
Molecular Mechanisms of Neuroinflammation in Aging and Alzheimer’s Disease Progression


 , ,
, ,
Abstract
:1. Introduction
2. Aging and Immunity
2.1. Neuroinflammation in the Aging Brain
2.1.1. Mitochondria in the Aging Brain
2.1.2. Oxidative Stress and Brain Aging
2.1.3. Astrocyte Senescence
2.1.4. Neuroinflammation and Defective Autophagy
3. Neuroinflammation in Alzheimer’s Disease
3.1. The Role of Microglia in Alzheimer’s Disease Pathogenesis
3.1.1. TREM2 Signaling
3.1.2. Scavenger Receptor Class A (SR-A) in Alzheimer’s Disease
3.1.3. CD33 Receptor in Alzheimer’s Disease
3.1.4. CD36 Receptor in Alzheimer’s Disease
3.1.5. Complement Receptor 3 (CR3) in Alzheimer’s Disease
3.1.6. TNF Signaling in Alzheimer’s Disease
3.2. Mitochondrial Dysfunction and Neuroinflammation in Alzheimer’s Disease Pathogenesis
3.3. Impaired Autophagy in Alzheimer’s Disease
3.4. Senescent Astrocytes and Alzheimer’s Disease
4. Therapeutic Strategies Targeting Neuroinflammation in Alzheimer’s Disease
4.1. Early Attempts of Anti-Inflammatory Treatment in AD
4.2. Anti-Inflammatory Molecules in Clinical Trials
4.2.1. Peroxisome Proliferator-Activated Receptor (PPAR)-γ Agonists
4.2.2. Tumor Necrosis Factor-α Inhibitors
4.2.3. Tyrosine Kinase Inhibitors
4.2.4. MAP Kinase Inhibition
4.2.5. Other Anti-Inflammatory Strategies
4.3. Anti-Inflammatory Strategies in Animal Models and In Vitro
4.3.1. Targeting TNF Signaling
4.3.2. Targeting the cGAS-STING Pathway
4.3.3. Targeting the Inflammasome
4.3.4. Targeting Immune Checkpoints
4.3.5. Targeting the Complement
4.3.6. Cell-Based and Cell-Derived Therapeutic Strategies
4.3.7. Nanotechnology-Based Anti-Inflammatory Approaches in AD
4.3.8. Other Anti-Inflammatory Strategies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ADAM | A disintegrin and metalloproteinase |
| AIM2 | absent in melanoma 2 |
| Akt | protein kinase B |
| AMP | adenosine monophosphate |
| AMPK | 5′-adenosine monophosphate (AMP)-activated protein kinase |
| AP1 | activator protein 1 |
| APOE | apolipoprotein E |
| APP | amyloid precursor protein |
| ARE | antioxidant response element |
| ASC | apoptosis-associated speck-like protein containing CARD |
| ATG | autophagy-related protein |
| ATP | adenosine triphosphate |
| BACE | β-site APP cleaving enzyme |
| BBB | blood brain barrier |
| BDNF | brain-derived neurotrophic factor |
| BER | base excision repair |
| CAMKII | calcium/calmodulin-dependent protein kinase II |
| CARD | caspase activation and recruitment domain |
| CAT | catalase |
| CBP | CREB binding protein |
| CCL | C-C motif chemokine ligand |
| CGAS | cyclic GMP-AMP synthase |
| CIP/KIP | CDK interacting protein/kinase inhibitory protein |
| CNS | central nervous system |
| COX | cyclooxygenase |
| CRISPR/Cas9 | clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 |
| CSF | cerebrospinal fluid |
| CXCL-CXCL | C-X-C motif ligand |
| CSF1R | colony stimulating factor 1 receptor |
| DAMPS | damage-associated molecular patterns |
| DAP12 | DNAX-activation protein of 12 kDa |
| DNA | deoxyribonucleic acid |
| Drp1 | dynamin-related protein 1 |
| EAAT | excitatory amino acid transporter |
| EMRE | essential MCU regulator |
| ER | endoplasmic reticulum |
| ERK | extracellular-regulated kinase |
| ETC | electron transport chain |
| EVs | extracellular vesicles |
| FGF21 | fibroblast growth factor 21 |
| FIP200 | focal adhesion kinase (FAK) family-interacting protein of 200 kDa |
| Fis1 | mitochondrial fission 1 protein |
| GABA | γ-aminobutyric acid |
| GDF15 | growth/differentiation factor 15 |
| GDNF | glial-derived neurotrophic factor |
| GFAP | glial fibrillary acidic protein |
| GMP | guanosine monophosphate |
| GPx | glutathione peroxidase |
| GSH | glutathione |
| GSK3β | glycogen synthase kinase-3 beta |
| GWAS | genome-wide association studies |
| HIF | hypoxia-inducible factor |
| HIV | human immunodeficiency virus |
| HMGB | high mobility box group |
| HMGCR | 3-hydroxy-3-methylglutaryl CoA reductase |
| HO-1 | heme oxygenase-1 |
| IC50 | half maximal inhibitory concentration |
| ICAM1 | intercellular adhesion molecule 1 |
| IF | interferon |
| IFNAR | interferon-α/β receptor |
| IκB | inhibitor of κB |
| IKK | IκB kinase |
| IL | interleukin |
| IMM | inner mitochondrial membrane |
| IP3Rs | inositol trisphosphate receptors |
| IRF | interferon regulatory factor |
| ISGF3 | interferon-stimulated gene factor-3 |
| JAK | Janus kinase |
| JIP | c-Jun N-terminal kinase-interacting protein |
| JNK pathway | Jun N-terminal kinase pathway |
| Keap1 | kelch like erythroid cell-derived protein with CNC homology (ECH)-associated protein 1 |
| LAMP1 | lysosomal-associated membrane protein 1 |
| LC3 | light chain 3 |
| MAMs | mitochondria-associated membranes |
| MAPK | mitogen-activated protein kinase |
| MCI | mild cognitive impairment |
| MCU | mitochondrial membrane Ca2+ uniporter |
| MCUR1 | MCU regulator 1 |
| Mfn | mitofusin |
| MHC | major histocompatibility complex |
| MICU 1 and 2 | mitochondrial calcium uptake 1 and 2 |
| miRNA | microRNA |
| MMP | matrix metalloproteinase |
| MPTP | mitochondrial permeability transition pore |
| MRI | magnetic resonance imaging |
| mRNA | messenger RNA |
| mTOR | mammalian target of rapamycin |
| mtDNA | mitochondrial DNA |
| NAD | nicotinamide adenine dinucleotide |
| NADH | reduced nicotinamide adenine dinucleotide |
| NCLX | Li+-permeable Na+/Ca2+ exchanger |
| NGF | nerve growth factor |
| NFATs | nuclear factor of activated T cells receptors |
| NF-κB | nuclear factor-kappa B |
| NFT | neurofibrillary tangle |
| NLR | nucleotide-binding oligomerization domain (NOD)-like receptor |
| NO | nitric oxide |
| NOS | nitric oxide synthase |
| NOX | NADPH oxidase |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| NSAIDs | non-steroidal anti-inflammatory drugs |
| NSF | N-ethylmaleimide sensitive fusion protein |
| OH8dG | nucleoside 8-hydroxy-2′-deoxyguanosine |
| OMM | outer mitochondrial membrane |
| OPA1 | optic atrophy 1 |
| OXPHOS | oxidative phosphorylation |
| PAMPs | pathogen-associated molecular patterns |
| PARP | poly(ADP-ribose) polymerase |
| PET | positron emission tomography |
| PGAM | phosphoglycerate mutase |
| PGC-1α | peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PI3K | phosphatidylinositol 3-kinase |
| PINK1 | PTEN-induced kinase 1 |
| POLG | DNA polymerase subunit gamma |
| PQBP1 | polyglutamine binding protein 1 |
| PRR | pattern recognition receptor |
| PSEN | presenilin |
| RAGE | receptor for advanced glycation endproducts |
| ROS | reactive oxygen species |
| RUNX1 | runt-related transcription factor 1 |
| RyR | ryanodine receptor |
| SASP | senescence-associated secretory phenotype |
| SNAREs | soluble N-ethylmaleimide sensitive factor attachment protein receptors |
| SOD | superoxide dismutase |
| SR-A | scavenger receptor class A |
| SR-B1 | scavenger receptor B1 |
| SREBP2 | sterol regulatory element-binding protein 2 |
| STAT | signal transducer and activator of transcription |
| STING | stimulator of interferon genes |
| Syk | spleen tyrosine kinase |
| TBK1 | TANK binding kinase 1 |
| TFAM | mitochondrial transcription factor A |
| Th | T helper lymphocytes |
| TIMP1 | tissue inhibitor of metalloproteinases 1 |
| TLR | toll-like receptor |
| TNF | tumor necrosis factor |
| TNFR | TNF-α receptor |
| TOM | translocase of the outer membrane |
| TRAIL | tumor necrosis factor-related apoptosis inducing ligand |
| Treg | regulatory T cells |
| TREM | triggering receptor expressed on myeloid cells |
| TREX1 | three prime repair exonuclease 1 |
| ULK | Unc-51-like kinase |
| VDAC | voltage-dependent anion channel |
| VGCC | voltage-gated calcium channel |
| VPS | vascular protein sorting |
| WIPI | tryptophan-aspartic acid (WD) repeat domain phosphoinositide-interacting proteins |
References
- Cao, X.; Hou, Y.; Zhang, X.; Xu, C.; Jia, P.; Sun, X.; Sun, L.; Gao, Y.; Yang, H.; Cui, Z.; et al. A comparative, correlate analysis and projection of global and regional life expectancy, healthy life expectancy, and their GAP: 1995–2025. J. Glob. Health 2020, 10, 020407. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.X.; Tian, Y.; Wang, Z.T.; Ma, Y.H.; Tan, L.; Yu, J.T. The epidemiology of Alzheimer’s disease modifiable risk factors and prevention. J. Prev. Alzheimers Dis. 2021, 8, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A.; Simion, A. Oxidative stress in the pathogenesis of Alzheimer’s disease and cerebrovascular disease with therapeutic implications. CNS Neurol. Disord. Drug Targets 2020, 19, 94–108. [Google Scholar]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Vetrano, D.L.; Triolo, F.; Maggi, S.; Malley, R.; Jackson, T.A.; Poscia, A.; Bernabei, R.; Ferrucci, L.; Fratiglioni, L. Fostering healthy aging: The interdependency of infections, immunity and frailty. Ageing Res. Rev. 2021, 69, 101351. [Google Scholar] [CrossRef]
- Cao, W.; Zheng, H. Peripheral immune system in aging and Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 51. [Google Scholar] [CrossRef]
- Mayne, K.; White, J.A.; McMurran, C.E.; Rivera, F.J.; de la Fuente, A.G. Aging and neurodegenerative disease: Is the adaptive immune system a friend or foe? Front. Aging Neurosci. 2020, 12, 57290. [Google Scholar] [CrossRef]
- Coder, B.D.; Wang, H.; Ruan, L.; Su, D.-M. Thymic involution perturbs negative selection leading to autoreactive T cells that induce chronic inflammation. J. Immunol. 2015, 194, 5825–5837. [Google Scholar] [CrossRef] [Green Version]
- Riley, R.L.; Khomtchouk, K.; Blomberg, B.B. Age-associated B cells (ABC) inhibit B lymphopoiesis and alter antibody repertoires in old age. Cell Immunol. 2017, 321, 61–67. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and inflammaging as two sides of the same coin: Friends or foes? Front. Immunol. 2018, 8, 1960. [Google Scholar] [CrossRef]
- Galea, I.; Bechmann, I.; Perry, V.H. What is immune privilege (not)? Trends Immunol. 2007, 28, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Enzmann, G.; Kargaran, S.; Engelhardt, B. Ischemia-reperfusion injury in stroke: Impact of the brain barriers and brain immune privilege on neutrophil function. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418794184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer’s disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Javanmehr, N.; Saleki, K.; Alijanizadeh, P.; Rezaei, N. Microglia dynamics in aging-related neurobehavioral and neuroinflammatory diseases. J. Neuroinflamm. 2022, 19, 273. [Google Scholar] [CrossRef]
- Szepesi, Z.; Manouchehrian, O.; Bachiller, S.; Deierborg, T. Bidirectional microglia–neuron communication in health and disease. Front. Cell. Neurosci. 2018, 12, 323. [Google Scholar] [CrossRef]
- Yan, J.; Yao, Y.; Yan, S.; Gao, R.; Lu, W.; He, W. Chiral protein supraparticles for tumor suppression and synergistic immunotherapy: An enabling strategy for bioactive supramolecular chirality construction. Nano Lett. 2020, 20, 5844–5852. [Google Scholar] [CrossRef]
- Rodríguez-Gómez, J.A.; Kavanagh, E.; Engskog-Vlachos, P.; Engskog, M.K.R.; Herrera, A.J.; Espinosa-Oliva, A.M.; Joseph, B.; Hajji, N.; Venero, J.L.; Burguillos, M.A. Microglia: Agents of the CNS pro-inflammatory response. Cells 2020, 9, 1717. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef] [Green Version]
- Lyu, J.; Jiang, X.; Leak, R.K.; Shi, Y.; Hu, X.; Chen, J. Microglial responses to brain injury and disease: Functional diversity and new opportunities. Transl. Stroke Res. 2021, 12, 474–495. [Google Scholar] [CrossRef]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Cheng, X.; Zhong, S.; Liu, C.; Jolkkonen, J.; Zhang, X.; Liang, Y.; Liu, Z.; Zhao, C. Communications between peripheral and the brain-resident immune system in neuronal regeneration after stroke. Front. Immunol. 2020, 11, 1931. [Google Scholar] [CrossRef]
- Jurcau, A.; Simion, A. Neuroinflammation in cerebral ischemia and ischemia/reperfusion injuries: From pathophysiology to therapeutic strategies. Int. J. Mol. Sci. 2021, 23, 14. [Google Scholar] [CrossRef]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Spittau, B. Aging microglia—Phenotypes, functions and implications for age-related neurodegenerative diseases. Front. Aging Neurosci. 2017, 9, 194. [Google Scholar] [CrossRef] [Green Version]
- Haas, R.H. Mitochondrial dysfunction in aging and diseases of aging. Biology 2019, 8, 48. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F.; Bernabei, R.; Marzetti, E. Fueling inflammaging through mitochondrial dysfunction: Mechanisms and molecular targets. Int. J. Mol. Sci. 2017, 18, 933. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, M.C.; Grosso, R.A.; Fader, C.M. Hallmarks of aging: An autophagic perspective. Front. Endocrinol. 2019, 9, 790. [Google Scholar] [CrossRef]
- Hegde, A.N.; Smith, S.G.; Duke, L.M.; Pourquoi, A.; Vaz, S. Perturbations of ubiquitin-proteasome-mediated proteolysis in aging and Alzheimer’s disease. Front. Aging Neurosci. 2019, 11, 324. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Yung, R. Meta-inflammaging at the crossroad of geroscience. Aging Med. 2019, 2, 157–161. [Google Scholar] [CrossRef] [Green Version]
- Shintouo, C.M.; Mets, T.; Beckwee, D.; Bautmans, I.; Ghogomu, S.M.; Souopgui, J.; Leemans, L.; Meriki, H.D.; Njemini, R. Is inflammageing influenced by the microbiota in the aged gut? A systematic review. Exp. Gerontol. 2020, 141, 111079. [Google Scholar] [CrossRef] [PubMed]
- Onyango, I.G.; Jauregui, G.V.; Čarná, M.; Bennett, J.P., Jr.; Stokin, G.B. Neuroinflammation in Alzheimer’s disease. Biomedicines 2021, 9, 524. [Google Scholar] [CrossRef] [PubMed]
- Bradburn, S.; Sarginson, J.; Murgatroyd, C.A. Association of peripheral interleukin-6 with global cognitive decline in non-demented adults: A meta-analysis of prospective studies. Front. Aging Neurosci. 2018, 9, 438. [Google Scholar] [CrossRef] [Green Version]
- Marcos-Pérez, D.; Sánchez-Flores, M.; Proietti, S.; Bonassi, S.; Costa, S.; Teixeira, J.P.; Fernández-Tajes, J.; Pásaro, E.; Laffon, B.; Valdiglesias, V. Association of inflammatory mediators with frailty status in older adults: Results from a systematic review and meta-analysis. GeroScience 2020, 42, 1451–1473. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho-Júnior, H.J.; Marini, F.; Landi, F.; Marzetti, E. Circulating inflammatory, mitochondrial dysfunction, and senescence-related markers in older adults with physical frailty and sarcopenia: A BIOSPHERE exploratory study. Int. J. Mol. Sci. 2022, 23, 14006. [Google Scholar] [CrossRef]
- Harré, E.-M.; Roth, J.; Gerstberger, R.; Hübschle, T. Interleukin-6 mediates lipopolysaccharide-induced nuclear STAT3 translocation in astrocytes of rat sensory circumventricular organs. Brain Res. 2003, 980, 151–155. [Google Scholar] [CrossRef]
- Jurcau, A. Insights into the pathogenesis of neurodegenerative diseases: Focus on mitochondrial dysfunction and oxidative stress. Int. J. Mol. Sci. 2021, 22, 11847. [Google Scholar] [CrossRef]
- Bolaños, J.P.; Almeida, A.; Moncada, S. Glycolysis: A bioenergetic or a survival pathway? Trends Biochem. Sci. 2010, 35, 145–149. [Google Scholar] [CrossRef]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef]
- Stefanatos, R.; Sanz, A. The role of mitochondrial ROS in the aging brain. FEBS Lett. 2017, 592, 743–758. [Google Scholar] [CrossRef] [Green Version]
- Jurcau, A.; Ardelean, A.I. Oxidative stress in ischemia/reperfusion injuries following acute ischemic stroke. Biomedicines 2022, 10, 574. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Milenkovic, D.; Blaza, J.N.; Larsson, N.G.; Hirst, J. The enigma of the respiratory chain supercomplex. Cell Metab. 2017, 25, 765–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paß, T.; Wiesner, R.J.; Pla-Martin, D. Selective neuron vulnerability in common and rare diseases–mitochondria in the focus. Front. Mol. Biosci. 2021, 8, 676187. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, K.; Yokota, T.; Hasan-Olive, M.M.; Sherazi, N.; Fakouri, N.B.; Desler, C.; Regnell, C.E.; Larsen, S.; Rasmussen, L.J.; Dela, F.; et al. Initial brain aging: Heterogeneity of mitochondrial size is associated with decline in complex I-linked respiration in cortex and hippocampus. Neurobiol. Aging 2018, 61, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Stahon, K.E.; Bastian, C.; Griffith, S.; Kidd, G.J.; Brunet, S.; Baltan, S. Age-related changes in axonal and mitochondrial ultrastructure and function in white matter. J. Neurosci. 2016, 36, 9990–10001. [Google Scholar] [CrossRef] [Green Version]
- Pollard, A.K.; Craig, E.L.; Chakrabarti, L. Mitochondrial complex I activity measured by spectrophotometry is reduced across all brain regions in ageing and more specifically in neurodegeneration. PLoS ONE 2016, 11, e0157405. [Google Scholar] [CrossRef] [Green Version]
- Hiona, A.; Sanz, A.; Kujoth, G.C.; Pamplona, R.; Seo, A.Y.; Hofer, T.; Someya, S.; Miyakawa, T.; Nakayama, C.; Samhan-Arias, A.K.; et al. Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS ONE 2010, 5, e11468. [Google Scholar] [CrossRef] [PubMed]
- Zia, A.; Pourbagher-Shahri, A.M.; Farkhondeh, T.; Samarghandian, S. Molecular and cellular pathways contributing to brain aging. Behav. Brain Funct. 2021, 17, 6. [Google Scholar] [CrossRef]
- Sharma, C.; Kim, S.; Nam, Y.; Jung, U.J.; Kim, S.R. Mitochondrial dysfunction as a driver of cognitive impairment in Alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 4850. [Google Scholar] [CrossRef]
- Kowald, A.; Kirkwood, T.B.L. Resolving the enigma of the clonal expansion of mtDNA deletions. Genes 2018, 9, 126. [Google Scholar] [CrossRef] [Green Version]
- Jurcău, M.C.; Andronie-Cioara, F.L.; Jurcău, A.; Marcu, F.; Ţiț, D.M.; Pașcalău, N.; Nistor-Cseppentö, D.C. The link between oxidative stress, mitochondrial dysfunction and neuroinflammation in the pathophysiology of Alzheimer’s disease: Therapeutic implications and future perspectives. Antioxidants 2022, 11, 2167. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.; Moraes, C.T. Mechanisms linking mtDNA damage and aging. Free Radic. Biol. Med. 2015, 85, 250–258. [Google Scholar] [CrossRef] [Green Version]
- Jurk, D.; Wang, C.; Miwa, S.; Maddick, M.; Korolchuk, V.; Tsolou, A.; Gonos, E.S.; Thrasivoulou, C.; Saffrey, M.J.; Cameron, K.; et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 2012, 11, 996–1004. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.; Sagara, Y.; Liu, Y.; Maher, P.; Schubert, D. The regulation of reactive oxygen species production during programmed cell death. J. Cell. Biol. 1998, 141, 1423–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holper, L.; Ben-Shachar, D.; Mann, J.J. Multivariate meta-analyses of mitochondrial complex I and IV in major depressive disorder, bipolar disorder, schizophrenia, Alzheimer disease, and Parkinson disease. Neuropsychopharmacology 2019, 44, 837–849. [Google Scholar] [CrossRef]
- Beckervordersandforth, R.; Ebert, B.; Schäffner, I.; Moss, J.; Fiebig, C.; Shin, J.; Moore, D.L.; Ghosh, L.; Trinchero, M.F.; Stockburger, C.; et al. Role of mitochondrial metabolism in the control of early lineage progression and aging phenotypes in adult hippocampal neurogenesis. Neuron 2017, 93, 560–573. [Google Scholar] [CrossRef] [Green Version]
- Farris, S.; Ward, J.M.; Carstens, K.E.; Samadi, M.; Wang, Y.; Dudek, S.M. Hippocampal subregions express distinct dendritic transcriptomes that reveal differences in mitochondrial function in CA2. Cell Rep. 2019, 29, 522–539. [Google Scholar] [CrossRef] [PubMed]
- Godoy, J.A.; Rios, J.A.; Picón-Pagès, P.; Herrera-Fernández, V.; Swaby, B.; Crepin, G.; Vicente, R.; Fernández-Fernández, J.M.; Muñoz, F.J. Mitostasis, calcium and free radicals in health, aging and neurodegeneration. Biomolecules 2021, 11, 1012. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schampel, A.; Kuerten, S. Danger: High voltage—The role of voltage-gated calcium channels in central nervous system pathology. Cells 2017, 6, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagelhus, E.A.; Amiry-Moghaddam, M.; Bergersen, L.H.; Bjaalie, J.G.; Eriksson, J.; Gundersen, V.; Leergaard, T.B.; Morth, J.P.; Storm-Mathisen, J.; Torp, R.; et al. The glia doctrine: Addressing the role of glial cells in healthy brain ageing. Mech. Ageing Dev. 2013, 134, 449–459. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef] [PubMed]
- Sagan, L. On the origin of mitosing cells. J. Theor. Biol. 1967, 14, 255–274. [Google Scholar] [CrossRef]
- Bajwa, E.; Pointer, C.B.; Klegeris, A. The role of mitochondrial damage-associated mitochondrial patterns in chronic neuroinflammation. Mediators Inflamm. 2019, 2019, 4050796. [Google Scholar] [CrossRef] [Green Version]
- Gouveia, A.; Bajwa, E.; Klegeris, A. Extracellular cytochrome c as an intercellular signaling molecule regulating microglial functions. Biochim. Biophys. Acta 2017, 1861, 2274–2281. [Google Scholar] [CrossRef]
- Ahmad, A.; Crupi, R.; Campolo, M.; Genovese, T.; Esposito, E.; Cuzzocrea, S. Absence of TLR4 reduces neurovascular unit and secondary inflammatory process after traumatic brain injury in mice. PLoS ONE 2013, 8, e57208. [Google Scholar] [CrossRef] [Green Version]
- Balasubramanian, K.; Maeda, A.; Lee, J.S.; Mohammadyani, D.; Dar, H.H.; Jiang, J.F.; St Croix, C.M.; Watkins, S.; Tyurin, V.A.; Tyurina, Y.Y.; et al. Dichotomous roles for externalized cardiolipin in extracellular signaling: Promotion of phagocytosis and attenuation of innate immunity. Sci. Signal. 2015, 8, ra95. [Google Scholar] [CrossRef] [PubMed]
- Grazioli, S.; Pugin, J. Mitochondrial damage-associated molecular patterns: From inflammatory signaling to human diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef] [Green Version]
- Schindler, S.M.; Frank, M.G.; Annis, J.L.; Maier, S.F.; Klegeris, A. Pattern recognition receptors mediate pro-inflammatory effects of extracellular mitochondrial transcription factor A (TFAM). Mol. Cell. Neurosci. 2018, 89, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Mecocci, P.; MacGarvey, U.; Kaufman, A.E.; Koontz, D.; Shoffner, J.M.; Wallace, D.C.; Beal, M.F. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann. Neurol. 1993, 34, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Mecocci, P.L.; MacGarvey, U.; Beal, M.F. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol. 1994, 36, 747–751. [Google Scholar] [CrossRef]
- Takahashi, A.; Loo, T.M.; Okada, R.; Kamachi, F.; Watanabe, Y.; Wakita, M.; Watanabe, S.; Kawamoto, S.; Miyata, K.; Barber, G.N.; et al. Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat. Commun. 2018, 9, 1249. [Google Scholar] [CrossRef]
- Panov, A.V.; Dikalov. S.I. Cardiolipin, perhydroxyl radicals, and lipid peroxidation in mitochondrial dysfunctions and aging. Oxid. Med. Cell. Longev. 2020, 2020, 1323028. [Google Scholar] [CrossRef]
- Kehm, R.; Baldensperger, T.; Raupbach, J.; Höhn, A. Protein oxidation—Formation mechanisms, detection and relevance as biomarkers in human diseases. Redox Biol. 2021, 42, 101901. [Google Scholar] [CrossRef]
- Mecocci, P.; Cherubini, A.; Beal, M.F.; Cecchetti, R.; Chionne, F.; Polidori, M.C.; Romano, G.; Senin, U. Altered mitochondrial membrane fluidity in AD brain. Neurosci. Lett. 1996, 207, 129–132. [Google Scholar] [CrossRef]
- Mangialasche, F.; Baglioni, M.; Cecchetti, R.; Kivipelto, M.; Ruggiero, C.; Piobbico, D.; Kussmaul, L.; Monastero, R.; Brancorsini, S.; Mecocci, P. Lymphocytic mitochondrial aconitase activity is reduced in Alzheimer’s disease and mild cognitive impairment. J. Alzheimers Dis. 2015, 44, 649–660. [Google Scholar] [CrossRef]
- Mecocci, P.; Boccardi, V.; Cecchetti, R.; Bastiani, P.; Scamosci, M.; Ruggiero, C.; Baroni, M. A long journey into aging, brain aging, and Alzheimer’s disease following the oxidative stress tracks. J. Alzheimers Dis. 2018, 62, 1319–1335. [Google Scholar] [CrossRef] [PubMed]
- Michalska, P.; León, R. When it comes to an end: Oxidative stress crosstalk with protein aggregation and neuroinflammation induce neurodegeneration. Antioxidants 2020, 9, 740. [Google Scholar] [CrossRef] [PubMed]
- Groeger, G.; Quiney, C.; Cotter, T.G. Hydrogen peroxide as a cell-survival signaling molecule. Antioxid. Redox Signal. 2009, 11, 2655–2671. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A.; Ardelean, I.A. Molecular pathophysiological mechanisms of ischemia/reperfusion injuries after recanalization therapy for acute ischemic stroke. J. Integr. Neurosci. 2021, 20, 727–744. [Google Scholar] [PubMed]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 system: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [Green Version]
- Zgorzynska, E.; Dziedzic, B.; Walczewska, A. An overview of the Nrf2/ARE pathway and its role in neurodegenerative diseases. Int. J. Mol. Sci. 2021, 22, 9592. [Google Scholar] [CrossRef]
- Bloom, D.A.; Jaiswal, A.K. Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element. J. Biol. Chem. 2003, 278, 44675–44682. [Google Scholar] [CrossRef] [Green Version]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [Green Version]
- Higashida, H.; Salmina, A.B.; Olovyannikova, R.Y.; Hashii, M.; Yokoyama, S.; Koizumi, K.; Jin, D.; Liu, H.-X.; Lopatina, O.; Amina, S.; et al. Cyclic ADP-ribose as a universal calcium signal molecule in the nervous system. Neurochem. Int. 2007, 51, 192–199. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Wu, D.; Ding, X.; Ying, W. CD38 plays key roles in both antioxidation and cell survival of H2O2-treated primary rodent astrocytes. Int. J. Physiol. Pathophysiol. Pharmacol. 2014, 6, 102–108. [Google Scholar]
- Guerreiro, S.; Privat, A.-L.; Bressac, L.; Toulorge, D. CD38 in neurodegeneration and neuroinflammation. Cells 2020, 9, 471. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zou, L.; Zhang, X.; Branco, V.; Wang, J.; Carvalho, C.; Holmgren, A.; Lu, J. Redox signaling mediated by thioredoxin and glutathione systems in the central nervous system. Antioxid. Redox Signal. 2017, 27, 989–1010. [Google Scholar] [CrossRef] [Green Version]
- Zerbinati, C.; Iuliano, L. Cholesterol and related sterols autoxidation. Free Radic. Biol. Med. 2017, 111, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J. Searching for harmony in transition-metal signaling. Nat. Chem. Biol. 2015, 11, 744–747. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Kobylarek, D.; Iwanowski, P.; Lewandowska, Z.; Limphaibool, N.; Szafranek, S.; Labryczka, A.; Kozubski, W. Advances in the potential biomarkers of epilepsy. Front. Neurol. 2019, 10, 685. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.D.; Snyder, S.H.; Bohr, V.A. Signaling by cGAS-STING in neurodegeneration, neuroinflammation, and aging. Trends Neurosci. 2021, 44, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.S.; Sohn, D.H. Damage-associated molecular patterns in inflammatory diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Lewcock, J.W.; Schlepckow, K.; Di Paolo, G.; Tahirovic, S.; Monroe, K.M.; Haass, C. Emerging microglial biology defines novel therapeutic approaches for Alzheimer’s disease. Neuron 2020, 108, 801–821. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Hooli, B.; Mullin, K.; Jin, S.C.; Cella, M.; Ulland, T.K.; Wang, Y.; Tanzi, R.E.; Colonna, M. Alzheimer’s disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimers Dement. 2017, 13, 381–387. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, J.D.; Ulland, T.K.; Colonna, M.; Holtzman, D.M. Elucidating the role of TREM2 in Alzheimer’s disease. Neuron 2017, 94, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colonna, M.; Wang, Y. TREM2 variants: New keys to decipher Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2016, 17, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Jay, T.R.; Miller, C.M.; Cheng, P.J.; Graham, L.C.; Bemiller, S.; Broihier, M.L.; Xu, G.; Margevicius, D.; Karlo, J.C.; Sousa, G.L.; et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med. 2015, 212, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, J.; Du, F.; Xu, H.; Sun, L.; Chen, Z.; Brautigam, C.A.; Zhang, X.; Chen, Z.J. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep. 2014, 6, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Jiang, F.; Kong, L.; Li, B.; Yang, Y.; Zhang, L.; Liu, B.; Zheng, Y.; Gao, C. Cutting edge: USP27X deubiquitinates and stabilizes the DNA sensor cGAS to regulate cytosolic DNA-mediated signaling. J. Immunol. 2019, 203, 2049–2054. [Google Scholar] [CrossRef]
- de Weerd, N.A.; Nguyen, T. The interferons and their receptors—Distribution and regulation. Immunol. Cell. Biol. 2012, 90, 483–491. [Google Scholar] [CrossRef]
- Owens, T.; Khorooshi, R.; Wlodarczyk, A.; Asgari, N. Interferons in the central nervous system: A few instruments play many tunes. Glia 2014, 62, 339–355. [Google Scholar] [CrossRef]
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory response in the CNS: Friend or foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J.R. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Yabal, M.; Calleja, D.J.; Simpson, D.S.; Lawlor, K.E. Stressing out the mitochondria: Mechanistic insights into NLRP3 inflammasome activation. J. Leukoc. Biol. 2019, 10, 377–399. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, B.; Xu, L.; Yu, S.; Fu, J.; Wang, J.; Yan, X.; Su, J. ROS-Induced mtDNA Release: The emerging messenger for communication between neurons and innate immune cells during neurodegenerative disorder progression. Antioxidants 2021, 10, 1917. [Google Scholar] [CrossRef]
- Verhratsky, A.; Nedergaard, M. Physiology of astroglia. Physiol. Rev. 2018, 98, 239–398. [Google Scholar] [CrossRef] [PubMed]
- Hussaini, S.M.Q.; Jang, M.H. New roles for old glue: Astrocyte function in synaptic plasticity and neurological disorders. Int. Neurourol. J. 2018, 22, S106–S114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.; Torres, C. Astrocyte senescence: Evidence and significance. Aging Cell 2019, 18, e12937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, M.H.; Fatima, M.; Mondal, A.C. Influence of microglia and astrocyte activation in the neuroinflammatory pathogenesis of Alzheimer’s disease: Rational insights for the therapeutic approaches. J. Clin. Neurosci. 2018, 59, 6–11. [Google Scholar] [CrossRef]
- Han, X.; Zhang, T.; Liu, H.; Mi, Y.; Gou, X. Astrocyte senescence and Alzheimer’s disease: A review. Front. Aging Neurosci. 2020, 12, 148. [Google Scholar] [CrossRef]
- Evans, R.J.; Wyllie, F.S.; Wynford-Thomas, D.; Kipling, D.; Jones, C.J. A P53-dependent, telomere-independent proliferative life span barrier in human astrocytes consistent with the molecular genetics of glioma development. Cancer Res. 2003, 63, 4854–4861. [Google Scholar]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of cellular senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Laberge, R.M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular senescence: Defining a path forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Sompol, P.; Norris, C.M. Ca2+, astrocyte activation and calcineurin/NFAT signaling in age-related neurodegenerative diseases. Front. Aging Neurosci. 2018, 10, 199. [Google Scholar] [CrossRef] [Green Version]
- Manocha, G.D.; Ghatak, A.; Puig, K.L.; Kraner, S.D.; Norris, C.M.; Combs, C.K. NFATc2 modulates microglial activation in the AβPP/PS1 mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2017, 58, 775–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sompol, P.; Furman, J.L.; Pleiss, M.M.; Kraner, S.D.; Artiushin, I.A.; Batten, S.R.; Quintero, J.E.; Simmerman, L.A.; Beckett, T.L.; Lovell, M.A.; et al. Calcineurin/NFAT signaling in activated astrocytes drives network hyperexcitability in Aβ-bearing mice. J. Neurosci. 2017, 37, 6132–6148. [Google Scholar] [CrossRef] [Green Version]
- Vasileiou, P.V.S.; Evangelou, K.; Vlasis, K.; Fildisis, G.; Panayiotidis, M.I.; Chronopoulos, E.; Passias, P.G.; Kouloukoussa, M.; Gorgoulis, V.G.; Havaki, S. Mitochondrial homeostasis and cellular senescence. Cells 2019, 8, 686. [Google Scholar] [CrossRef] [Green Version]
- Mombach, J.C.; Vendrusculo, B.; Bugs, C.A. A model for p38/MAPK-induced astrocyte senescence. PLoS ONE 2015, 10, e0125217. [Google Scholar] [CrossRef]
- Davalos, A.R.; Kawahara, M.; Malhotra, G.K.; Schaum, N.; Huang, J.; Ved, U.; Beausejour, C.M.; Coppe, J.P.; Rodier, F.; Campisi, J. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell. Biol. 2013, 201, 613–629. [Google Scholar] [CrossRef]
- Zhang, X.; Lao, K.; Qiu, Z.; Rahman, M.S.; Zhang, Y.; Gou, X. Potential astrocytic receptors and transporters in the pathogenesis of Alzheimer’s disease. J. Alzheimers Dis. 2019, 67, 1109–1122. [Google Scholar] [CrossRef]
- Pajarillo, E.; Rizor, A.; Lee, J.; Aschner, M.; Lee, E. The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: Potential targets for neurotherapeutics. Neuropharmacology 2019, 161, 107559. [Google Scholar] [CrossRef]
- Bellaver, B.; Souza, D.G.; Souza, D.O.; Quincozes-Santos, A. Hippocampal astrocyte cultures from adult and aged rats reproduce changes in glial functionality observed in the aging brain. Mol. Neurobiol. 2017, 54, 2969–2985. [Google Scholar] [CrossRef]
- Jurcau, A.; Simion, A. Cognition, statins, and cholesterol in elderly ischemic stroke patients: A neurologist’s perspective. Medicina 2021, 57, 616. [Google Scholar] [CrossRef]
- Li, W.; He, P.; Huang, Y.; Li, Y.F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M.; et al. Selective autophagy of intracellular organelles: Recent research advances. Theranostics 2021, 11, 222–256. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and neurodegeneration: Pathogenic mechanisms and therapeutic opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheon, S.Y.; Kim, H.; Rubinsztein, D.C.; Lee, J.E. Autophagy, cellular aging and age-related human diseases. Exp. Neurobiol. 2019, 28, 643–657. [Google Scholar] [CrossRef] [PubMed]
- Moreau, K.; Renna, M.; Rubinsztein, D.C. Connections between SNAREs and autophagy. Trends Biochem. Sci. 2013, 38, 57–63. [Google Scholar] [CrossRef]
- Lőrincz, P.; Juhász, G. Autophagosome-Lysosome Fusion. J. Mol. Biol. 2020, 432, 2462–2482. [Google Scholar] [CrossRef]
- Baker, R.W.; Hughson, F.M. Chaperoning SNARE assembly and disassembly. Nat. Rev. Mol. Cell Biol. 2016, 17, 465–479. [Google Scholar] [CrossRef] [Green Version]
- Ahmadpour, D.; Babazadeh, R.; Nystrom, T. Hitchhiking on vesicles: A way to harness age-related proteopathies? FEBS J. 2020, 287, 5068–5079. [Google Scholar] [CrossRef]
- Guebel, D.V.; Torres, N.V. Sexual dimorphism and aging in the human hippocampus: Identification, validation, and impact of differentially expressed genes by factorial microarray and network analysis. Front. Aging Neurosci. 2016, 8, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, N.; Zhang, L.; Li, R.; Fu, W.; Ma, K.; Li, X.; Wang, L.; Wang, J.; Zhang, H.; et al. Autophagy regulates chromatin ubiquitination in DNA damage response through elimination of SQSTM1/p62. Mol. Cell 2016, 63, 34–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- Sun, Y.; Coppé, J.P.; Lam, E.W. Cellular senescence: The sought or the unwanted? Trends Mol. Med. 2018, 24, 871–885. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–990. [Google Scholar] [CrossRef]
- Kang, J.; Lemaire, H.G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Müller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736. [Google Scholar] [CrossRef]
- Maia, M.A.; Sousa, E. BACE-1 and γ-secretase as therapeutic targets for Alzheimer’s disease. Pharmaceuticals 2019, 12, 41. [Google Scholar] [CrossRef] [Green Version]
- Bursavich, M.G.; Harrison, B.A.; Blain, J.F. Gamma secretase modulators: New Alzheimer’s drugs on the horizon? J. Med. Chem. 2016, 59, 7389–7409. [Google Scholar] [CrossRef]
- Yang, L.B.; Lindholm, K.; Yan, R.; Citron, M.; Xia, W.; Yang, X.L.; Beach, T.; Sue, L.; Wong, P.; Price, D.; et al. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 2003, 9, 3–4. [Google Scholar] [CrossRef]
- Coulson, D.T.; Beyer, N.; Quinn, J.G.; Brockbank, S.; Hellemans, J.; Irvine, G.B.; Ravid, R.; Johnston, J.A. BACE1 mRNA expression in Alzheimer’s disease postmortem brain tissue. J. Alzheimers Dis. 2010, 22, 1111–1122. [Google Scholar] [CrossRef]
- Bahn, G.; Park, J.-S.; Yun, U.J.; Lee, Y.J.; Choi, Y.; Park, J.S.; Baek, S.H.; Choi, B.Y.; Cho, Y.S.; Kim, H.K.; et al. NRF2/ARE pathway negatively regulates BACE1 expression and ameliorates cognitive deficits in mouse Alzheimer’s models. Proc. Natl. Acad. Sci. USA 2019, 116, 12516–12523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, O.M.; Reiche, J.; Schmidt, V.; Gotthardt, M.; Spoelgen, R.; Behlke, J.; von Arnim, C.A.; Breiderhoff, T.; Jansen, P.; Wu, X.; et al. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2005, 102, 13461–13466. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer disease: An update on pathobiology and treatment strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Gremer, L.; Schölzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.G.; Tusche, M.; Lopez-Iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril structure of amyloid-β(1-42) by cryo-electron microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosna, J.; Philipp, S.; Albay, R., 3rd; Reyes-Ruiz, J.M.; Baglietto-Vargas, D.; LaFerla, F.M.; Glabe, C.G. Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 11. [Google Scholar] [PubMed]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef]
- Rabinovici, G.D.; Gatsonis, C.; Apgar, C.; Chaudhary, K.; Gareen, I.; Hanna, L.; Hendrix, J.; Hillner, B.E.; Olson, C.; Lesman-Segev, O.H.; et al. Association of amyloid positron emission tomography with subsequent change in clinical management among Medicare beneficiaries with mild cognitive impairment or dementia. JAMA 2019, 321, 1286–1294. [Google Scholar] [CrossRef]
- Jagust, W. Imaging the evolution and pathophysiology of Alzheimer disease. Nat. Rev. Neurosci. 2018, 19, 687–700. [Google Scholar] [CrossRef]
- Vergara, C.; Houben, S.; Suain, V.; Yilmaz, Z.; De Decker, R.; Vanden Dries, V.; Boom, A.; Mansour, S.; Leroy, K.; Ando, K.; et al. Amyloid-β pathology enhances pathological fibrillary tau seeding induced by Alzheimer PHF in vivo. Acta Neuropathol. 2019, 137, 397–412. [Google Scholar] [CrossRef] [Green Version]
- Pickett, E.K.; Herrmann, A.G.; McQueen, J.; Abt, K.; Dando, O.; Tulloch, J.; Jain, P.; Dunnett, S.; Sohrabi, S.; Fjeldstad, M.P.; et al. Amyloid beta and tau cooperate to cause reversible behavioral and transcriptional deficits in a model of Alzheimer’s disease. Cell Rep. 2019, 29, 3592–3604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolós, M.; Llorens-Martín, M.; Jurado-Arjona, J.; Hernández, F.; Rábano, A.; Avila, J. Direct evidence of internalization of Tau by microglia in vitro and in vivo. J. Alzheimers Dis. 2016, 50, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Cserép, C.; Pósfai, B.; Lénárt, N.; Fekete, R.; László, Z.I.; Lele, Z.; Orsolits, B.; Molnár, G.; Heindl, S.; Schwarcz, A.D.; et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 2020, 367, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Efthymiou, A.G.; Goate, A.M. Late onset Alzheimer’s disease genetics implicate microglial pathways in disease risk. Mol. Neurodegener. 2017, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Bisceglia, P.; Lo Vecchio, F.; Latino, R.R.; Gravina, C.; Urbano, M.; la Torre, A.; Desina, G.; Greco, A.; Leone, M.; Antonioni, A. Italian Case Report with a Double Mutation in PSEN1 (K311R and E318G). Neurol. Int. 2022, 14, 417–422. [Google Scholar] [CrossRef]
- Escott-Price, V.; Myers, A.J.; Huentelman, M.; Hardy, J. Polygenic risk score analysis of pathologically confirmed Alzheimer disease. Ann. Neurol. 2017, 82, 311–314. [Google Scholar] [CrossRef]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Da Mesquita, S.; Louveau, A.; Vaccari, A.; Smirnov, I.; Cornelison, R.C.; Kingsmore, K.M.; Contarino, C.; Onengut-Gumuscu, S.; Farber, E.; Raper, D.; et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature 2018, 560, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Gate, D.; Saligrama, N.; Leventhal, O.; Yang, A.C.; Unger, M.S.; Middeldorp, J.; Chen, K.; Lehallier, B.; Channappa, D.; De Los Santos, M.B.; et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 2020, 577, 399–404. [Google Scholar] [CrossRef]
- Dodiya, H.B.; Frith, M.; Sidebottom, A.; Cao, Y.; Koval, J.; Chang, E.; Sisodia, S.S. Synergistic depletion of gut microbial consortia, but not individual antibiotics, reduces amyloidosis in APPPS1-21 Alzheimer’s transgenic mice. Sci. Rep. 2020, 10, 8183. [Google Scholar] [CrossRef] [PubMed]
- Doens, D.; Fernandez, P.L. Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2014, 11, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Revilla, J.; Alonso-Bellido, I.M.; Burguillos, M.A.; Herrera, A.J.; Espinosa-Oliva, A.M.; Ruiz, R.; Cruz-Hernández, L.; García-Domínguez, I.; Roca-Ceballos, M.A.; Santiago, M.; et al. Reformulating pro-oxidant microglia in neurodegeneration. J. Clin. Med. 2019, 8, 1719. [Google Scholar] [CrossRef] [PubMed]
- Stancu, I.C.; Cremers, N.; Vanrusselt, H.; Couturier, J.; Vanoosthuyse, A.; Kessels, S.; Lodder, C.; Brône, B.; Huaux, F.; Octave, J.N.; et al. Aggregated Tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019, 137, 599–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, M.; Shiwaku, H.; Tanaka, H.; Obita, T.; Ohuchi, S.; Yoshioka, Y.; Jin, X.; Kondo, K.; Fujita, K.; Homma, H.; et al. Tau activates microglia via the PQBP1-cGAS-STING pathway to promote brain inflammation. Nat. Commun. 2021, 12, 6565. [Google Scholar] [CrossRef]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Microglia and aging: The role of the TREM2–DAP12 and CX3CL1-CX3CR1 axes. Int. J. Mol. Sci. 2018, 19, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; Chen, X.-F.; Wang, T.; Wang, Z.; Liao, C.; Wang, Z.; Huang, R.; Wang, D.; Li, X.; Wu, L.; et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 2017, 214, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Suárez-Calvet, M.; Kleinberger, G.; Araque Caballero, M.Á.; Brendel, M.; Rominger, A.; Alcolea, D.; Fortea, J.; Lleó, A.; Blesa, R.; Gispert, J.D.; et al. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol. Med. 2016, 8, 466–476. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Piña-Crespo, J.C.; Zhang, M.; et al. TREM2 is a receptor for β-amyloid that mediates microglial function. Neuron 2018, 97, 1023–1031. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Jia, L.; Liu, C.-C.; Rong, Z.; Zhong, L.; Yang, L.; Chen, X.-F.; Fryer, J.D.; Wang, X.; Zhang, Y.-W.; et al. TREM2 promotes microglial survival by activating wnt/β-catenin pathway. J. Neurosci. 2017, 37, 1772–1784. [Google Scholar] [CrossRef] [Green Version]
- Ulland, T.K.; Song, W.M.; Huang, S.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 2017, 170, 649–663. [Google Scholar] [CrossRef]
- Lee, C.Y.D.; Daggett, A.; Gu, X.; Jiang, L.-L.; Langfelder, P.; Li, X.; Wang, N.; Zhao, Y.; Park, C.S.; Cooper, Y.; et al. Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer’s disease models. Neuron 2018, 97, 1032–1048. [Google Scholar] [CrossRef] [Green Version]
- Yuan, C.; Aierken, A.; Xie, Z.; Li, N.; Zhao, J.; Qing, H. The age-related microglial transformation in Alzheimer’s disease pathogenesis. Neurobiol. Aging 2020, 92, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Cornejo, F.; Vruwink, M.; Metz, C.; Muñoz, P.; Salgado, N.; Poblete, J.; Andrés, M.E.; Eugenín, J.; von Bernhardi, R. Scavenger receptor-A deficiency impairs immune response of microglia and astrocytes potentiating Alzheimer’s disease pathophysiology. Brain Behav. Immun. 2018, 69, 336–350. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.D.; Zhu, Y.G.; Lin, N.; Zhang, J.; Ye, Q.Y.; Huang, H.P.; Chen, X.C. Microglial phagocytosis induced by fibrillar β-amyloid is attenuated by oligomeric β-amyloid: Implications for Alzheimer’s disease. Mol. Neurodegener. 2011, 6, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L. CD33 in Alzheimer’s disease: Biology, pathogenesis, and therapeutics: A mini-review. Gerontology 2019, 65, 323–331. [Google Scholar] [CrossRef]
- Griciuk, A.; Patel, S.; Federico, A.N.; Choi, S.H.; Innes, B.J.; Oram, M.K.; Cereghetti, G.; McGinty, D.; Anselmo, A.; Sadreyev, R.I.; et al. TREM2 acts downstream of CD33 in modulating microglial pathology in Alzheimer’s disease. Neuron 2019, 103, 820–835. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; El Khoury, J.; Medeiros, L.A.; Terada, K.; Geula, C.; Luster, A.D.; Freeman, M.W. A CD36-initiated signaling cascade mediates inflammatory effects of β-amyloid. J. Biol. Chem. 2002, 277, 47373–47379. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Wang, J.; Yao, L.; Li, C.; Wang, J.; Liu, Y.; Tao, X.; Sun, H.; Liao, H. The adhesion and migration of microglia to β-amyloid (Aβ) is decreased with aging and inhibited by Nogo/NgR pathway. J. Neuroinflamm. 2018, 15, 210. [Google Scholar] [CrossRef] [Green Version]
- Czirr, E.; Castello, N.A.; Mosher, K.I.; Castellano, J.M.; Hinkson, I.V.; Lucin, K.M.; Baeza-Raja, B.; Ryu, J.K.; Li, L.; Farina, S.N.; et al. Microglial complement receptor 3 regulates brain Aβ levels through secreted proteolytic activity. J. Exp. Med. 2017, 214, 1081–1092. [Google Scholar] [CrossRef] [Green Version]
- Silverman, S.M.; Ma, W.; Wang, X.; Zhao, L.; Wong, W.T. C3- and CR3-dependent microglial clearance protects photoreceptors in retinitis pigmentosa. J. Exp. Med. 2019, 216, 1925–1943. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [Green Version]
- Streit, W.J.; Braak, H.; Del Tredici, K.; Leyh, J.; Lier, J.; Khoshbouei, H.; Eisenlöffel, C.; Müller, W.; Bechmann, I. Microglial activation occurs late during preclinical Alzheimer’s disease. Glia 2018, 66, 2550–2562. [Google Scholar] [CrossRef]
- Hopp, S.C.; Lin, Y.; Oakley, D.; Roe, A.D.; DeVos, S.L.; Hanlon, D.; Hyman, B.T. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 269. [Google Scholar] [CrossRef] [Green Version]
- Davies, D.S.; Ma, J.; Jegathees, T.; Goldsbury, C. Microglia show altered morphology and reduced arborization in human brain during aging and Alzheimer’s disease. Brain Pathol. 2017, 27, 795–808. [Google Scholar] [CrossRef]
- Rivera-Escalera, F.; Pinney, J.J.; Owlett, L.; Ahmed, H.; Thakar, J.; Olschowka, J.A.; Elliott, M.R.; O’Banion, M.K. IL-1beta-driven amyloid plaque clearance is associated with an expansion of transcriptionally reprogrammed microglia. J. Neuroinflamm. 2019, 16, 261. [Google Scholar] [CrossRef]
- Lau, S.F.; Fu, A.K.Y.; Ip, N.Y. Cytokine signaling convergence regulates the microglial state transition in Alzheimer’s disease. Cell. Mol. Life Sci. 2021, 78, 4703–4712. [Google Scholar] [CrossRef] [PubMed]
- Banyopadhyay, S.; Hartley, D.M.; Cahill, C.M.; Lahiri, D.K.; Chattopadhyay, N.; Rogers, J.T. Interleukin-1alpha stimulates non-amyloidogenic pathway by alpha-secretase (ADAM-10 and ADAM-17) cleavage of APP in human astrocytic cells involving p38 MAP kinase. J. Neurosci. Res. 2006, 84, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Koper, O.M.; Kaminska, J.; Sawicki, K.; Kemona, H. CXCL9, CXCL10, CXCL11, and their receptor (CXCR3) in neuroinflammation and neurodegeneration. Adv. Clin. Exp. Med. 2018, 27, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Xu, G.; Jay, T.R.; Bhatta, S.; Kim, K.W.; Jung, S.; Landreth, G.E.; Ransohoff, R.M.; Lamb, B.T. Opposing effects of membrane-anchored CX3CL1 on amyloid and tau pathologies via the p38 MAPK pathway. J. Neurosci. 2014, 34, 12538–12546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Zhang, Y.; Sterling, K.; Song, W. Brain-derived neurotrophic factor in Alzheimer’s disease and its pharmaceutical potential. Transl. Neurodegener. 2022, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Sharif, M.; Noroozian, M.; Hashemian, F. Do serum GDNF levels correlate with severity of Alzheimer’s disease? Neurol. Sci. 2021, 42, 2865–2872. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, C.; Tiberi, A.; Giustizieri, M.; Marrone, M.C.; Gobbo, F.; Carucci, N.M.; Meli, G.; Arisi, I.; D’Onofrio, M.; Marinelli, S.; et al. NGF steers microglia toward a neuroprotective phenotype. Glia 2018, 66, 1395–1416. [Google Scholar] [CrossRef]
- Schindowski, K.; Belarbi, K.; Buee, L. Neurotrophic factors in Alzheimer’s disease: Role of axonal transport. Genes Brain Behav. 2008, 7, 43–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.L.; Li, W.W.; Xu, Y.L.; Gao, S.H.; Xu, M.Y.; Bu, X.L.; Liu, Y.H.; Wang, J.; Zhu, J.; Zeng, F.; et al. Neurotrophin receptor p75 mediates amyloid beta-induced tau pathology. Neurobiol. Dis. 2019, 132, 104567. [Google Scholar] [CrossRef] [PubMed]
- Ogunmokun, G.; Dewanjee, S.; Chakraborty, P.; Valupadas, C.; Chaudhary, A.; Kolli, V.; Anand, U.; Vallamkondu, J.; Goel, P.; Paluru, H.P.R.; et al. The potential role of cytokines and growth factors in the pathogenesis of Alzheimer’s disease. Cells 2021, 10, 2790. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Lyra, E.; Silva, N.M.; Gonçalves, R.A.; Pascoal, T.A.; Lima-Filho, R.A.S.; Resende, E.P.F.; Vieira, E.L.M.; Teixeira, A.L.; de Souza, L.C.; Peny, J.A.; et al. Pro-inflammatory interleukin-6 signaling links cognitive impairments and peripheral metabolic alterations in Alzheimer’s disease. Transl. Psychiatry 2021, 11, 251. [Google Scholar] [CrossRef]
- Fakhoury, M. Inflammation in Alzheimer’s disease. Curr. Alzheimer Res. 2020, 17, 959–961. [Google Scholar] [CrossRef]
- D’Anna, L.; Abu-Rumeileh, S.; Fabris, M.; Pistis, C.; Baldi, A.; Sanvilli, N.; Curcio, F.; Gigli, G.L.; D’Anna, S.; Valente, M. Serum interleukin-10 levels correlate with cerebrospinal fluid amyloid beta deposition in Alzheimer disease patients. Neurodegener. Dis. 2017, 17, 227–234. [Google Scholar] [CrossRef]
- Ojala, J.O.; Sutinen, E.M. The role of interleukin-18, oxidative stress and metabolic syndrome in Alzheimer’s disease. J. Clin. Med. 2017, 6, 55. [Google Scholar] [CrossRef] [Green Version]
- Culjak, M.; Perkovic, M.N.; Uzun, S.; Strac, D.S.; Erjavec, G.N.; Leko, M.B.; Simic, G.; Tudor, L.; Konjevod, M.; Kozumplik, O.; et al. The association between TNF-alpha, IL-1 alpha and IL-10 with Alzheimer’s disease. Curr. Alzheimer Res. 2020, 17, 972–984. [Google Scholar] [CrossRef] [PubMed]
- Estrada, L.D.; Oliveira-Cruz, L.; Cabrera, D. Transforming growth factor beta type 1 role in neurodegeneration: Implications for Alzheimer’s disease. Curr. Protein Pept. Sci. 2018, 34, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Joly-Amado, A.; Hunter, J.; Quadri, Z.; Zamudio, F.; Rocha-Rangel, P.V.; Chan, D.; Kesarwani, A.; Nash, K.; Lee, D.C.; Morgan, D.; et al. CCL2 overexpression in the brain promotes glial activation and accelerates tau pathology in a mouse model of tauopathy. Front. Immunol. 2020, 11, 997. [Google Scholar] [CrossRef]
- Park, J.; Baik, S.H.; Mook-Jung, I.; Irimia, D.; Cho, H. Mimicry of central-peripheral immunity in Alzheimer’s disease and discovery of neurodegenerative roles in neutrophil. Front. Immunol. 2019, 10, 2231. [Google Scholar] [CrossRef]
- Wojcieszak, J.; Kuczynska, K.; Zawilska, J.B. role of chemokines in the development and progression of Alzheimer’s disease. J. Mol. Neurosci. 2022, 72, 1929–1951. [Google Scholar] [CrossRef] [PubMed]
- Guedes, J.R.; Lao, T.; Cardoso, A.L.; El Khoury, J. Roles of microglial and monocyte chemokines and their receptors in regulating Alzheimer’s disease-associated amyloid-β and tau pathologies. Front. Neurol. 2018, 9, 549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taipa, R.; das Neves, S.P.; Sousa, A.L.; Fernandes, J.; Pinto, C.; Correia, A.P.; Santos, E.; Pinto, P.S.; Carneiro, P.; Costa, P.; et al. Pro-inflammatory and anti-inflammatory cytokines in the CSF of patients with Alzheimer’s disease and their correlation with cognitive decline. Neurobiol. Aging 2019, 76, 125–132. [Google Scholar] [CrossRef]
- Agrawal, I.; Jha, S. Mitochondrial dysfunction and Alzheimer’s disease: Role of microglia. Front. Aging Neurosci. 2020, 12, 252. [Google Scholar] [CrossRef]
- Van Horssen, J.; van Schaik, P.; Witte, M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci. Lett. 2019, 710, 132931. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, P.; Sripada, L.; Singh, K.; Bhatelia, K.; Singh, R.; Singh, R. TNF-α regulates miRNA targeting mitochondrial complex-I and induces cell death in dopaminergic cells. Biochim. Biophys. Acta 2015, 1852, 451–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, K.; Wagner, P.D.; Breen, E.C. TNF-α-mediated reduction in PGC-1α may impair skeletal muscle function after cigarette smoke exposure. J. Cell. Physiol. 2010, 222, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Cioffi, F.; Adam, R.H.I.; Broersen, K. Molecular mechanisms and genetics of oxidative stress in Alzheimer’s disease. J. Alzheimer’s Dis. 2019, 72, 981–1017. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell., B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Sultana, R.; Poon, H.F.; Cai, J.; Pierce, W.M.; Merchant, M.; Klein, J.B.; Markesbery, W.R.; Butterfield, D.A. Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiol. Dis. 2006, 22, 76–87. [Google Scholar] [CrossRef]
- Reed, T.; Perluigi, M.; Sultana, R.; Pierce, W.M.; Klein, J.B.; Turner, D.M.; Coccia, R.; Markesbery, W.R.; Butterfield, D.A. Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: Insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiol. Dis. 2008, 30, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; van Horssen, J. Oxidative stress and its impact on neurons and glia in multiple sclerosis lesions. Biochim. Biophys. Acta 2016, 1862, 506–510. [Google Scholar] [CrossRef]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Yan, S.D. Mitochondrial Abeta: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Tamagno, E.; Guglielmotto, M.; Vasciaveo, V.; Tabaton, M. Oxidative Stress and Beta Amyloid in Alzheimer’s Disease. Which Comes First: The Chicken or the Egg? Antioxidants 2021, 10, 1479. [Google Scholar] [CrossRef]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [Green Version]
- Shafiei, S.M.; Guerrero-Muñoz, M.J.; Castillo-Caranza, D.L. Tau oligomers: Cytotoxicity, propagation, and mitochondrial damage. Front. Aging Neurosci. 2017, 9, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takashima, A. Tauopathies and tau oligomers. J. Alzheimers Dis. 2013, 37, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Castillo-Caranza, D.L.; Sengupta, U.; Clos, A.L.; Jackson, G.R.; Kayed, R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 2011, 6, 39. [Google Scholar] [CrossRef] [Green Version]
- Pérez, M.J.; Jara, C.; Quintanilla, R.A. Contribution of tau pathology to mitochondrial impairment in neurodegeneration. Front. Neurosci. 2018, 12, 441. [Google Scholar] [CrossRef]
- Veitch, D.P.; Weiner, M.W.; Aisen, P.S.; Beckett, L.A.; Cairns, N.J.; Green, R.C.; Harvey, D.; Jack, C.R., Jr.; Jagust, W.; Morris, J.C.; et al. Alzheimer’s Disease Neuroimaging Initiative. Understanding disease progression and improving Alzheimer’s disease clinical trials: Recent highlights from the Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement. 2019, 15, 106–152. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Haroutunian, V.; Katsel, P.; Cardozo, C.P.; Ho, L.; Buxbaum, J.D.; Pasinetti, G.M. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 2009, 66, 352–361. [Google Scholar] [CrossRef]
- Wiedemann, N.; Pfanner, N. Mitochondrial machineries for protein import and assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef] [Green Version]
- Chai, Y.L.; Xing, H.; Chong, J.R.; Francis, P.T.; Ballard, C.G.; Chen, C.P.; Lai, M.K.P. Mitochondrial translocase of the outer membrane alterations may underlie dysfunctional oxidative phosphorylation in Alzheimer’s disease. J. Alzheimers Dis. 2018, 61, 793–801. [Google Scholar] [CrossRef]
- Wright, G.; Terada, K.; Yano, M.; Sergeev, I.; Mori, M. Oxidative stress inhibits the mitochondrial import of preproteins and leads to their degradation. Exp. Cell. Res. 2001, 263, 107–117. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Trushin, S.; Christensen, T.A.; Bachmeier, B.V.; Gateno, B.; Schroeder, A.; Yao, J.; Itoh, K.; Sesaki, H.; Poon, W.W.; et al. Altered brain energetics induces mitochondrial fission arrest in Alzheimer’s Disease. Sci. Rep. 2016, 6, 18725. [Google Scholar] [CrossRef] [Green Version]
- Jurcau, A.; Nunkoo, V.S. Tau-targeted therapy in Alzheimer’s disease: History and current state. In Frontiers in Clinical Drug Research; Ibarra Arias, J.J.J., Ed.; Bentham Science Publishers: Singapore, 2021; Volume 2, pp. 56–138. [Google Scholar]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Jiang, S.; Shao, C.; Tang, F.; Wang, W.; Zhu, X. Dynamin-like protein 1 cleavage by calpain in Alzheimer’s disease. Aging Cell 2019, 18, e12912. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.I.; Lee, K.H.; Gabr, A.A.; Choi, G.E.; Kim, J.S.; Ko, S.H.; Han, H.J. Abeta-induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis. Biochim. Biophys. Acta 2016, 1863, 2820–2834. [Google Scholar] [CrossRef]
- Kim, B.; Park, J.; Chang, K.T.; Lee, D.S. Peroxiredoxin 5 prevents amyloid-beta oligomer-induced neuronal cell death by inhibiting ERK-Drp1-mediated mitochondrial fragmentation. Free Radic. Biol. Med. 2016, 90, 184–194. [Google Scholar] [CrossRef]
- Ishihara, N.; Fujita, Y.; Oka, T.; Mihara, K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006, 25, 2966–2977. [Google Scholar] [CrossRef] [Green Version]
- Jara, C.; Aránguiz, A.; Cerpa, W.; Tapia-Rojas, C.; Quintanilla, R.A. Genetic ablation of tau improves mitochondrial function and cognitive abilities in the hippocampus. Redox Biol. 2018, 18, 279–294. [Google Scholar] [CrossRef]
- Wang, W.; Yin, J.; Ma, X.; Zhao, F.; Siedlak, S.L.; Wang, Z.; Torres, S.; Fujioka, H.; Xu, Y.; Perry, G.; et al. Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum. Mol. Genet. 2017, 26, 4118–4131. [Google Scholar] [CrossRef] [Green Version]
- Szabo, L.; Eckert, A.; Grimm, A. Insights into Disease-Associated Tau Impact on Mitochondria. Int. J. Mol. Sci. 2020, 21, 6344. [Google Scholar] [CrossRef]
- Hu, Y.; Li, X.C.; Wang, Z.H.; Luo, Y.; Zhang, X.; Liu, X.P.; Feng, Q.; Wang, Q.; Yue, Z.; Chen, Z.; et al. Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget 2016, 7, 17356–17368. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.M. Neuronal lysosomes. Neurosci. Lett. 2019, 697, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tammineni, P.; Ye, X.; Feng, T.; Aikal, D.; Cai, Q. Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer’s disease neurons. Elife 2017, 6, e21776. [Google Scholar] [CrossRef] [PubMed]
- Gowrishankar, S.; Yuan, P.; Wu, Y.; Schrag, M.; Paradise, S.; Grutzendler, J.; De Camilli, P.; Ferguson, S.M. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc. Natl. Acad. Sci. USA 2015, 112, E3699–E3708. [Google Scholar] [CrossRef] [Green Version]
- Flannery, P.J.; Trushina, E. Mitochondrial dynamics and transport in Alzheimer’s disease. Mol. Cell. Neurosci. 2019, 98, 109–120. [Google Scholar] [CrossRef]
- Zheng, Y.R.; Zhang, X.N.; Chen, Z. Mitochondrial transport serves as a mitochondrial quality control strategy in axons: Implications for central nervous system disorders. CNS Neurosci. Ther. 2019, 25, 876–886. [Google Scholar] [CrossRef] [Green Version]
- Jurcau, A. Molecular pathophysiological mechanisms in Huntington’s disease. Biomedicines 2022, 10, 1432. [Google Scholar] [CrossRef]
- Wang, Q.; Tian, J.; Chen, H.; Du, H.; Guo, L. Amyloid beta-mediated KIF5A deficiency disrupts anterograde axonal mitochondrial movement. Neurobiol. Dis. 2019, 127, 410–418. [Google Scholar] [CrossRef]
- Stavoe, A.K.H.; Holzbaur, E.L.F. Axonal autophagy: Mini-review for autophagy in the CNS. Neurosci. Lett. 2019, 697, 17–23. [Google Scholar] [CrossRef]
- Ebneth, A.; Godemann, R.; Stamer, K.; Illenberger, S.; Trinczek, B.; Mandelkow, E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: Implications for Alzheimer’s disease. J. Cell Biol. 1998, 143, 777–794. [Google Scholar] [CrossRef] [Green Version]
- Ittner, L.M.; Ke, Y.D.; Götz, J. Phosphorylated tau interacts with c-Jun N-terminal kinase-interacting protein 1 (JIP1) in Alzheimer disease. J. Biol. Chem. 2009, 284, 20909–20916. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; De Groof, A.J.C.; Boldogh, I.; Bird, T.D.; Gibson, G.E.; Koehler, C.M.; Yu, W.H.; Duff, K.E.; Yaffe, M.P.; Pon, L.A.; et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 2009, 175, 1810–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.L.; Mayne, M.; Holden, C.P.; Geiger, J.D.; Mattson, M.P. Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J. Biol. Chem. 2000, 275, 18195–18200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 2146. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Schon, E.A. On the pathogenesis of Alzheimer’s disease: The MAM hypothesis. FASEB J. 2017, 31, 864–867. [Google Scholar] [CrossRef] [Green Version]
- Britti, E.; Ros, J.; Esteras, N.; Abramov, A.Y. Tau inhibits mitochondrial calcium efflux and makes neurons vulnerable to calcium-induced cell death. Cell. Calcium 2020, 86, 102150. [Google Scholar] [CrossRef]
- Khan, M.S.H.; Hegde, V. Obesity and diabetes mediated chronic inflammation: A potential biomarker in Alzheimer’s disease. J. Pers. Med. 2020, 10, 42. [Google Scholar] [CrossRef]
- Xie, N.; Wu, C.; Wang, C.; Cheng, X.; Zhang, L.; Zhang, H.; Lian, Y. Inhibition of the mitochondrial calcium uniporter inhibits Aβ-induced apoptosis by reducing reactive oxygen species-mediated endoplasmic reticulum stress in cultured microglia. Brain Res. 2017, 1676, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Chiozzi, P.; Sarti, A.C.; Sanz, J.M.; Giuliani, A.L.; Adinolfi, E.; Vultaggio-Poma, V.; Falzoni, S.; Di Virgilio, F. Amyloid β-dependent mitochondrial toxicity in mouse microglia requires P2X7 receptor expression and is prevented by nimodipine. Sci. Rep. 2019, 9, 6475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.M.; Moore, Z.; Minter, M.R.; Crack, P.J. Type-I interferon pathway in neuroinflammation and neurodegeneration: Focus on Alzheimer’s disease. J. Neural. Transm. 2018, 125, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef]
- Rohn, T.T.; Wirawan, E.; Brown, R.J.; Harris, J.R.; Masliah, E.; Vandenabeele, P. Depletion of Beclin-1 due to proteolytic cleavage by caspases in the Alzheimer’s disease brain. Neurobiol. Dis. 2011, 43, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Wooten, M.C.; Gearing, M.; Wooten, M.W. Age-associated oxidative damage to the p62 promoter: Implications for Alzheimer disease. Free Radic. Biol. Med. 2009, 46, 492–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef] [Green Version]
- Nixon, R.A.; Yang, D.S. Autophagy failure in Alzheimer’s disease—Locating the primary defect. Neurobiol. Dis. 2011, 43, 38–45. [Google Scholar] [CrossRef] [Green Version]
- Lim, F.; Hernandez, F.; Lucas, J.J.; Gomez-Ramos, P.; Moran, M.A.; Avila, J. FTDP-17 mutations in tau transgenic mice provoke lysosomal abnormalities and Tau filaments in forebrain. Mol. Cell. Neurosci. 2001, 18, 702–714. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; Van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Zhou, B.; Lin, M.Y.; Sheng, Z.H. Progressive endolysosomal deficits impair autophagic clearance beginning at early asymptomatic stages in fALS mice. Autophagy 2015, 11, 1934–1936. [Google Scholar] [CrossRef]
- Liu, J.; Li, L. Targeting Autophagy for the Treatment of Alzheimer’s Disease: Challenges and Opportunities. Front. Mol. Neurosci. 2019, 12, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.; Burré, J.; Südhof, T.C. Proteasome inhibition alleviates SNARE-dependent neurodegeneration. Sci. Transl. Med. 2012, 4, 147ra113. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kim, J.; Kim, H.Y.; Ryoo, N.; Lee, S.; Kim, Y.; Rhim, H.; Shin, Y.K. Amyloid-β oligomers may impair SNARE-mediated exocytosis by direct binding to syntaxin 1a. Cell Rep. 2015, 12, 1244–1251. [Google Scholar] [CrossRef]
- Margiotta, A. Role of SNAREs in Neurodegenerative Diseases. Cells 2021, 10, 991. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sanchez, K.; Ariza-Salamanca, D.; Mora-Munoz, L. Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef] [Green Version]
- Acosta, C.; Anderson, H.D.; Anderson, C.M. Astrocyte dysfunction in Alzheimer disease. J. Neurosci. Res. 2017, 95, 2430–2447. [Google Scholar] [CrossRef] [PubMed]
- Ries, M.; Sastre, M. Mechanisms of abeta clearance and degradation by glial cells. Front. Aging Neurosci. 2016, 8, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iram, T.; Trudler, D.; Kain, D.; Kanner, S.; Galron, R.; Vassar, R.; Barzilai, A.; Blinder, P.; Fishelson, Z.; Frenkel, D. Astrocytes from old Alzheimer’s disease mice are impaired in Aβ uptake and in neuroprotection. Neurobiol. Dis. 2016, 96, 84–94. [Google Scholar] [CrossRef]
- Garwood, C.J.; Ratcliffe, L.E.; Simpson, J.E.; Heath, P.R.; Ince, P.G.; Wharton, S.B. Review: Astrocytes in Alzheimer’s disease and other age-associated dementias: A supporting player with a central role. Neuropathol. Appl. Neurobiol. 2017, 43, 281–298. [Google Scholar] [CrossRef]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef]
- Kawano, H.; Katsurabayashi, S.; Kakazu, Y.; Yamashita, Y.; Kubo, N.; Kubo, M.; Okuda, H.; Takasaki, K.; Kubota, K.; Mishima, K.; et al. Long-term culture of astrocytes attenuates the readily releasable pool of synaptic vesicles. PLoS ONE 2012, 7, e48034. [Google Scholar] [CrossRef]
- Abdelnour, C.; Agosta, F.; Bozzali, M.; Fougère, B.; Iwata, A.; Nilforooshan, R.; Takada, L.T.; Viñuela, F.; Traber, M. Perspectives and challenges in patient stratification in Alzheimer’s disease. Alzheimers Res. Ther. 2022, 14, 112. [Google Scholar] [CrossRef]
- Fornari, C.; Mori, F.; Zoppi, N.; Libri, I.; Silvestri, C.; Cosseddu, M.; Turrone, R.; Maffi, M.; Caratozzolo, S.; Borroni, B.; et al. Diagnostic accuracy of the five-word test for mild cognitive impairment due to Alzheimer’s disease. Neurol. Int. 2022, 14, 357–367. [Google Scholar] [CrossRef] [PubMed]
- de Wilde, A.; van der Flier, W.M.; Pelkmans, W.; Bouwman, F.; Verwer, J.; Groot, C.; van Buchem, M.M.; Zwan, M.; Ossenkoppele, R.; Yaqub, M.; et al. Association of amyloid positron emission tomography with changes in diagnosis and patient treatment in an unselected memory clinic cohort: The ABIDE project. JAMA Neurol. 2018, 75, 1062–1070. [Google Scholar] [CrossRef] [PubMed]
- Harrison, T.M.; La Joie, R.; Maass, A.; Baker, S.L.; Swinnerton, K.; Fenton, L.; Mellinger, T.J.; Edwards, L.; Pham, J.; Miller, B.L.; et al. Longitudinal tau accumulation and atrophy in aging and Alzheimer disease. Ann. Neurol. 2019, 85, 229–240. [Google Scholar] [CrossRef]
- Cai, Z.; Li, S.; Matuskey, D.; Nabulsi, N.; Huang, Y. PET imaging of synaptic density: A new tool for investigation of neuropsychiatric diseases. Neurosci. Lett. 2019, 691, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Shaw, L.M.; Stomrud, E.; Mattsson, N.; Toledo, J.B.; Buck, K.; Wahl, S.; Eichenlaub, U.; Lifke, V.; Simon, M.; et al. Predicting clinical decline and conversion to Alzheimer’s disease or dementia using novel Elecsys Aβ(1-42), pTau and tTau CSF immunoassays. Sci. Rep. 2019, 9, 19024. [Google Scholar] [CrossRef] [Green Version]
- Galasko, D.; Xiao, M.; Xu, D.; Smirnov, D.; Salmon, D.P.; Dewit, N.; Vanbrabant, J.; Jacobs, D.; Vanderstichele, H.; Vanmechelen, E. Alzheimer’s Disease Neuroimaging Initiative (ADNI). Synaptic biomarkers in CSF aid in diagnosis, correlate with cognition and predict progression in MCI and Alzheimer’s disease. Alzheimers Dement. 2019, 5, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Janelidze, S.; Mattsson, N.; Palmqvist, S.; Smith, R.; Beach, T.G.; Serrano, G.E.; Chai, X.; Proctor, N.K.; Eichenlaub, U.; Zetterberg, H.; et al. Plasma p-tau181 in Alzheimer’s disease: Relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat. Med. 2020, 26, 379–386. [Google Scholar] [CrossRef]
- Palmqvist, S.; Janelidze, S.; Quiroz, Y.T.; Zetterberg, H.; Lopera, F.; Stomrud, E.; Su, Y.; Chen, Y.; Serrano, G.E.; Leuzy, A.; et al. Discriminative accuracy of plasma phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA 2020, 324, 772–781. [Google Scholar] [CrossRef]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS–ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [Green Version]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging–Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Villain, N.; Frisoni, G.B.; Rabinovici, G.D.; Sabbagh, M.; Cappa, S.; Bejanin, A.; Bombois, S.; Epelbaum, S.; Teichmann, M.; et al. Clinical diagnosis of Alzheimer’s disease: Recommendations of the International Working Group. Lancet Neurol. 2021, 20, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Aisen, P.; Apostolova, L.G.; Atri, A.; Salloway, S.; Weiner, M. Aducanumab: Appropriate use recommendations. J. Prev. Alzheimers Dis. 2021, 8, 398–410. [Google Scholar] [CrossRef]
- Slota, J.A.; Booth, S.A. MicroRNAs in neuroinflammation: Implications in disease pathogenesis, biomarker discovery and therapeutic applications. Non-Coding RNA 2019, 5, 35. [Google Scholar] [CrossRef] [Green Version]
- Stewart, W.F.; Kawas, C.; Corrada, M.; Metter, E.J. Risk of Alzheimer’s disease and duration of NSAID use. Neurology 1997, 48, 626–632. [Google Scholar] [CrossRef]
- Shadfar, S.; Hwang, C.J.; Lim, M.-S.; Choi, D.-Y.; Hong, J.T. Involvement of inflammation in Alzheimer’s disease pathogenesis and therapeutic potential of anti-inflammatory agents. Arch. Pharm. Res. 2015, 38, 2106–2119. [Google Scholar] [CrossRef]
- Rogers, J.; Kirby, L.C.; Hempelman, S.R.; Berry, D.L.; McGeer, P.L.; Kaszniak, A.W.; Zalinski, J.; Cofield, M.; Mansukhani, L.; Willson, P.; et al. Clinical trial of indomethacin in Alzheimer’s disease. Neurology 1993, 43, 1609–1611. [Google Scholar] [CrossRef]
- Breitner, J.C.; Baker, L.D.; Montine, T.J.; Meinert, C.L.; Lyketsos, C.G.; Ashe, K.H.; Brandt, J.; Craft, S.; Evans, D.E.; ADAPT Research Group; et al. Extended results of the Alzheimer’s disease anti-inflammatory prevention trial. Alzheimers Dement. 2011, 7, 402–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porrini, V.; Lanzillotta, A.; Branca, C.; Benarese, M.; Parrella, E.; Lorenzini, L.; Calza, L.; Flaibani, R.; Spano, P.; Imbimbo, B.; et al. CHF5074 (CSP-1103) induces microglia alternative activation in plaque-free Tg2576 mice and primary glial cultures exposed to beta-amyloid. Neuroscience 2015, 302, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Clinicaltrials.Gov, Homepage on the Internet. Available online: ClinicalTrials.gov (accessed on 31 October 2022).
- Landreth, G.E.; Heneka, M.T. Anti-inflammatory actions of peroxisome proliferator-activated receptor gamma agonists in Alzheimer’s disease. Neurobiol. Aging. 2001, 22, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Denner, L.A.; Rodriguez-Rivera, J.; Haidacher, S.J.; Jahrling, J.B.; Carmical, J.R.; Hernandez, C.M.; Zhao, Y.; Sadygov, R.G.; Starkey, J.M.; Spratt, H.; et al. Cognitive enhancement with rosiglitazone links the hippocampal PPARγ and ERK MAPK signaling pathways. J. Neurosci. 2012, 32, 16725–16735. [Google Scholar] [CrossRef] [Green Version]
- Watson, G.S.; Cholerton, B.A.; Reger, M.A.; Baker, L.D.; Plymate, S.R.; Asthana, S.; Fishel, M.A.; Kulstad, J.J.; Green, P.S.; Cook, D.G.; et al. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: A preliminary study. Am. J. Geriatr. Psychiatry 2005, 13, 950–958. [Google Scholar] [CrossRef]
- Harrington, C.; Sawchak, S.; Chiang, C.; Davies, J.; Donovan, C.; Saunders, A.M.; Irizarry, M.; Jeter, B.; Zvartau-Hind, M.; van Dyck, C.H.; et al. Rosiglitazone does not improve cognition or global function when used as adjunctive therapy to AChE inhibitors in mild-to-moderate Alzheimer’s disease: Two phase 3 studies. Curr. Alzheimer Res. 2011, 8, 592–606. [Google Scholar] [CrossRef]
- Heneka, M.T.; Fink, A.; Doblhammer, G. Effect of pioglitazone medication on the incidence of dementia. Ann. Neurol. 2015, 78, 284–294. [Google Scholar] [CrossRef]
- Burns, D.K.; Alexander, R.C.; Welsh-Bohmer, K.A.; Culp, M.; Chiang, C.; O’Neil, J.; Evans, R.M.; Harrigan, P.; Plassman, B.L.; Burke, J.R.; et al. Safety and efficacy of pioglitazone for the delay of cognitive impairment in people at risk of Alzheimer’s disease (TOMMORROW): A prognostic biomarker study and a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2021, 20, 537–547. [Google Scholar] [CrossRef]
- Shi, J.Q.; Shen, W.; Chen, J.; Wang, B.R.; Zhong, L.L.; Zhu, Y.W.; Zhu, H.-Q.; Zhang, Q.-Q.; Zhang, Y.-D.; Xu, J. Anti-TNF-α reduces amyloid plaques and tau phosphorylation and induces CD11c-positive dendritic-like cell in the APP/PS1 transgenic mouse brains. Brain Res. 2011, 1368, 239–247. [Google Scholar] [CrossRef]
- Camargo, C.H.F.; Justus, F.F.; Retzlaff, G.; Blood, M.R.Y.; Schafranski, M.D. Action of anti-TNF-α drugs on the progression of Alzheimer’s disease: A case report. Dement. Neuropsychol. 2015, 9, 196–200. [Google Scholar] [CrossRef]
- Tobinick, E.; Gross, H.; Weinberger, A.; Cohen, H. TNF-alpha modulation for treatment of Alzheimer’s disease: A 6-month pilot study. MedGenMed. 2006, 8, 25. [Google Scholar] [PubMed]
- Butchart, J.; Brook, L.; Hopkins, V.; Teeling, J.; Puntener, U.; Culliford, D.; Sharples, R.; Sharif, S.; McFarlane, B.; Raybould, R.; et al. Etanercept in Alzheimer disease: A randomized, placebo-controlled, double-blind, phase 2 trial. Neurology 2015, 84, 2161–2168. [Google Scholar] [CrossRef] [Green Version]
- Barnum, C.J.; Chen, X.; Chung, J.; Chang, J.; Williams, M.; Grigoryan, N.; Tesi, R.J.; Tansey, M.G. Peripheral administration of the selective inhibitor of soluble tumor necrosis factor (TNF) XPro®1595 attenuates nigral cell loss and glial activation in 6-OHDA hemiparkinsonian rats. J. Parkinsons Dis. 2014, 4, 349–360. [Google Scholar] [CrossRef]
- MacPherson, K.P.; Sompol, P.; Kannarkat, G.T.; Chang, J.; Sniffen, L.; Wildner, M.E.; Norris, C.M.; Tansey, M.G. Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol. Dis. 2017, 102, 81–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piette, F.; Belmin, J.; Vincent, H.; Schmidt, N.; Pariel, S.; Verny, M.; Marquis, C.; Mely, J.; Hugonot-Diener, L.; Kinet, J.P.; et al. Masitinib as an adjunct therapy for mild-to-moderate Alzheimer’s disease: A randomised, placebo-controlled phase 2 trial. Alzheimers Res. Ther. 2011, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Liu, M.; Nguyen, X.V.; Bing, G. P38 MAP kinase is activated at early stages in Alzheimer’s disease brain. Exp. Neurol. 2003, 183, 394–405. [Google Scholar] [CrossRef]
- Duffy, J.P.; Harrington, E.M.; Salituro, F.G.; Cochran, J.E.; Green, J.; Gao, H.; Bemis, G.W.; Evindar, G.; Galullo, V.P.; Ford, P.J.; et al. The discovery of VX-745: A novel and selective p38alpha kinase inhibitor. ACS Med. Chem. Lett. 2011, 2, 758–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.M.; Minasov, G.; Arancio, O.; Chico, L.W.; Van Eldik, L.J.; Anderson, W.F.; Pelletier, J.C.; Watterson, D.M. A selective and brain penetrant p38alphaMAPK inhibitor candidate for neurologic and neuropsychiatric disorders that attenuates neuroinflammation and cognitive dysfunction. J. Med. Chem. 2019, 62, 5298–5311. [Google Scholar] [CrossRef] [PubMed]
- Prins, N.D.; Harrison, J.E.; Chu, H.M.; Blackburn, K.; Alam, J.J.; Scheltens, P.; REVERSE-SD Study Investigators. A phase 2 double-blind placebo-controlled 24-week treatment clinical study of the p38 alpha kinase inhibitor neflamapimod in mild Alzheimer’s disease. Alzheimers Res. Ther. 2021, 13, 106. [Google Scholar] [CrossRef]
- Tormählen, N.M.; Martorelli, M.; Kuhn, A.; Maier, F.; Guezguez, J.; Burnet, M.; Albrecht, W.; Laufer, S.A.; Koch, P. Design and synthesis of highly selective brain penetrant p38α mitogen-activated protein kinase inhibitors. J. Med. Chem. 2022, 65, 1225–1242. [Google Scholar] [CrossRef]
- Reading, C.L.; Ahlem, C.N.; Murphy, M.F. NM101 Phase III study of NE3107 in Alzheimer’s disease: Rationale, design and therapeutic modulation of neuroinflammation and insulin resistance. Neurodegener. Dis. Manag. 2021, 11, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Baricitinib: First global approval. Drugs 2017, 77, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Urbina, F.; Puhl, A.C.; Ekins, S. Recent advances in drug repurposing using machine learning. Curr. Opin. Chem. Biol. 2021, 65, 74–84. [Google Scholar] [CrossRef]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The multi-faceted ecto-enzyme CD38: Roles in immunomodulation, cancer, aging, and metabolic diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potter, H.; Woodcock, J.H.; Boyd, T.D.; Coughlan, C.M.; O’Shaughnessy, J.R.; Borges, M.T.; Thaker, A.A.; Raj, B.A.; Adamszuk, K.; Scott, D.; et al. Safety and efficacy of sagramostim (GM-CSF) in the treatment of Alzheimer’s disease. Alzheimers Dement. 2021, 7, e12158. [Google Scholar]
- Toguchi, M.; Gonzalez, D.; Furukawa, S.; Inagaki, S. Involvement of Sema4D in the control of microglia activation. Neurochem. Int. 2009, 55, 573–580. [Google Scholar] [CrossRef]
- Nordengen, K.; Kirsebom, B.E.; Henjum, K.; Selnes, P.; Gísladóttir, B.; Wettergreen, M.; Torsetnes, S.B.; Grøntvedt, G.R.; Waterloo, K.K.; Aarsland, D.; et al. Glial activation and inflammation along the Alzheimer’s disease continuum. J. Neuroinflamm. 2019, 16, 46. [Google Scholar] [CrossRef] [Green Version]
- Merlini, M.; Kirabali, T.; Kulic, L.; Nitsch, R.M.; Ferretti, M.T. Extravascular CD3+ T cells in brains of Alzheimer disease patients correlate with tau but not with amyloid pathology: An immunohistochemical study. Neurodegener. Dis. 2018, 18, 49–56. [Google Scholar] [CrossRef]
- Ciccocioppo, F.; Lanuti, P.; Pierdomenico, L.; Simeone, P.; Bologna, G.; Ercolino, E.; Buttari, F.; Fantozzi, R.; Thomas, A.; Onofrj, M.; et al. The characterization of regulatory T-cell profiles in Alzheimer’s disease and multiple sclerosis. Sci. Rep. 2019, 9, 8788. [Google Scholar] [CrossRef] [Green Version]
- Oberstein, T.J.; Taha, L.; Spitzer, P.; Hellstern, J.; Herrmann, M.; Kornhuber, J.; Maler, J.M. Imbalance of circulating Th17 and regulatory T cells in Alzheimer’s disease: A case control study. Front. Immunol. 2018, 9, 1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dansokho, C.; Ait Ahmed, D.; Aid, S.; Toly-Ndour, C.; Chaigneau, T.; Calle, V.; Cagnard, N.; Holzenberger, M.; Piaggio, E.; Aucouturier, P.; et al. Regulatory T cells delay disease progression in Alzheimer-like pathology. Brain 2016, 139, 1237–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Fischer, R.; Naudé, P.J.; Maier, O.; Nyakas, C.; Duffey, M.; Van der Zee, E.A.; Dekens, D.; Douwenga, W.; Herrmann, A.; et al. Essential protective role of tumor necrosis factor receptor 2 in neurodegeneration. Proc. Natl. Acad. Sci. USA 2016, 113, 12304–12309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, R.; Maier, O.; Siegemund, M.; Wajant, H.; Scheurich, P.; Pfizenmaier, K. A TNF receptor 2 selective agonist rescues human neurons from oxidative stress-induced cell death. PLoS ONE 2011, 6, e27621. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, S.-Y.; Shon, J.; Kim, K.; Lee, H.J.; Kim, K.A.; Lee, B.-Y.; Oh, S.-H.; Kim, N.K.; Kim, O.J. Adalimumab improves cognitive impairment, exerts neuroprotective effects and attenuates neuroinflammation in an Aβ1-40-injected mouse model of Alzheimer’s disease. Cytotherapy 2019, 21, 671–682. [Google Scholar] [CrossRef]
- Wang, M.; Sooreshjani, M.A.; Mikek, C.; Opoku-Temeng, C.; Sintim, H.O. Suramin potently inhibits cGAMP synthase, cGAS, in THP1 cells to modulate IFN-β levels. Future Med. Chem. 2018, 10, 1301–1317. [Google Scholar] [CrossRef] [Green Version]
- An, J.; Woodward, J.J.; Sasaki, T.; Minie, M.; Elkon, K.B. Cutting edge: Antimalarial drugs inhibit IFN-β production through blockade of cyclic GMP-AMP synthase-DNA interaction. J. Immunol. 2015, 194, 4089–4093. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.; Brault, A.; Vincent, F.; Weng, S.; Wang, H.; Dumlao, D.; Aulabaugh, A.; Aivazian, D.; Castro, D.; Chen, M.; et al. Discovery of PF-06928215 as a high affinity inhibitor of cGAS enabled by a novel fluorescence polarization assay. PLoS ONE 2017, 12, e0184843. [Google Scholar] [CrossRef]
- Vincent, J.; Adura, C.; Gao, P.; Luz, A.; Lama, L.; Asano, Y.; Okamoto, R.; Imaeda, T.; Aida, J.; Rothamel, K.; et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat. Commun. 2017, 8, 750. [Google Scholar] [CrossRef] [Green Version]
- Lai, J.; Luo, X.; Tian, S.; Zhang, X.; Huang, S.; Wang, H.; Li, Q.; Cai, S.; Chen, Q. Compound C reducing interferon expression by inhibiting cGAMP accumulation. Front. Pharmacol. 2020, 11, 88. [Google Scholar] [CrossRef] [Green Version]
- Hansen, A.L.; Mukai, K.; Schopfer, F.J.; Taguchi, T.; Holm, C.K. STING palmitoylation as a therapeutic target. Cell. Mol. Immunol. 2019, 16, 236–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haag, S.M.; Gulen, M.F.; Reymond, L.; Gibelin, A.; Abrami, L.; Decout, A.; Heymann, M.; van der Goot, F.G.; Turcatti, G.; Behrendt, R.; et al. Targeting STING with covalent small-molecule inhibitors. Nature 2018, 559, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hong, Z.; Wang, Z.; Li, F.; Mei, J.; Huang, L.; Lou, X.; Zhao, S.; Song, L.; Chen, W.; et al. The cyclopeptide astin C specifically inhibits the innate immune CDN sensor STING. Cell Rep. 2018, 25, 3405–3421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siu, T.; Altman, M.D.; Baltus, G.A.; Childers, M.; Ellis, J.M.; Gunaydin, H.; Hatch, H.; Ho, T.; Jewell, J.; Lacey, B.M.; et al. Discovery of a novel cGAMP competitive ligand of the inactive form of STING. ACS Med. Chem. Lett. 2018, 10, 92–97. [Google Scholar] [CrossRef]
- Larrick, J.W.; Mendelsohn, A.R. Modulation of cGAS-STING Pathway by Nicotinamide Riboside in Alzheimer’s Disease. Rejuvenation Res. 2021, 24, 397–402. [Google Scholar] [CrossRef]
- Hou, Y.; Wei, Y.; Lautrup, S.; Yang, B.; Wang, Y.; Cordonnier, S.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. NAD(+) supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc. Natl. Acad. Sci. USA 2021, 118, e2011226118. [Google Scholar] [CrossRef]
- Li, T.; Lu, L.; Pember, E.; Li, X.; Zhang, B.; Zhu, Z. New insights into neuroinflammation involved in pathogenic mechanism of Alzheimer’s disease and its potential for therapeutic intervention. Cells 2022, 11, 1925. [Google Scholar] [CrossRef]
- Li, J.; Huang, J.; Jeong, J.H.; Park, S.J.; Wei, R.; Peng, J.; Luo, Z.; Chen, Y.T.; Feng, Y.; Luo, J.L. Selective TBK1/IKKi dual inhibitors with anticancer potency. Int. J. Cancer 2014, 134, 1972–1980. [Google Scholar] [CrossRef] [Green Version]
- Thomson, D.W.; Poeckel, D.; Zinn, N.; Rau, C.; Strohmer, K.; Wagner, A.J.; Graves, A.P.; Perrin, J.; Bantscheff, M.; Duempelfeld, B.; et al. Discovery of GSK8612, a highly selective and potent TBK1 inhibitor. ACS Med. Chem. Lett. 2019, 10, 780–785. [Google Scholar] [CrossRef]
- Bailly, C. The potential value of amlexanox in the treatment of cancer: Molecular targets and therapeutic perspectives. Biochem. Pharmacol. 2022, 197, 114895. [Google Scholar] [CrossRef]
- Goldberg, E.L.; Dixit, V.D. Drivers of age-related inflammation and strategies for healthspan extension. Immunol. Rev. 2015, 265, 63–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dempsey, C.; Rubio Araiz, A.; Bryson, K.J.; Finucane, O.; Larkin, C.; Mills, E.L.; Robertson, A.A.; Cooper, M.A.; O’Neill, L.A.; Lynch, M.A. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curdy, N.; Lanvin, O.; Laurent, C.; Fournié, J.-J.; Franchini, D.-M. Regulatory mechanisms of inhibitory immune checkpoint receptors expression. Trends Cell Biol. 2019, 29, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, M.; González-Cao, M.; Viteri, S.; Karachaliou, N.; Altavilla, G.; Rosell, R. Programmed cell death protein-1/programmed cell death ligand-1 pathway inhibition and predictive biomarkers: Understanding transforming growth factor-beta role. Transl. Lung Cancer Res. 2015, 4, 728–742. [Google Scholar]
- Munafó, A.; Burgaletto, C.; Di Benedetto, G.; Di Mauro, M.; Di Mauro, R.; Bernardini, R.; Cantarella, G. Repositioning of immunomodulators: A ray of hope for Alzheimer’s disease? Front. Neurosci. 2020, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, N.; Dvir-Szternfeld, R.; Tsitsou-Kampeli, A.; Keren-Shaul, H.; Ben-Yehuda, H.; Weill-Raynal, P.; Cahalon, L.; Kertser, A.; Baruch, K.; Amit, I.; et al. PD-1/PD-L1 checkpoint blockade harnesses monocyte-derived macrophages to combat cognitive impairment in a tauopathy mouse model. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Latta-Mahieu, M.; Elmer, B.; Bretteville, A.; Wang, Y.; Lopez-Grancha, M.; Goniot, P.; Moindrot, N.; Ferrari, P.; Blanc, V.; Schussler, N.; et al. Systemic immune-checkpoint blockade with anti-PD1 antibodies does not alter cerebral amyloid-β burden in several amyloid transgenic mouse models. Glia 2017, 66, 492–504. [Google Scholar] [CrossRef]
- Lansita, J.A.; Mease, K.M.; Qiu, H.; Yednock, T.; Sankaranarayan, S.; Kramer, S. Nonclinical development of ANX005: A humanized anti-C1q antibody for treatment of autoimmune and neurodegenerative diseases. Int. J. Toxicol. 2017, 36, 449–462. [Google Scholar] [CrossRef] [Green Version]
- Lian, H.; Litvinchuk, A.; Chiang, A.C.; Aithmitti, N.; Jankowsky, J.L.; Zheng, H. Astrocyte-Microglia Cross Talk through Complement Activation Modulates Amyloid Pathology in Mouse Models of Alzheimer’s Disease. J. Neurosci. 2016, 36, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, M.-C.; Sawyer, N.; Greig, G.M.; Hamel, M.; Kargman, S.; Ducharme, Y.; Lau, C.K.; Friesen, R.W.; O’Neill, G.P.; Gervais, F.G.; et al. The C3a receptor antagonist SB 290,157 has agonist activity. Immunol. Lett. 2005, 100, 139–145. [Google Scholar] [CrossRef]
- Hu, J.; Yang, Y.; Wang, M.; Yao, Y.; Chang, Y.; He, Q.; Ma, R.; Li, G. Complement C3a receptor antagonist attenuates tau hyperphosphorylation via glycogen synthase kinase 3β signaling pathways. Eur. J. Pharmacol. 2019, 850, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, M.I.; Ager, R.R.; Chu, S.-H.; Yazan, O.; Sanderson, S.D.; LaFerla, F.M.; Taylor, S.M.; Woodruff, T.M.; Tenner, A.J. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J. Immunol. 2009, 183, 1375–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbaszadeh, H.; Ghorbani, F.; Derakhshani, M.; Movassaghpour, A.; Yousefi, M. Human umbilical cord mesenchymal stem cell-derived extracellular vesicles: A novel therapeutic paradigm. J. Cell. Physiol. 2019, 235, 706–717. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Shen, Y.; Wang, P.; Xie, Z.; Xu, S.; Zhu, Z.; Wang, Y.; Lyu, Y.; Wang, D.; Xu, L.; et al. Exosomes isolated from human umbilical cord mesenchymal stem cells alleviate neuroinflammation and reduce amyloid-beta deposition by modulating microglial activation in Alzheimer’s disease. Neurochem. Res. 2018, 43, 2165–2177. [Google Scholar] [CrossRef]
- Garcia-Contreras, M.; Thakor, A.S. Extracellular vesicles in Alzheimer’s disease: From pathology to therapeutic approaches. Neural. Regen. Res. 2023, 18, 18–22. [Google Scholar]
- Cai, Z.-Y.; Xiao, M.; Quazi, S.H.; Ke, Z.-Y. Exosomes: A novel therapeutic target for Alzheimer’s disease? Neural. Regen. Res. 2018, 13, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A.; Jurcau, M.C. Therapeutic strategies in Huntington’s disease: From genetic defect to gene therapy. Biomedicines 2022, 10, 1895. [Google Scholar] [CrossRef]
- Nistor-Cseppentö, D.C.; Jurcău, M.C.; Jurcău, A.; Andronie-Cioară, F.L.; Marcu, F. Stem Cell- and Cell-Based Therapies for Ischemic Stroke. Bioengineering 2022, 9, 717. [Google Scholar] [CrossRef]
- Derakshankhah, H.; Sajadimajd, S.; Jafari, S.; Izadi, Z.; Sarvari, A.; Sharifi, M.; Falahati, M.; Moakedi, F.; Muganda, W.C.A.; Muller, M.; et al. Novel therapeutic strategies for Alzheimer’s disease: Implications from cell-based therapy and nanotherapy. Nanomedicine 2020, 24, 102149. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Kouyoumdjian, H.; Zhu, D.C.; El-Dakdouki, M.H.; Lorenz, K.; Chen, J.; Li, W.; Huang, X. Glyconanoparticle aided detection of β-amyloid by magnetic resonance imaging and attenuation of β-amyloid induced toxicity. ACS Chem. Neurosci. 2013, 4, 575–584. [Google Scholar] [CrossRef] [Green Version]
- Hajipour, M.J.; Ghasemi, F.; Aghaverdi, H.; Raoufi, M.; Linne, U.; Atyabi, F.; Nabipour, I.; Azhdarzadeh, M.; Derakhshankhah, H.; Lotfabadi, A.; et al. Sensing of Alzheimer’s disease and multiple sclerosis using nano-bio interfaces. J. Alzheimers Dis. 2017, 59, 1187–1202. [Google Scholar] [CrossRef] [PubMed]
- Frozza, R.L.; Bernardi, A.; Hoppe, J.B.; Meneghetti, A.B.; Matté, A.; Battastini, A.M.; Pohlmann, A.R.; Guterres, S.S.; Salbego, C. Neuroprotective effects of resveratrol against Aβ administration in rats are improved by lipid-core nanocapsules. Mol. Neurobiol. 2013, 47, 1066–1080. [Google Scholar] [CrossRef] [PubMed]
- Sadegh Malvajerd, S.; Izadi, Z.; Azadi, A.; Kurd, M.; Derakhshankhah, H.; Sharifzadeh, M.; Akbari Javar, H.; Hamidi, M. Neuroprotective potential of curcumin-loaded nanostructured lipid carrier in an animal model of Alzheimer’s disease: Behavioral and biochemical evidence. J. Alzheimers Dis. 2019, 69, 671–686. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Zhang, M.; Gong, D.; Chen, R.; Hu, X.; Sun, T. The size-effect of gold nanoparticles and nanoclusters in the inhibition of amyloid-β fibrillation. Nanoscale 2017, 9, 4107–4113. [Google Scholar] [CrossRef]
- de Oliveira, J.; Kucharska, E.; Lima Garcez, M.; Scarpatto Rodrigues, M.; Quevedo, J.; Moreno-Gonzalez, I.; Budni, J. Inflammatory cascade in Alzheimer’s disease pathogenesis: A review of experimental findings. Cells 2021, 10, 2581. [Google Scholar] [CrossRef]
- Muller, A.P.; Ferreira, G.K.; Pires, A.J.; de Bem Silveira, G.; de Souza, D.L.; Brandolfi, J.A.; de Souza, C.T.; Paula, M.M.S.; Silveira, P.C.L. Gold nanoparticles prevent cognitive deficits, oxidative stress and inflammation in a rat model of sporadic dementia of Alzheimer’s type. Mater. Sci. Eng. Mater. Biol. Appl. 2017, 77, 476–483. [Google Scholar] [CrossRef]
- Rodrigues, M.S.; de Paula, G.C.; Duarte, M.B.; de Rezende, V.L.; Possato, J.C.; Farias, H.R.; Medeiros, E.B.; Feuser, P.E.; Streck, E.L.; de Ávila, R.A.M.; et al. Nanotechnology as a therapeutic strategy to prevent neuropsychomotor alterations associated with hypercholesterolemia. Colloids Surf. Biointerfaces 2021, 201, 111608. [Google Scholar] [CrossRef]
- Di Francesco, A.; Di Germanio, C.; Bernier, M.; de Cabo, R. A time to fast. Science 2018, 362, 770–775. [Google Scholar] [CrossRef] [Green Version]
- Behl, T.; Kaur, G.; Sehgal, A.; Singh, S.; Bhatia, S.; Al-Harrasi, A.; Zengin, G.; Bungau, S.G.; Munteanu, M.A.; Brisc, M.C.; et al. Elucidating the multi-targeted role of nutraceuticals: A complementary therapy to starve neurodegenerative diseases. Int. J. Mol. Sci. 2021, 22, 4045. [Google Scholar] [CrossRef]
- Jurcau, A. The role of natural antioxidants in the prevention of dementia—Where do we stand and future perspectives. Nutrients 2021, 13, 282. [Google Scholar] [CrossRef] [PubMed]
- Man, A.W.C.; Zhou, Y.; Xia, N.; Li, H. Involvement of gut microbiota, microbial metabolites and interaction with polyphenol in host immunometabolism. Nutrients 2020, 12, 3054. [Google Scholar] [CrossRef] [PubMed]
- Kesika, P.; Suganthy, N.; Sivamaruthi, B.S.; Chaiyasut, C. Role of gut-brain axis, gut microbial composition, and probiotic intervention in Alzheimer’s disease. Life Sci. 2021, 264, 118627. [Google Scholar] [CrossRef] [PubMed]
- Shabbir, U.; Arshad, M.; Sameen, A.; Oh, D.-H. Crosstalk between gut and brain in Alzheimer’s disease. The role of gut microbiota modulation strategies. Nutrients 2021, 13, 690. [Google Scholar] [CrossRef]




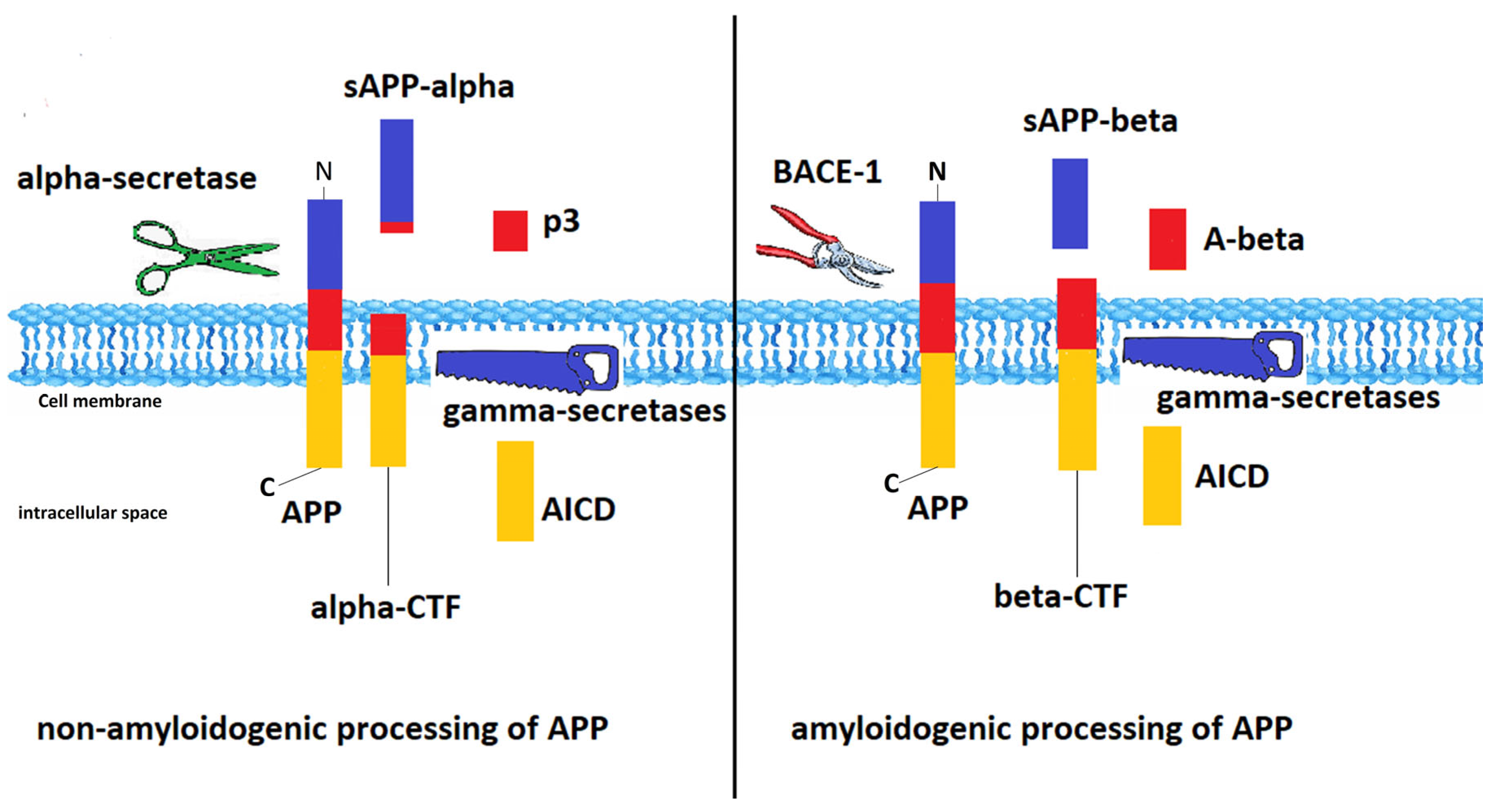{kind=link}
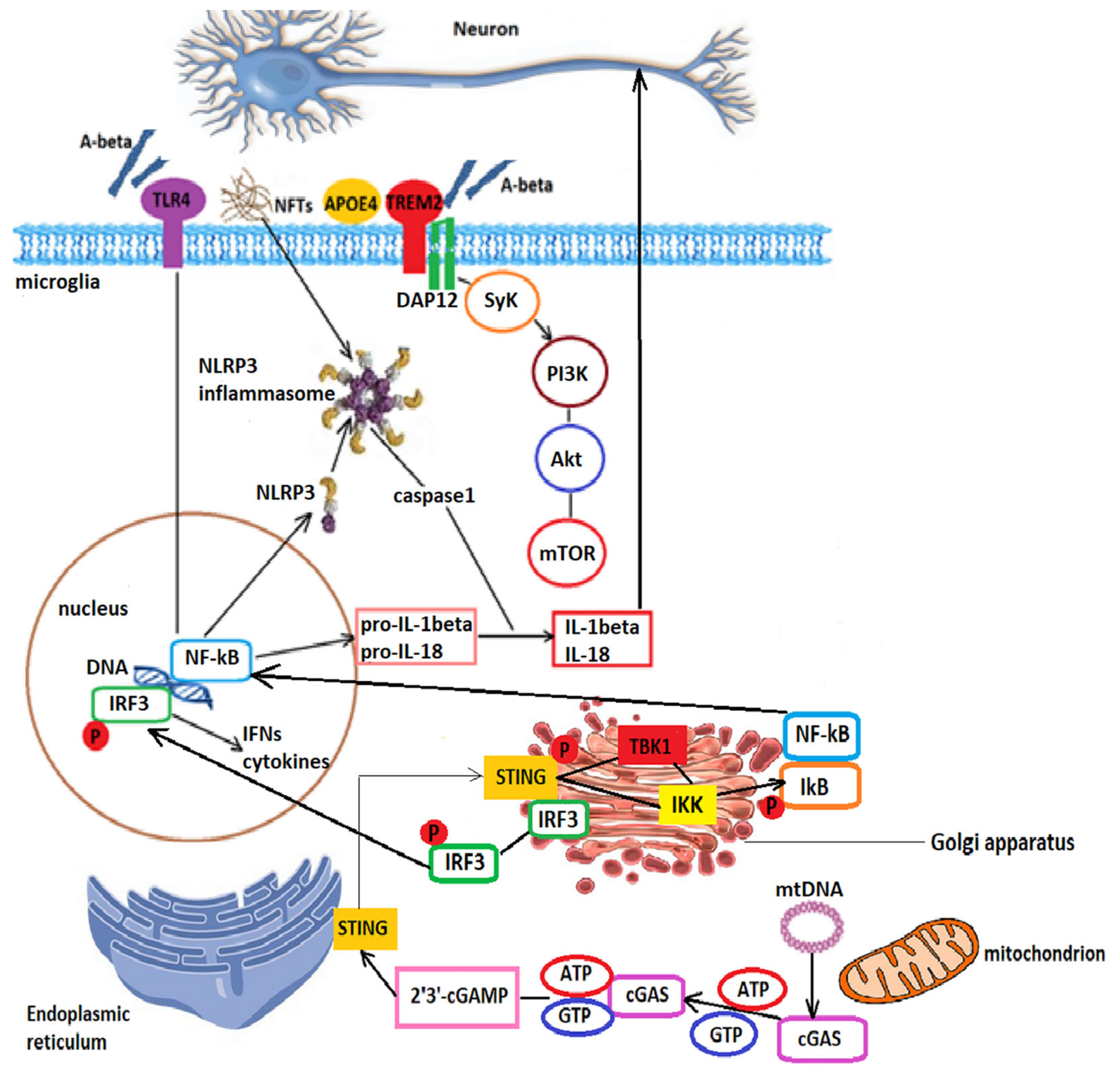{kind=link}
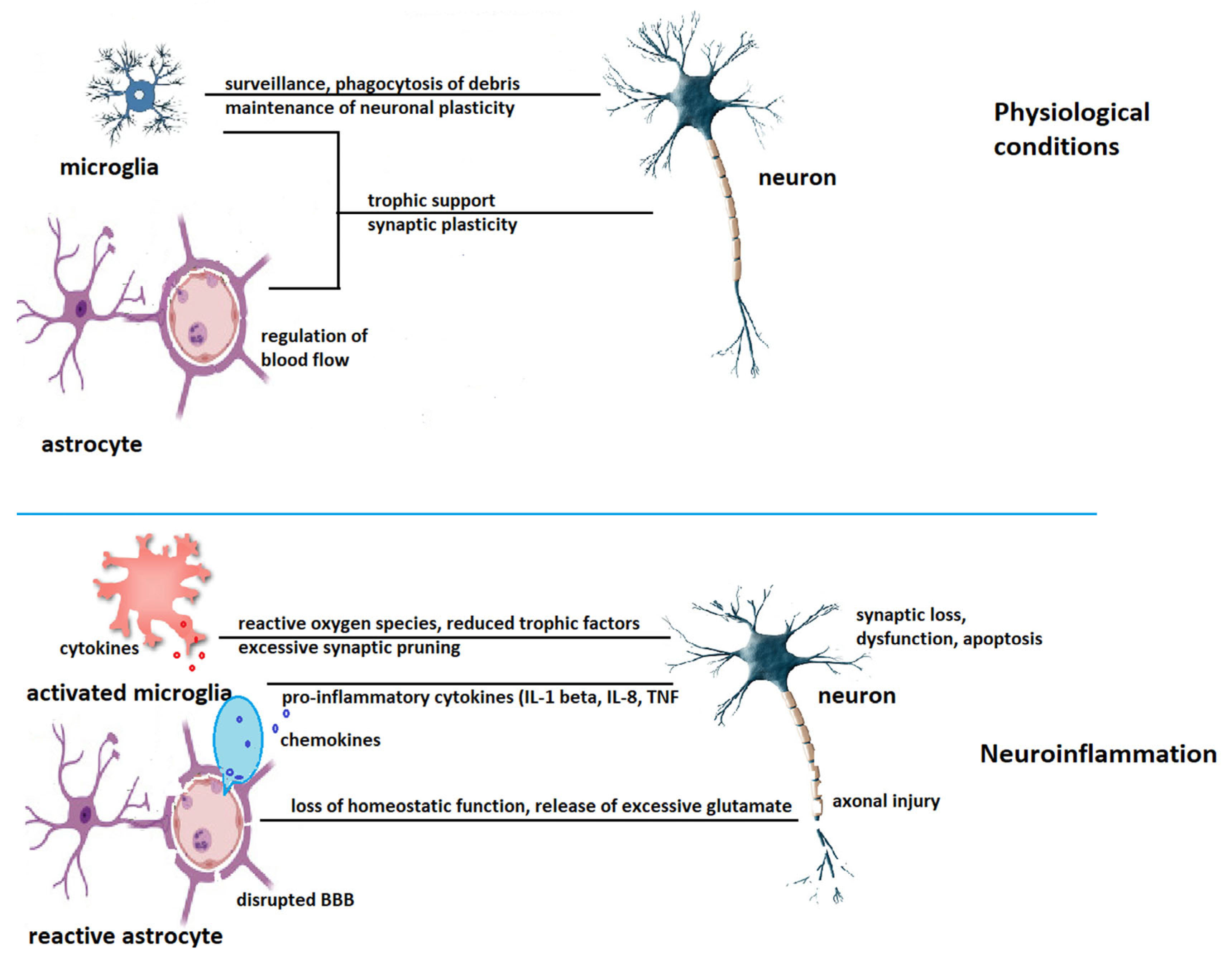{kind=link}
| Mediator | Functions | References |
|---|---|---|
| Protective effects | ||
| IL-1β | Increases α- and γ-secretases, downregulates BACE-1, promotes Aβ clearance | [196,197] |
| IL-1α | Increases α-secretase, increases sAPPα, decreases amyloidogenic APP processing | [198] |
| CXCL10 | Decreases Aβ deposition | [199] |
| CX3CL1 | Decreases Aβ deposition, upregulates phosphorylated tau | [200] |
| Brain derived neurotrophic factor (BDNF) | Promotes the non-amyloidogenic pathway, upregulates sAPPα, dephosphorylates tau via TrkB and PI3K signaling, improves memory performance | [201] |
| Glial derived neurotrophic factor (GDNF) | neuroprotection | [202] |
| Nerve growth factor (NGF) Neurotrophin 3 Neurotrophin 4 | Modulates microglial polarization toward a non-inflammatory phenotype Limits cleavage of caspases 3,8, and 9, upregulates neuronal apoptosis inhibitory protein Regulates tau dephosphorylation | [203] [204,205] [206] |
| Enhance AD pathology | ||
| IL-4 | Upregulates Aβ production, increases tau phosphorylation | [207] |
| IL-6 | Increases tau phosphorylation, increases amyloid plaque burden | [208] |
| IL-8 | Increases tau phosphorylation, promotes Aβ deposition | [209] |
| IL-10 | Promotes Aβ deposition | [210] |
| IL-18 | Upregulates BACE-1 and γ-secretase, enhances Aβ formation | [211] |
| TNF-α | Upregulates BACE-1 and γ-secretase, increases sAPPβ | [212] |
| Transforming growth factor (TGF)-β | Increases Aβ deposition | [213] |
| CCL2 | Enhances amyloid production and deposition, accelerate tau pathology | [214] |
| CCL3 | Promotes infiltration with T lymphocytes, upregulates BACE-1 | [215,216] |
| CCL5 | Promotes T cell infiltration in the brain, enhances microglial proliferation | [217] |
| Interferon-γ | Upregulates BACE-1 and γ-secretases, increases amyloid accumulation | [218] |
| Stages | Clinical Picture |
|---|---|
| Early onset AD/MCI | Difficulty in word-finding, impairment in reasoning and judgement |
| Mild AD | Memory loss, misplacing items, restlessness, anxiety, altered personality, episodes of aggression |
| Moderate AD | Attention deficit, recognition problems, confusion, delusions, paranoia, hallucinations, impulsive behavior |
| Severe AD | Severe dementia, functional limitations, swallowing difficulties, loss of bladder/bowel control, weight loss, seizures, enhanced diurnal sleep time with nocturnal insomnia |
| NINCDS-ADRDA Criteria, 1984 [302] | NIA-AA, 2011 [303] | IWG-AA, 2016 [304] | NIA-AA, 2018 [305] | IWG, 2021 [306] | |
|---|---|---|---|---|---|
| Setting | Research and clinical | Research and clinical | Research | Research | Research and clinical |
| Clinical requirements | Memory changes and other cognitive impairments | Amnestic or non-amnestic mild cognitive impairment, or dementia | None | None | Amnestic syndrome of hippocampal type, primary progressive aphasia of the semantic, non-fluent, or logopenic variant, corticobasal syndrome, behavioral or dysexecutive frontal syndromes |
| Biological requirements | None | Amyloid β marker (CSF or PET) or marker of degeneration (CSF tau, phosphorylated tau, 18F-fluorodeoxyglucose-PET, and T1-weighted MRI) | Amyloid β marker (CSF or PET) and tau marker (CSF or PET) | Amyloid β marker (CSF or PET) and tau marker (CSF or PET) | Amyloid β marker (CSF or PET) and tau marker (CSF or PET) |
| Phase, Status | Molecule | Trial Identifier | Number of Participants | Estimated Date of Completion | Sponsor |
|---|---|---|---|---|---|
| Not applicable, recruiting | AL002 | NCT03671880 | 30 | December 2024 | InSightec |
| Phase 1, recruiting | TB006 | NCT04920786 (TB006SAD) | 48 | January 2023 | TrueBinding, Inc. |
| Phase 1, recruiting | VT301 (regulatory T cells) | NCT05016427 | 12 | April 2022 | VTBIO Co., LTD |
| Phase 1, recruiting | emtricitabine | NCT04500847 (LINE-AD) | 35 | August 2023 | Butler Hospital, The Miriam Hospital, Alzheimer’s Association, Brown University |
| Phase 1/2, active, not recruiting | Dasatinib + quercetin | NCT04063124 | 5 | December 2023 | University of Texas Health Science center at San Antonio, Mayo Clinic |
| Phase 1/2, enrolling by invitation | Dasatinib + quercetin | NCT04785300, ALSENLITE | 20 | December 2023 | Mayo Clinic |
| Phase 1/2, recruiting | Dasatinib + quercetin | NCT05422885 STAMINA | 12 | June 2023 | Hebrew Senior Life |
| Phase 1/2, recruiting | TB006 | NCT05074498 | 140 | October 2022 | TrueBinding, Inc. |
| Phase 1/2, recruiting | Baricitinib | NCT05189106 (NADALS) | 265 | January 2024 | Alector Inc; AbbVie |
| Phase 1/2, recruiting | Pepinemab | NCT04381468 SIGNAL-AD | 40 | February 2024 | Vaccinex Inc; Alzheimer’s Drug Discovery Foundation; Alzheimer’s Association |
| Phase 2, recruiting, open-label | XPro-1595 | NCT05522387 | 261 | December 2025 | Immune Bio, Inc. |
| Phase 2, not yet recruiting | XPro-1595 | NCT05321498 | 60 | January 2023 | Immune Bio, Inc. |
| Phase 2, recruiting | XPro-1595 | NCT05318976 MINDFuL | 201 | June 2023 | Immune Bio, Inc. |
| Phase 2, recruiting | Dasatinib + quercetin | NCT04685590SToMP-AD | 48 | January 2023 | University of Texas, Wake Forest health Sciences |
| Phase 2, recruiting | VX-745, Neflamapimod | NCT03435861 | 40 | June 2021 | University Hospital of Toulouse |
| Phase 2, long term extension study, active, not recruiting | TB006 | NCT05476783 | 180 | October 2024 | TrueBinding, Inc. |
| Phase 2, recruiting | AL002 | NCT04592874INVOKE-2 | 265 | January 2024 | Alector Inc; AbbVie |
| Phase 2, recruiting | Daratumumab | NCT04070378 DARZAD | 15 | June 2023 | Janssen Scientific Affairs, LLC |
| Phase 2, recruiting | Canakinumab | NCT04795466 | 90 | February 2026 | Novartis Pharmaceuticals |
| Phase 2, recruiting | Sargramostim | NCT04902703 SESAD | 42 | July 2024 | University of Colorado, National Institute of Aging; Alzheimer’s Association; Partner Therapeutics, Inc. |
| Phase 2, recruiting | Lenalidomide | NCT04032626 MCLENA-1 | 30 | September 2024 | St Joseph’s Hospital and Medical Center Phoenix; National Institute of Aging; The Cleveland Clinic |
| Phase 3, recruiting | NE3107 | NCT04669028 | 316 | January 2023 | BioVie, Inc. |
| Phase 3, not yet recruiting | Masitinib | NCT05564169 | 600 | December 2025 | AB Science |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andronie-Cioara, F.L.; Ardelean, A.I.; Nistor-Cseppento, C.D.; Jurcau, A.; Jurcau, M.C.; Pascalau, N.; Marcu, F. Molecular Mechanisms of Neuroinflammation in Aging and Alzheimer’s Disease Progression. Int. J. Mol. Sci. 2023, 24, 1869. https://doi.org/10.3390/ijms24031869
Andronie-Cioara FL, Ardelean AI, Nistor-Cseppento CD, Jurcau A, Jurcau MC, Pascalau N, Marcu F. Molecular Mechanisms of Neuroinflammation in Aging and Alzheimer’s Disease Progression. International Journal of Molecular Sciences. 2023; 24(3):1869. https://doi.org/10.3390/ijms24031869
Chicago/Turabian StyleAndronie-Cioara, Felicia Liana, Adriana Ioana Ardelean, Carmen Delia Nistor-Cseppento, Anamaria Jurcau, Maria Carolina Jurcau, Nicoleta Pascalau, and Florin Marcu. 2023. "Molecular Mechanisms of Neuroinflammation in Aging and Alzheimer’s Disease Progression" International Journal of Molecular Sciences 24, no. 3: 1869. https://doi.org/10.3390/ijms24031869
APA StyleAndronie-Cioara, F. L., Ardelean, A. I., Nistor-Cseppento, C. D., Jurcau, A., Jurcau, M. C., Pascalau, N., & Marcu, F. (2023). Molecular Mechanisms of Neuroinflammation in Aging and Alzheimer’s Disease Progression. International Journal of Molecular Sciences, 24(3), 1869. https://doi.org/10.3390/ijms24031869