20, 1878–1912. [Google Scholar] [CrossRef] [Green Version]
- Worley, B.; Powers, R. PCA as a practical indicator of OPLS-DA model reliability. Curr. Metab. 2016, 4, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Avci, O.; Abdeljaber, O.; Kiranyaz, S.; Hussein, M.; Gabbouj, M.; Inman, D.J. A review of vibration-based damage detection in civil structures: From traditional methods to Machine Learning and Deep Learning applications. Mech. Syst. Signal Process. 2021, 147, 107077. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef]
- Bickel, U. How to measure drug transport across the blood-brain barrier. Neurotherapeutics 2005, 2, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; Zhang, L.; Feng, H.W.; Li, S.M.; Liu, M.; Zhao, J.; Liu, H.S. Prediction of the Blood-Brain Barrier (BBB) Permeability of Chemicals Based on Machine-Learning and Ensemble Methods. Chem. Res. Toxicol. 2021, 34, 1456–1467. [Google Scholar] [CrossRef] [PubMed]
- Shaker, B.; Yu, M.S.; Song, J.S.; Ahn, S.; Ryu, J.Y.; Oh, K.S.; Na, D. LightBBB: Computational prediction model of blood-brain-barrier penetration based on LightGBM. Bioinformatics 2021, 37, 1135–1139. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Divakaran, R.; Konda, L.S.K.; Kristam, R. A classification model for blood brain barrier penetration. J. Mol. Graph. Model. 2020, 96, 107516. [Google Scholar] [CrossRef] [PubMed]
- Plisson, F.; Piggott, A.M. Predicting blood–brain barrier permeability of marine-derived kinase inhibitors using ensemble classifiers reveals potential hits for neurodegenerative disorders. Mar. Drugs 2019, 17, 81. [Google Scholar] [CrossRef] [Green Version]
- Miao, R.; Xia, L.Y.; Chen, H.H.; Huang, H.H.; Liang, Y. Improved Classification of Blood-Brain-Barrier Drugs Using Deep Learning. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Cherian Parakkal, S.; Datta, R.; Das, D. DeepBBBP: High Accuracy Blood-Brain-Barrier Permeability Prediction with a Mixed Deep Learning Model. Mol. Inform. 2022, 41, 2100315. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Venkatraman, V. FP-ADMET: A compendium of fingerprint-based ADMET prediction models. J. Cheminformatics 2021, 13, 75. [Google Scholar] [CrossRef]
- Wang, S.; Li, Z.; Zhang, S.G.; Jiang, M.J.; Wang, X.F.; Wei, Z.Q. Molecular Property Prediction Based on a Multichannel Substructure Graph (vol 8, pg 18601, 2020). IEEE Access 2020, 8, 127968. [Google Scholar] [CrossRef]
- Chen, J.-H.; Tseng, Y.J. A general optimization protocol for molecular property prediction using a deep learning network. Brief. Bioinform. 2022, 23, bbab367. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.T.; Yang, Y.W.; Huang, S.H.; Chen, L.X.; Kuang, Z.Y.; Heng, Y.; Mei, H. Molecular image-based convolutional neural network for the prediction of ADMET properties. Chemom. Intell. Lab. Syst. 2019, 194, 103853. [Google Scholar] [CrossRef]
- Wu, Z.; Xian, Z.; Ma, W.; Liu, Q.; Huang, X.; Xiong, B.; He, S.; Zhang, W. Artificial neural network approach for predicting blood brain barrier permeability based on a group contribution method. Comput. Methods Programs Biomed. 2021, 200, 105943. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.H.; Su, B.H.; Battalora, L.C.; Liu, S.; Tseng, Y.J. Ensemble modeling with machine learning and deep learning to provide interpretable generalized rules for classifying CNS drugs with high prediction power. Brief. Bioinform. 2022, 23, bbab377. [Google Scholar] [CrossRef] [PubMed]
- Achiaa Atwereboannah, A.; Wu, W.-P.; Nanor, E. Prediction of Drug Permeability to the Blood-Brain Barrier using Deep Learning. In Proceedings of the 4th International Conference on Biometric Engineering and Applications, Taiyuan, China, 25–27 May 2021; pp. 104–109. [Google Scholar]
- Alsenan, S.; Al-Turaiki, I.; Hafez, A. A Recurrent Neural Network model to predict blood-brain barrier permeability. Comput. Biol. Chem. 2020, 89, 107377. [Google Scholar] [CrossRef]
- Alsenan, S.A.; Al-Turaiki, I.M.; Hafez, A.M. Feature extraction methods in quantitative structure–activity relationship modeling: A comparative study. IEEE Access 2020, 8, 78737–78752. [Google Scholar] [CrossRef]
- Deng, D.G.; Chen, X.W.; Zhang, R.C.; Lei, Z.R.; Wang, X.J.; Zhou, F.F. XGraphBoost: Extracting Graph Neural Network-Based Features for a Better Prediction of Molecular Properties (vol 61, pg 2697, 2021). J. Chem. Inf. Model. 2021, 61, 4820–4822. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, H.; Wu, Z.; Wang, T.; Li, W.; Tang, Y.; Liu, G. In silico prediction of blood–brain barrier permeability of compounds by machine learning and resampling methods. ChemMedChem 2018, 13, 2189–2201. [Google Scholar] [CrossRef]
- Wang, W.Y.; Kim, M.T.; Sedykh, A.; Zhu, H. Developing Enhanced Blood-Brain Barrier Permeability Models: Integrating External Bio-Assay Data in QSAR Modeling. Pharm. Res. 2015, 32, 3055–3065. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.H.; Abraham, M.H.; Ibrahim, A.; Fish, P.V.; Cole, S.; Lewis, M.L.; de Groot, M.J.; Reynolds, D.P. Predicting penetration across the blood-brain barrier from simple descriptors and fragmentation schemes. J. Chem. Inf. Model. 2007, 47, 170–175. [Google Scholar] [CrossRef]
- Li, H.; Yap, C.W.; Ung, C.Y.; Xue, Y.; Cao, Z.W.; Chen, Y.Z. Effect of selection of molecular descriptors on the prediction of blood-brain barrier penetrating and nonpenetrating agents by statistical learning methods. J. Chem. Inf. Model. 2005, 45, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Adenot, M.; Lahana, R. Blood-brain barrier permeation models: Discriminating between potential CNS and non-CNS drugs including P-glycoprotein substrates. J. Chem. Inf. Comput. Sci. 2004, 44, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.F.; Teixeira, A.L.; Pinheiro, L.; Falcao, A.O. A Bayesian approach to in silico blood-brain barrier penetration modeling. J. Chem. Inf. Model. 2012, 52, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: A benchmark for molecular machine learning. Chem. Sci. 2018, 9, 513–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Zheng, F.; Zhan, C.-G. Improved prediction of blood–brain barrier permeability through machine learning with combined use of molecular property-based descriptors and fingerprints. AAPS J. 2018, 20, 54. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Chen, Y.; Cai, X.; Xu, R. Predict drug permeability to blood-brain-barrier from clinical phenotypes: Drug side effects and drug indications. Bioinformatics 2017, 33, 901–908. [Google Scholar] [CrossRef] [Green Version]
- Brito-Sanchez, Y.; Marrero-Ponce, Y.; Barigye, S.J.; Yaber-Goenaga, I.; Perez, C.M.; Huong, L.T.T.; Cherkasov, A. Towards Better BBB Passage Prediction Using an Extensive and Curated Data Set. Mol. Inform. 2015, 34, 308–330. [Google Scholar] [CrossRef]
- Chico, L.K.; Van Eldik, L.J.; Watterson, D.M. Targeting protein kinases in central nervous system disorders. Nat. Rev. Drug Discov. 2009, 8, 892–909. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, M.; Letunic, I.; Jensen, L.J.; Bork, P. The SIDER database of drugs and side effects. Nucleic Acids Res. 2016, 44, D1075–D1079. [Google Scholar] [CrossRef]
- Shen, J.; Cheng, F.; Xu, Y.; Li, W.; Tang, Y. Estimation of ADME properties with substructure pattern recognition. J. Chem. Inf. Model. 2010, 50, 1034–1041. [Google Scholar] [CrossRef]
- Tang, Q.; Nie, F.; Zhao, Q.; Chen, W. A merged molecular representation deep learning method for blood–brain barrier permeability prediction. Brief. Bioinform. 2022, 23, bbac357. [Google Scholar] [CrossRef] [PubMed]
- Kouskoura, M.G.; Piteni, A.I.; Markopoulou, C.K. A new descriptor via bio-mimetic chromatography and modeling for the blood brain barrier (Part II). J. Pharm. Biomed. Anal. 2019, 164, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Toropov, A.A.; Toropova, A.P.; Beeg, M.; Gobbi, M.; Salmona, M. QSAR model for blood-brain barrier permeation. J. Pharmacol. Toxicol. Methods 2017, 88, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Herbertz, T.; Hudkins, R.L.; Dorsey, B.D.; Mallamo, J.P. Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chem. Neurosci. 2012, 3, 50–68. [Google Scholar] [CrossRef] [Green Version]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Steijaert, M.; Wegner, J.K.; Ceulemans, H.; Clevert, D.-A.; Hochreiter, S. Large-scale comparison of machine learning methods for drug target prediction on ChEMBL. Chem. Sci. 2018, 9, 5441–5451. [Google Scholar] [CrossRef] [Green Version]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Venkateswarlu, V. Distribution of Drugs. In Biopharmaceutics and Pharmacokinetics; PharmaMed Press: Watford, UK, 2008; p. 77. [Google Scholar]
- Toma, C.; Gadaleta, D.; Roncaglioni, A.; Toropov, A.; Toropova, A.; Marzo, M.; Benfenati, E. QSAR Development for Plasma Protein Binding: Influence of the Ionization State. Pharm. Res. 2019, 36, 28. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.W.; Chang, S.; Zhang, Z.; Li, Z.G.; Li, S.Z.; Xie, P.; Yau, W.P.; Lin, H.S.; Cai, W.M.; Zhang, Y.C.; et al. A novel strategy for prediction of human plasma protein binding using machine learning techniques. Chemom. Intell. Lab. Syst. 2020, 199, 103962. [Google Scholar] [CrossRef]
- Peng, Y.Z.; Lin, Y.M.; Jing, X.Y.; Zhang, H.; Huang, Y.R.; Luo, G.S. Enhanced Graph Isomorphism Network for Molecular ADMET Properties Prediction. IEEE Access 2020, 8, 168344–168360. [Google Scholar] [CrossRef]
- Wang, N.-N.; Deng, Z.-K.; Huang, C.; Dong, J.; Zhu, M.-F.; Yao, Z.-J.; Chen, A.F.; Lu, A.-P.; Mi, Q.; Cao, D.-S. ADME properties evaluation in drug discovery: Prediction of plasma protein binding using NSGA-II combining PLS and consensus modeling. Chemom. Intell. Lab. Syst. 2017, 170, 84–95. [Google Scholar] [CrossRef]
- Lou, C.; Yang, H.; Wang, J.; Huang, M.; Li, W.; Liu, G.; Lee, P.W.; Tang, Y. IDL-PPBopt: A Strategy for Prediction and Optimization of Human Plasma Protein Binding of Compounds via an Interpretable Deep Learning Method. J. Chem. Inf. Model. 2022, 62, 2788–2799. [Google Scholar] [CrossRef] [PubMed]
- Obach, R.S.; Lombardo, F.; Waters, N.J. Trend analysis of a database of intravenous pharmacokinetic parameters in humans for 670 drug compounds. Drug Metab. Dispos. 2008, 36, 1385–1405. [Google Scholar] [CrossRef] [Green Version]
- Sushko, I.; Novotarskyi, S.; Körner, R.; Pandey, A.K.; Rupp, M.; Teetz, W.; Brandmaier, S.; Abdelaziz, A.; Prokopenko, V.V.; Tanchuk, V.Y. Online chemical modeling environment (OCHEM): Web platform for data storage, model development and publishing of chemical information. J. Comput.-Aided Mol. Des. 2011, 25, 533–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oprisiu, I.; Winiwarter, S. In Silico ADME Modeling; Academic Press: Cambridge, MA, USA, 2021; pp. 208–222. [Google Scholar] [CrossRef]
- Zhivkova, Z. Quantitative Structure—Pharmacokinetics Relationships for Plasma Protein Binding of Basic Drugs. J. Pharm. Pharm. Sci. 2017, 20, 349–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Votano, J.R.; Parham, M.; Hall, L.M.; Hall, L.H.; Kier, L.B.; Oloff, S.; Tropsha, A. QSAR modeling of human serum protein binding with several modeling techniques utilizing structure− information representation. J. Med. Chem. 2006, 49, 7169–7181. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Yang, H.; Li, J.; Wang, T.; Li, W.; Liu, G.; Tang, Y. In silico prediction of compounds binding to human plasma proteins by QSAR models. ChemMedChem 2018, 13, 572–581. [Google Scholar] [CrossRef]
- Zhu, X.-W.; Sedykh, A.; Zhu, H.; Liu, S.-S.; Tropsha, A. The use of pseudo-equilibrium constant affords improved QSAR models of human plasma protein binding. Pharm. Res. 2013, 30, 1790–1798. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, R.; Esaki, T.; Kawashima, H.; Natsume-Kitatani, Y.; Nagao, C.; Ohashi, R.; Mizuguchi, K. Predicting fraction unbound in human plasma from chemical structure: Improved accuracy in the low value ranges. Mol. Pharm. 2018, 15, 5302–5311. [Google Scholar] [CrossRef] [Green Version]
- Douguet, D. Data sets representative of the structures and experimental properties of FDA-approved drugs. ACS Med. Chem. Lett. 2018, 9, 204–209. [Google Scholar] [CrossRef]
- Tajimi, T.; Wakui, N.; Yanagisawa, K.; Yoshikawa, Y.; Ohue, M.; Akiyama, Y. Computational prediction of plasma protein binding of cyclic peptides from small molecule experimental data using sparse modeling techniques. BMC Bioinform. 2018, 19, 157–170. [Google Scholar] [CrossRef]
- Ingle, B.L.; Veber, B.C.; Nichols, J.W.; Tornero-Velez, R. Informing the human plasma protein binding of environmental chemicals by machine learning in the pharmaceutical space: Applicability domain and limits of predictability. J. Chem. Inf. Model. 2016, 56, 2243–2252. [Google Scholar] [CrossRef]
- Li, H.; Chen, Z.; Xu, X.; Sui, X.; Guo, T.; Liu, W.; Zhang, J. Predicting human plasma protein binding of drugs using plasma protein interaction QSAR analysis (PPI-QSAR). Biopharm. Drug Dispos. 2011, 32, 333–342. [Google Scholar] [CrossRef]
- Zhivkova, Z.; Doytchinova, I. Quantitative structure—Plasma protein binding relationships of acidic drugs. J. Pharm. Sci. 2012, 101, 4627–4641. [Google Scholar] [CrossRef]
- Zhang, S.; Yan, Z.; Huang, Y.; Liu, L.; He, D.; Wang, W.; Fang, X.; Zhang, X.; Wang, F.; Wu, H.; et al. HelixADMET: A robust and endpoint extensible ADMET system incorporating self-supervised knowledge transfer. Bioinformatics 2022. [Google Scholar] [CrossRef]
- Wei, Y.; Li, S.; Li, Z.; Wan, Z.; Lin, J. Interpretable-ADMET: A Web Service for ADMET Prediction and Optimization based on Deep Neural Representation. Bioinformatics 2022, 38, 2863–2871. [Google Scholar] [CrossRef]
- Roberts, J.A.; Pea, F.; Lipman, J. The Clinical Relevance of Plasma Protein Binding Changes. Clin. Pharmacokinet. 2013, 52, 1–8. [Google Scholar] [CrossRef]
- Seyfinejad, B.; Ozkan, S.A.; Jouyban, A. Recent advances in the determination of unbound concentration and plasma protein binding of drugs: Analytical methods. Talanta 2021, 225, 122052. [Google Scholar] [CrossRef]
- Bohnert, T.; Gan, L.-S. Plasma protein binding: From discovery to development. J. Pharm. Sci. 2013, 102, 2953–2994. [Google Scholar] [CrossRef]
- Wang, Y.C.; Liu, H.C.; Fan, Y.R.; Chen, X.Y.; Yang, Y.; Zhu, L.; Zhao, J.N.; Chen, Y.D.; Zhang, Y.M. In Silico Prediction of Human Intravenous Pharmacokinetic Parameters with Improved Accuracy. J. Chem. Inf. Model. 2019, 59, 3968–3980. [Google Scholar] [CrossRef]
- Lombardo, F.; Berellini, G.; Obach, R.S. Trend analysis of a database of intravenous pharmacokinetic parameters in humans for 1352 drug compounds. Drug Metab. Dispos. 2018, 46, 1466–1477. [Google Scholar] [CrossRef] [Green Version]
- Mulpuru, V.; Mishra, N. In Silico Prediction of Fraction Unbound in Human Plasma from Chemical Fingerprint Using Automated Machine Learning. Acs Omega 2021, 6, 6791–6797. [Google Scholar] [CrossRef]
- Zhou, Y.D.; Cahya, S.; Combs, S.A.; Nicolaou, C.A.; Wang, J.B.; Desai, P.V.; Shen, J. Exploring Tunable Hyperparameters for Deep Neural Networks with Industrial ADME Data Sets. J. Chem. Inf. Model. 2019, 59, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, E.N.; Joshi, E.; Pande, V.S.; Cheng, A.C. Improvement in ADMET Prediction with Multitask Deep Featurization. J. Med. Chem. 2020, 63, 8835–8848. [Google Scholar] [CrossRef]
- Yamagata, T.; Zanelli, U.; Gallemann, D.; Perrin, D.; Dolgos, H.; Petersson, C. Comparison of methods for the prediction of human clearance from hepatocyte intrinsic clearance for a set of reference compounds and an external evaluation set. Xenobiotica 2017, 47, 741–751. [Google Scholar] [CrossRef]
- Fagerholm, U.; Spjuth, O.; Hellberg, S. Comparison between lab variability and in silico prediction errors for the unbound fraction of drugs in human plasma. Xenobiotica 2021, 51, 1095–1100. [Google Scholar] [CrossRef]
- Mansoor, A.; Mahabadi, N. Volume of Distribution; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Smith, D.A.; Beaumont, K.; Maurer, T.S.; Di, L. Volume of Distribution in Drug Design. J. Med. Chem. 2015, 58, 5691–5698. [Google Scholar] [CrossRef]
- Hsu, F.; Chen, Y.C.; Broccatelli, F. Evaluation of Tissue Binding in Three Tissues across Five Species and Prediction of Volume of Distribution from Plasma Protein and Tissue Binding with an Existing Model. Drug Metab. Dispos. 2021, 49, 330–336. [Google Scholar] [CrossRef]
- Maxwell, S. How Are Drugs Distributed around the Body. Available online: https://vimeo.com/469366240 (accessed on 11 November 2022).
- Lombardo, F.; Bentzien, J.; Berellini, G.; Muegge, I. In Silico Models of Human PK Parameters. Prediction of Volume of Distribution Using an Extensive Data Set and a Reduced Number of Parameters. J. Pharm. Sci. 2021, 110, 500–509. [Google Scholar] [CrossRef]
- Simeon, S.; Montanari, D.; Gleeson, M.P. Investigation of Factors Affecting the Performance of in silico Volume Distribution QSAR Models for Human, Rat, Mouse, Dog & Monkey. Mol. Inform. 2019, 38, e1900059. [Google Scholar] [CrossRef]
- Ye, Z.; Yang, Y.; Li, X.; Cao, D.; Ouyang, D. An Integrated Transfer Learning and Multitask Learning Approach for Pharmacokinetic Parameter Prediction. Mol. Pharm. 2019, 16, 533–541. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, F.; Jing, Y.K. In Silico Prediction of Volume of Distribution in Humans. Extensive Data Set and the Exploration of Linear and Nonlinear Methods Coupled with Molecular Interaction Fields Descriptors. J. Chem. Inf. Model. 2016, 56, 2042–2052. [Google Scholar] [CrossRef]
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Krüger, F.A.; Light, Y.; Mak, L.; McGlinchey, S. The ChEMBL bioactivity database: An update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, T.W.; Diderich, R.; Kuseva, C.D.; Mekenyan, O.G. The OECD QSAR toolbox starts its second decade. In Computational Toxicology; Springer: Berlin/Heidelberg, Germany, 2018; pp. 55–77. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Schyman, P.; Liu, R.; Desai, V.; Wallqvist, A. vNN web server for ADMET predictions. Front. Pharmacol. 2017, 8, 889. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.; Zhang, X.; Pan, X.; Wang, B.; Ji, C.; Qi, Y.; Zhang, J.Z. HobPre: Accurate prediction of human oral bioavailability for small molecules. J. Cheminformatics 2022, 14, 1. [Google Scholar] [CrossRef]
- Wu, F.; Zhou, Y.; Li, L.; Shen, X.; Chen, G.; Wang, X.; Liang, X.; Tan, M.; Huang, Z. Computational Approaches in Preclinical Studies on Drug Discovery and Development. Front. Chem. 2020, 8, 726. [Google Scholar] [CrossRef]
- Jia, C.Y.; Li, J.Y.; Hao, G.F.; Yang, G.F. A drug-likeness toolbox facilitates ADMET study in drug discovery. Drug Discov. Today 2020, 25, 248–258. [Google Scholar] [CrossRef]
- Irwin, J.J.; Tang, K.G.; Young, J.; Dandarchuluun, C.; Wong, B.R.; Khurelbaatar, M.; Moroz, Y.S.; Mayfield, J.; Sayle, R.A. ZINC20—A free ultralarge-scale chemical database for ligand discovery. J. Chem. Inf. Model. 2020, 60, 6065–6073. [Google Scholar] [CrossRef]
- Pence, H.E.; Williams, A. ChemSpider: An Online Chemical Information Resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Fu, T.; Gao, W.; Zhao, Y.; Roohani, Y.; Leskovec, J.; Coley, C.W.; Xiao, C.; Sun, J.; Zitnik, M. Artificial intelligence foundation for therapeutic science. Nat. Chem. Biol. 2022, 18, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Kass-Hout, T.A.; Xu, Z.; Mohebbi, M.; Nelsen, H.; Baker, A.; Levine, J.; Johanson, E.; Bright, R.A. OpenFDA: An innovative platform providing access to a wealth of FDA’s publicly available data. J. Am. Med. Inform. Assoc. 2016, 23, 596–600. [Google Scholar] [CrossRef] [Green Version]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Erehman, J.; Gohlke, B.-O.; Wilhelm, T.; Preissner, R.; Dunkel, M. Super Natural II—A database of natural products. Nucleic Acids Res. 2015, 43, D935–D939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linstrom, P.J.; Mallard, W.G. The NIST Chemistry WebBook: A chemical data resource on the internet. J. Chem. Eng. Data 2001, 46, 1059–1063. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Mohanraj, K.; Karthikeyan, B.S.; Vivek-Ananth, R.; Chand, R.; Aparna, S.; Mangalapandi, P.; Samal, A. IMPPAT: A curated database of Indian medicinal plants, phytochemistry and therapeutics. Sci. Rep. 2018, 8, 4329. [Google Scholar] [CrossRef] [Green Version]
- Danishuddin; Kumar, V.; Faheem, M.; Woo Lee, K. A decade of machine learning-based predictive models for human pharmacokinetics: Advances and challenges. Drug Discov. Today 2022, 27, 529–537. [Google Scholar] [CrossRef]
- Kumar, A.; Kini, S.G.; Rathi, E. A Recent Appraisal of Artificial Intelligence and In Silico ADMET Prediction in the Early Stages of Drug Discovery. Mini Rev. Med. Chem. 2021, 21, 2788–2800. [Google Scholar] [CrossRef]
- Ferreira, L.L.G.; Andricopulo, A.D. ADMET modeling approaches in drug discovery. Drug Discov. Today 2019, 24, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Pantaleao, S.Q.; Fernandes, P.O.; Goncalves, J.E.; Maltarollo, V.G.; Honorio, K.M. Recent Advances in the Prediction of Pharmacokinetics Properties in Drug Design Studies: A Review. ChemMedChem 2022, 17, e202100542. [Google Scholar] [CrossRef] [PubMed]
- Pasrija, P.; Jha, P.; Upadhyaya, P.; Khan, M.S.; Chopra, M. Machine Learning and Artificial Intelligence: A Paradigm Shift in Big Data-Driven Drug Design and Discovery. Curr. Top. Med. Chem. 2022, 22, 1692–1727. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.Z.; Baber, J.C.; Bahmanyar, S.S. The challenges of generalizability in artificial intelligence for ADME/Tox endpoint and activity prediction. Expert Opin. Drug Discov. 2021, 16, 1045–1056. [Google Scholar] [CrossRef]
- Kantify. AI in Drug Discovery: ADMET Property Prediction. Available online: https://kantify.com/use-cases/ai-in-drug-discovery-admet-property-prediction (accessed on 14 October 2022).
- Bhhatarai, B.; Walters, W.P.; Hop, C.E.C.A.; Lanza, G.; Ekins, S. Opportunities and challenges using artificial intelligence in ADME/Tox. Nat. Mater. 2019, 18, 418–422. [Google Scholar] [CrossRef]
- Brown, N.; Cambruzzi, J.; Cox, P.J.; Davies, M.; Dunbar, J.; Plumbley, D.; Sellwood, M.A.; Sim, A.; Williams-Jones, B.I.; Zwierzyna, M. Big data in drug discovery. Prog. Med. Chem. 2018, 57, 277–356. [Google Scholar] [CrossRef]
- Lee, C.H.; Yoon, H.-J. Medical big data: Promise and challenges. Kidney Res. Clin. Pract. 2017, 36, 3. [Google Scholar] [CrossRef] [Green Version]
- Schneider, P.; Walters, W.P.; Plowright, A.T.; Sieroka, N.; Listgarten, J.; Goodnow, R.A.; Fisher, J.; Jansen, J.M.; Duca, J.S.; Rush, T.S. Rethinking drug design in the artificial intelligence era. Nat. Rev. Drug Discov. 2020, 19, 353–364. [Google Scholar] [CrossRef]
- Chicco, D. Ten quick tips for machine learning in computational biology. BioData Min. 2017, 10, 35. [Google Scholar] [CrossRef]
- Fourches, D.; Williams, A.J.; Patlewicz, G.; Shah, I.; Grulke, C.; Wambaugh, J.; Richard, A.; Tropsha, A. Computational Tools for ADMET Profiling. Comput. Toxicol. Risk Assess. Chem. 2018, 211–244. [Google Scholar] [CrossRef]
- Healthcare, G. It Will Take Years for AI Use to Peak in Drug Discovery and Development Process. Available online: https://www.pharmaceutical-technology.com/comment/ai-peak-drug-discovery-development/ (accessed on 1 November 2022).




| Name | No. of ADMET Prediction Models | Methods | Website * | Ref. |
|---|---|---|---|---|
| OECD QSAR Toolbox | 902 | QSAR | https://qsartoolbox.org/ | [91] |
| iDrug ADMET prediction | 60 | AI | https://drug.ai.tencent.com/console/en/admet | |
| AdmetSAR 2.0 | 52 | RF, SVM, k-NN | http://lmmd.ecust.edu.cn/admetsar2/ | [21] |
| ADMETlab 2.0 | 67 | GNN | https://admetmesh.scbdd.com/ | [50] |
| Interpretable-ADMET | 59 | GCNN GAT | http://cadd.pharmacy.nankai.edu.cn/interpretableadmet/ | [71] |
| HelixADMET | 52 | RF, GNN | https://paddlehelix.baidu.com/app/drug/admet/train | [70] |
| FP-ADMET | 50 | RF | https://gitlab.com/vishsoft/fpadmet | [22] |
| SwissADME | 35 | MLR, SVM, RNN, etc. | http://www.swissadme.ch/ | [92] |
| vNN-ADMET | 15 | k-NN | https://vnnadmet.bhsai.org/ | [93] |
| ICDrug ADMET | 14 | RF | www.icdrug.com/ICDrug/ADMET | [94] |
| Virtual Rat | 12 | RF, C5.0, DT | https://virtualrat.cmdm.tw/ | [3] |
| LightBBB | 1 (BBB) | Light GBM | http://ssbio.cau.ac.kr/software/bbb | [16] |
| Deep B3 | 1 (BBB) | CNN | http://cbcb.cdutcm.edu.cn/deepb3/ | [45] |
| Property | Tool | Methods | No. of Compounds | Performance | |
|---|---|---|---|---|---|
| AUC | R2 | ||||
| BBB | AdmetSAR 2.0 | SVM | 1839 | 0.944 | |
| ADMETLab 2.0 | GNN | 1601 | 0.908 | ||
| FP-ADMET | RF | 7236 | 0.92 | ||
| Interpretable-ADMET | GCNN & GAT | 1830 | 0.897 | ||
| HelixADMET | GNN | 1791 | 0.944 | ||
| PPB | AdmetSAR 2.0 | GCNN | 1209 | 0.668 | |
| ADMETLab 2.0 | GNN | 1573 | 0.733 | ||
| FP-ADMET | RF | 8103 | 0.92 | ||
| Interpretable-ADMET | GCNN & GAT | 2044 | 0.563 | ||
| HelixADMET | GNN | 1744 | 0.747 | ||
| Fu | AdmetSAR 2.0 | - | - | - | - |
| ADMETLab 2.0 | GNN | 1494 | 0.763 | ||
| FP-ADMET | RF | 2319 | 0.63 | ||
| Interpretable-ADMET | - | - | - | - | |
| HelixADMET | - | - | - | - | |
| Vd | AdmetSAR 2.0 | - | - | - | - |
| ADMETLab 2.0 | GNN | 1399 | 0.782 | ||
| FP-ADMET | RF | 1951 | 0.45 | ||
| Interpretable-ADMET | - | - | - | - | |
| HelixADMET | - | - | - | - | |
| Name | Data Size (Compounds) * | Website * | Ref. |
|---|---|---|---|
| ZINC20 | >750 million | https://zinc20.docking.org/ | [97] |
| ChemSpider | 115 million | http://www.chemspider.com/ | [98] |
| PubChem | >111 million | https://pubchem.ncbi.nlm.nih.gov/ | [99] |
| Therapeutics Data Commons | 4,264,939 | https://tdcommons.ai/ | [100] |
| OCHEM 4.2 | 3,791,680 | https://ochem.eu/home/show.do | [58] |
| openFDA | >3 million | https://open.fda.gov/ | [101] |
| ChEMBL | >2.2 million | www.ebi.ac.uk/chembl/ | [102] |
| GOSTAR | 1.76 million | https://www.gostardb.com/ | |
| BindingDB | >1 million | https://www.bindingdb.org/ | [103] |
| Supernatural II | 325,508 | http://bioinformatics.charite.de/supernatural | [104] |
| NIST Chemistry WebBook | >70,000 | http://webbook.nist.gov/ | [105] |
| SIDER 4.1 | 55,730 | http://sideeffects.embl.de/ | [43] |
| ContaminantDB | >54,000 | https://contaminantdb.ca/ | |
| DrugBank 5.1.9 | 14,665 | http://www.drugbank.ca/ | [106] |
| IMPPAT 2.0 | 17,967 | https://cb.imsc.res.in/imppat | [107] |
| KEGG | 12,000 | https://www.kegg.jp/ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, T.T.V.; Tayara, H.; Chong, K.T. Recent Studies of Artificial Intelligence on In Silico Drug Distribution Prediction. Int. J. Mol. Sci. 2023, 24, 1815. https://doi.org/10.3390/ijms24031815
Tran TTV, Tayara H, Chong KT. Recent Studies of Artificial Intelligence on In Silico Drug Distribution Prediction. International Journal of Molecular Sciences. 2023; 24(3):1815. https://doi.org/10.3390/ijms24031815
Chicago/Turabian StyleTran, Thi Tuyet Van, Hilal Tayara, and Kil To Chong. 2023. "Recent Studies of Artificial Intelligence on In Silico Drug Distribution Prediction" International Journal of Molecular Sciences 24, no. 3: 1815. https://doi.org/10.3390/ijms24031815
APA StyleTran, T. T. V., Tayara, H., & Chong, K. T. (2023). Recent Studies of Artificial Intelligence on In Silico Drug Distribution Prediction. International Journal of Molecular Sciences, 24(3), 1815. https://doi.org/10.3390/ijms24031815