
Unveiling IL-33/ST2 Pathway Unbalance in Cardiac Remodeling Due to Obesity in Zucker Fatty Rats

 ,
, 
 , , ,
, , ,  ,
, 
Abstract
:1. Introduction
2. Results
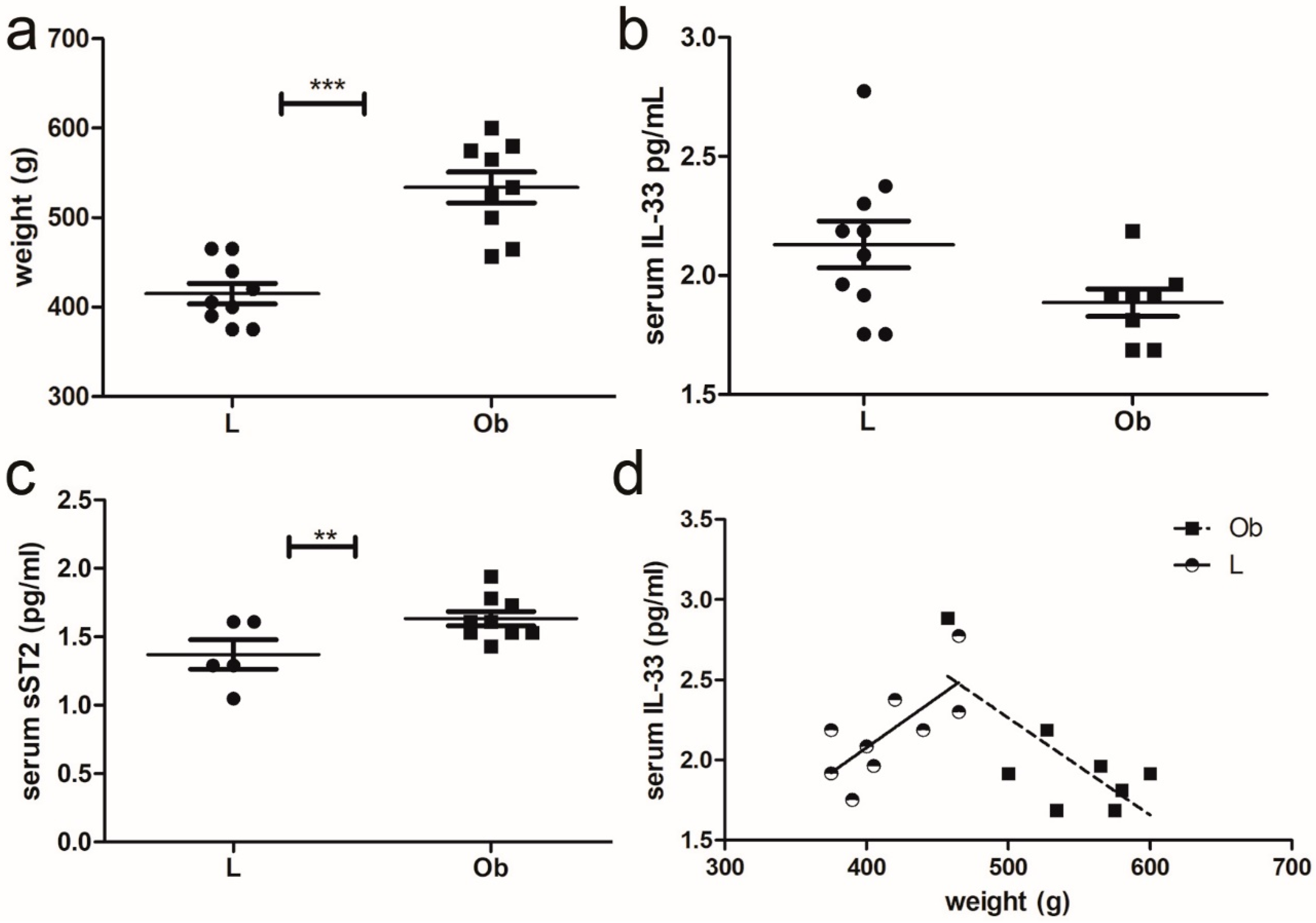
2.1. Serum Levels of IL-33 and sST2 in Relation to Obesity in ZR
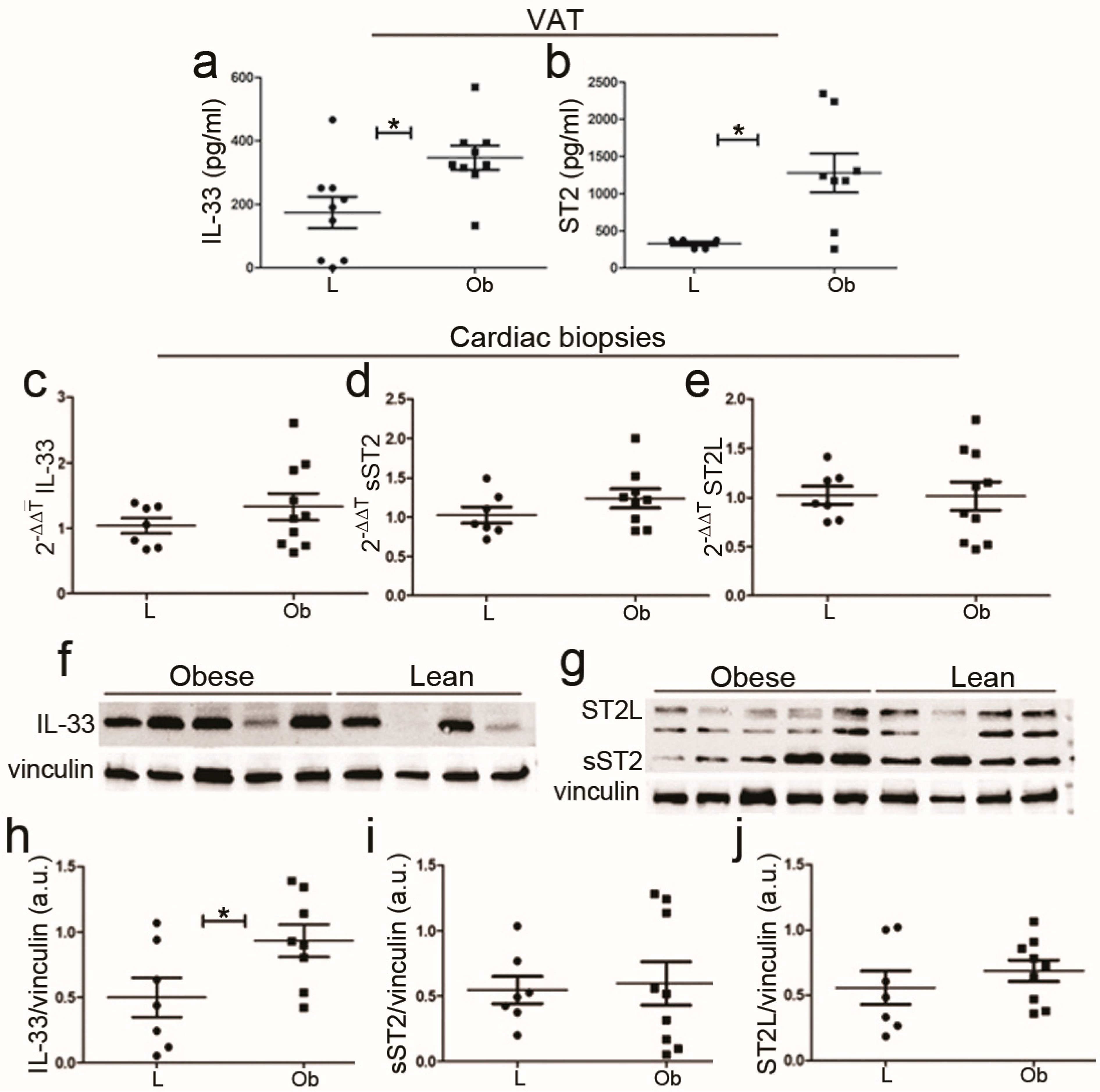
2.2. IL-33/ST2 Pathway Expression in Visceral Adipose and Cardiac Tissue
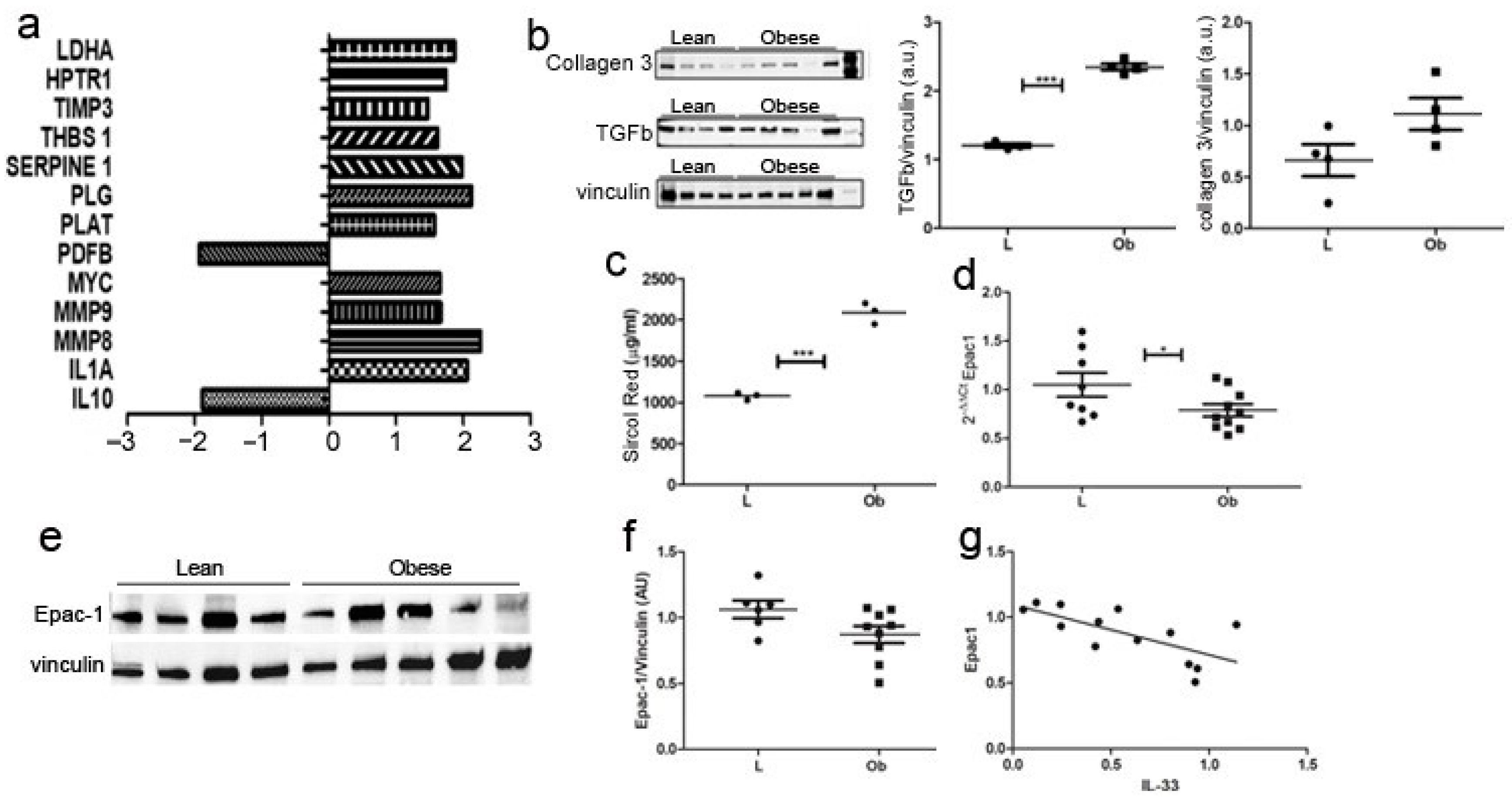
2.3. Characterization of Fibrotic Signalling in Cardiac Tissue of Obese Rats
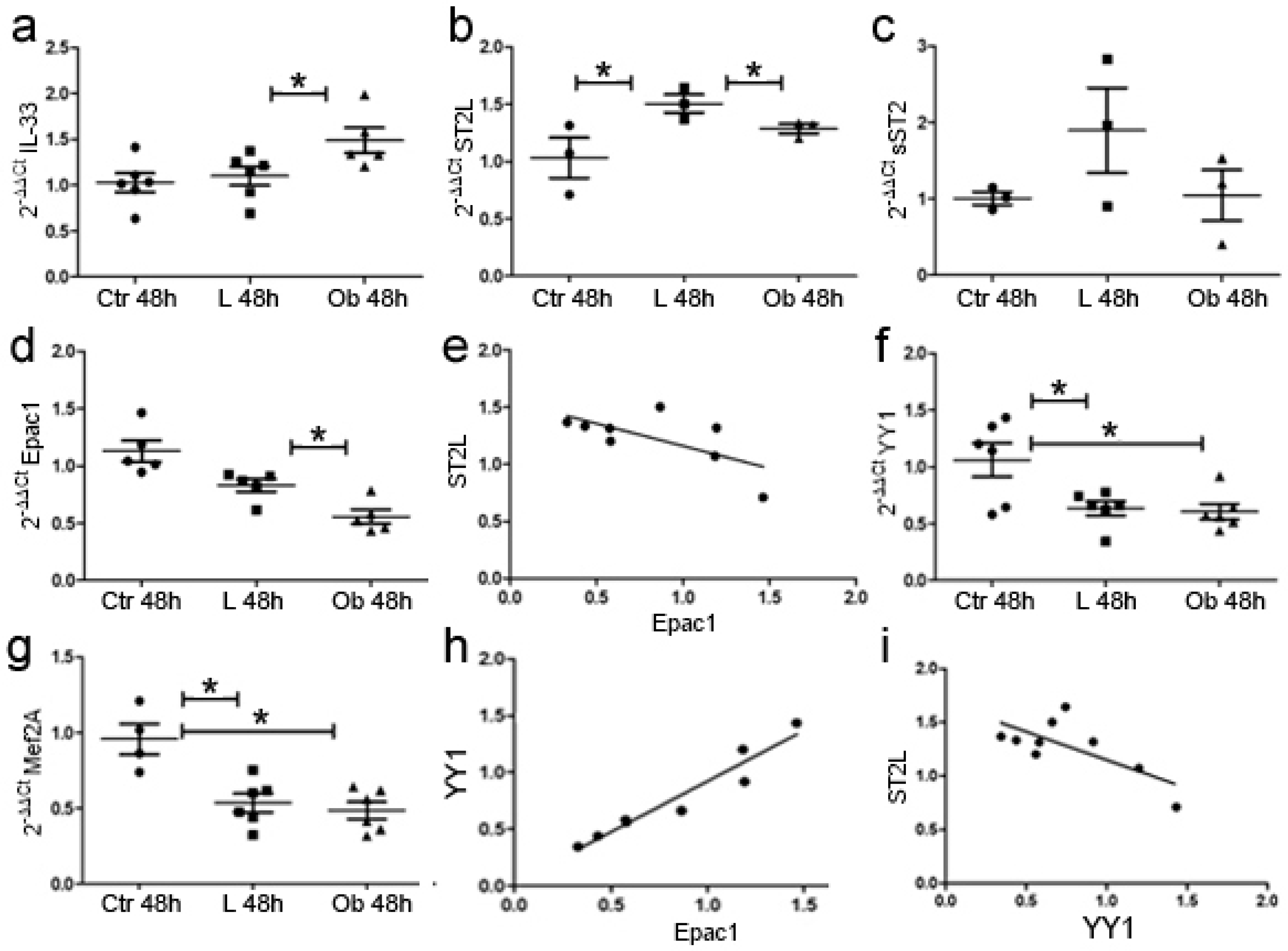
2.4. In Vitro Characterisation of Cardiomyocytes following VAT Stimulation
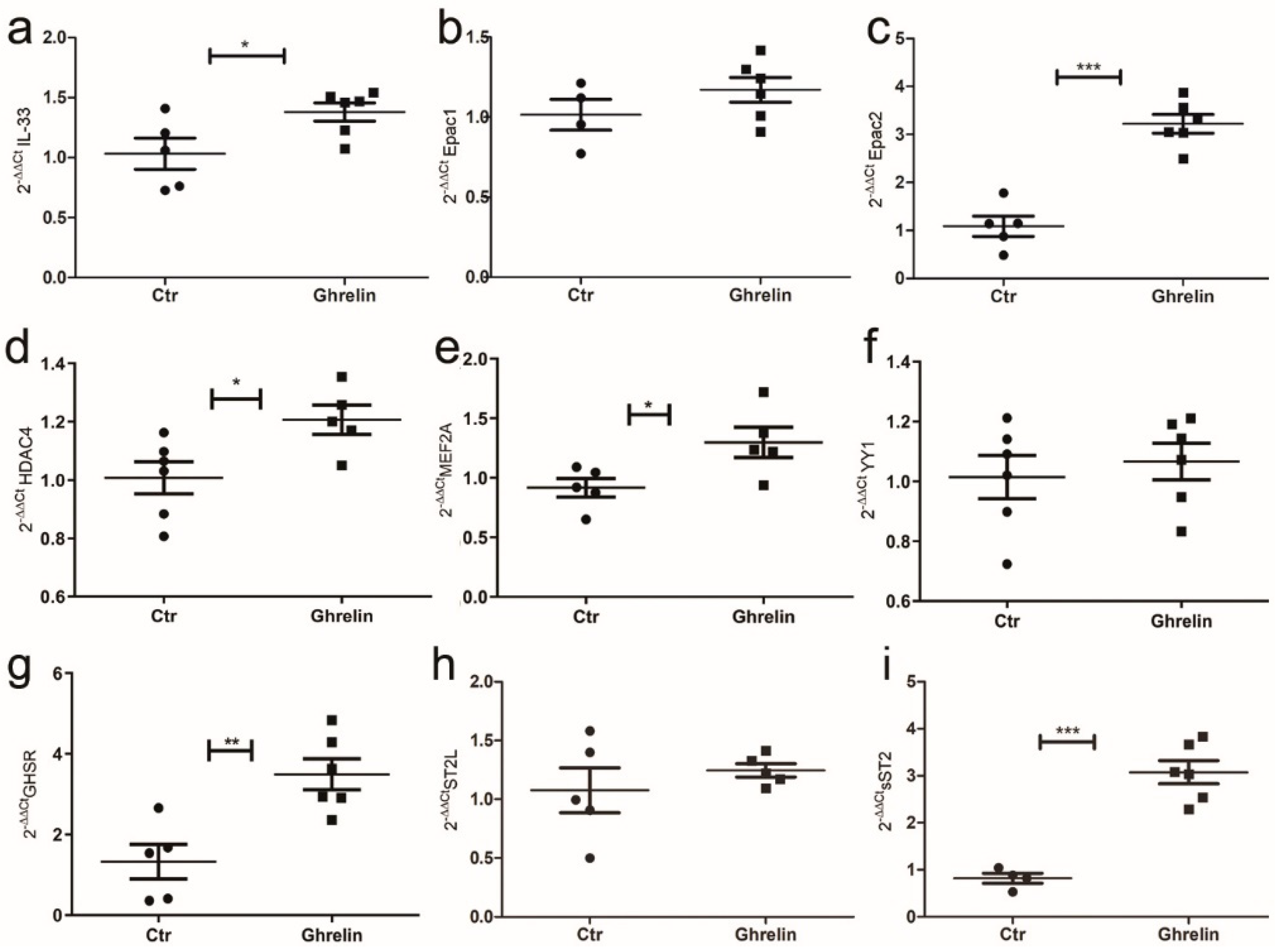
2.5. In Vitro Ghrelin Stimulation of Cardiomyocytes Modulated IL-33/ST2 Expression
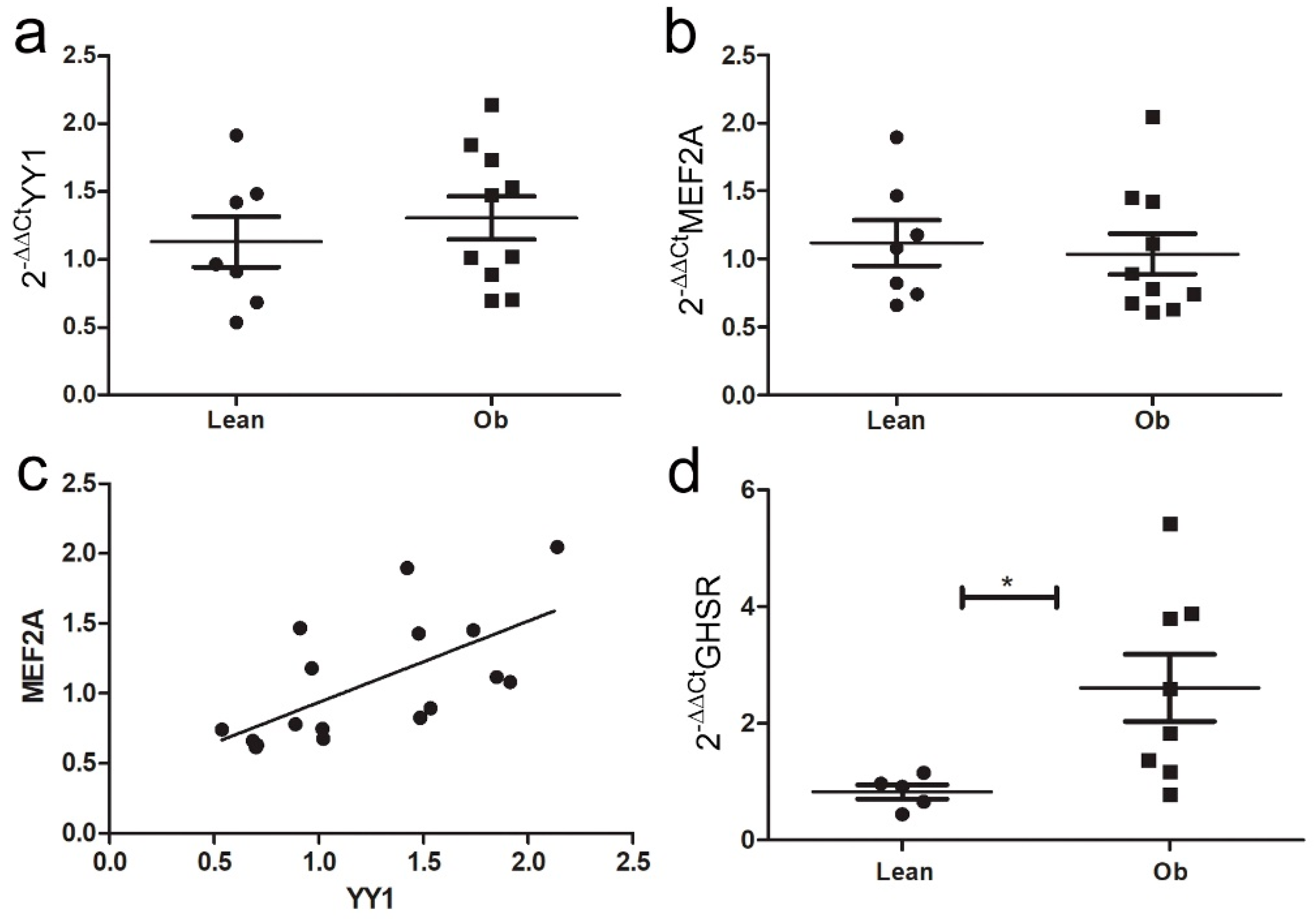
2.6. Characterization of Ghrelin-IL-33/ST2 Axis in Zucker Rat Cardiac Tissue
3. Discussion
4. Material and Methods
4.1. Animal Models Included in This Study
4.2. Total RNA Extraction and Reverse Transcription
4.3. RT2 Profiler PCR Arrays
4.4. Western Blot Analysis
4.5. Cell Culture and Transfection
4.6. ELISA Assay
4.7. Sircol Assay
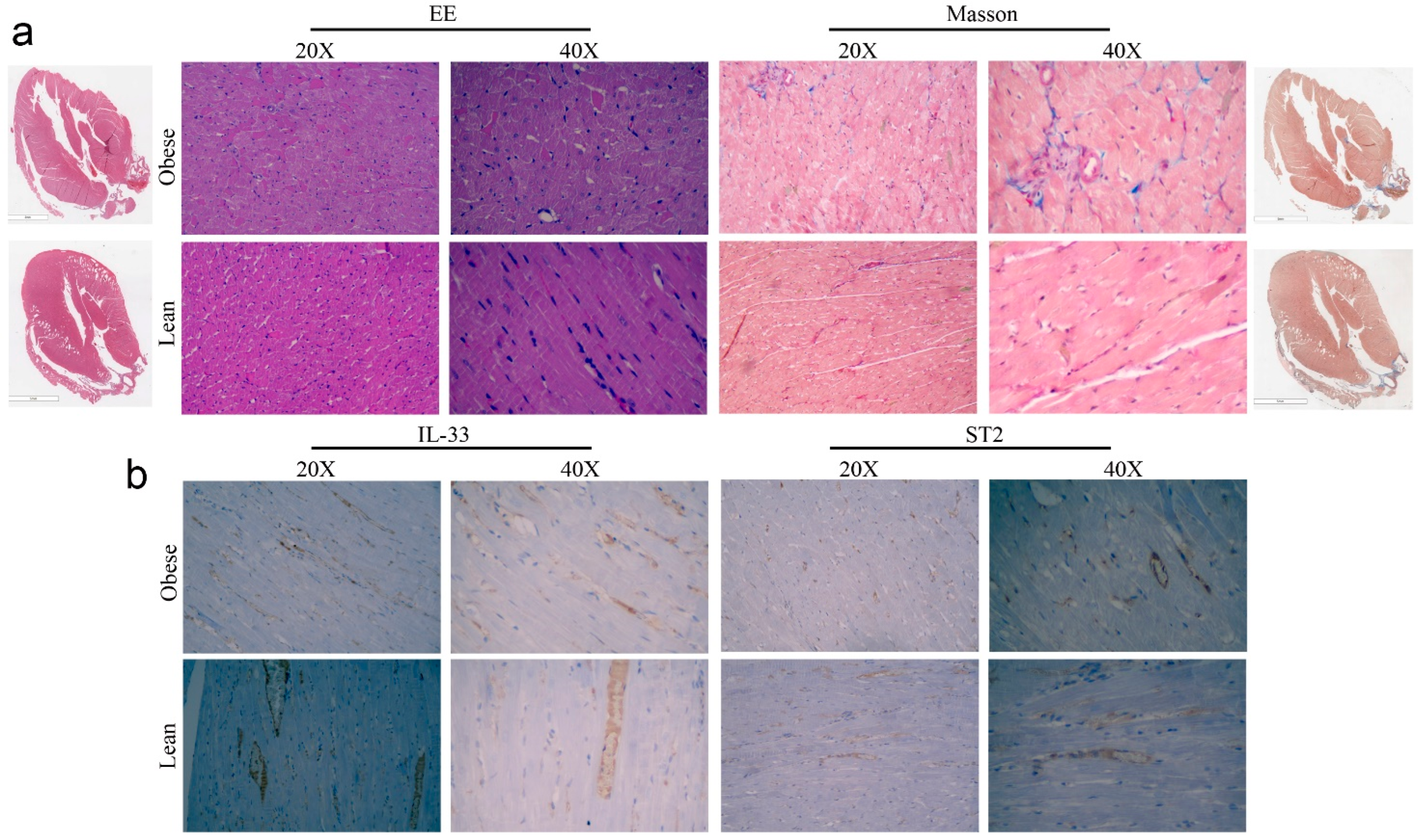
4.8. Histological Analysis
4.9. List of Included Antibodies
- Collagen 3 alpha1 antibody, Cohesion biosciences (London, UK), mouse monoclonal 1:1000 140 KDa.
- IL-33 Enzo, rabbit polyclonal antibody, 1:1000 25 kDa.
- Epac 1 (5D3), Cell signaling, Mouse mAb 1:1000 100 kDa.
- Epac 2 (5B1), Cell signaling Mouse, mAb 1:1000 115 kDa.
- FOXP3 (F-9) Santa Cruz (Dallas, Texas) Mouse monoclonal 1:200 48 kDa.
- ST2, Proteintech (Rosemont, Illinois, USA), polyclonal antibody Rabbit 63/37/30 kDa.
- ST2, Enzo, polyclonal antibody, 1:500.
- Tgf beta1, Cohesion biosciences, rabbit polyclonal, 1:500 43 kDa.
- Vinculin, Cell Signaling, Rabbit mAb 1:1000 124 kDa.
4.10. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mandviwala, T.; Khalid, U.; Deswal, A. Obesity and Cardiovascular Disease: A Risk Factor or a Risk Marker? Curr. Atheroscler. Rep. 2016, 18, 21. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tao, L.; Yuan, Y.; Lau, W.B.; Li, R.; Lopez, B.L.; Christopher, T.A.; Tian, R.; Ma, X.L. Cardioprotective effect of adiponectin is partially mediated by its AMPK-independent antinitrative action. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E384–E391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lage, R.; Moscoso, I.; Fernandez-Trasancos, A.; Cebro, M.; Couselo, M.; Fandino-Vaquero, R.; Bravo, S.B.; Sierra, J.; Gonzalez-Juanatey, J.R.; Eiras, S. Differential behaviour of epicardial adipose tissue-secretomes with high and low orosomucoid levels from patients with cardiovascular disease in H9C2 cells. Mol. Cell. Endocrinol. 2015, 416, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulos, A.S.; Antoniades, C. The role of epicardial adipose tissue in cardiac biology: Classic concepts and emerging roles. J. Physiol. 2017, 595, 3907–3917. [Google Scholar] [CrossRef] [Green Version]
- Karastergiou, K.; Evans, I.; Ogston, N.; Miheisi, N.; Nair, D.; Kaski, J.C.; Jahangiri, M.; Mohamed-Ali, V. Epicardial adipokines in obesity and coronary artery disease induce atherogenic changes in monocytes and endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1340–1346. [Google Scholar] [CrossRef] [Green Version]
- Uchida, Y.; Uchida, Y.; Shimoyama, E.; Hiruta, N.; Kishimoto, T.; Watanabe, S. Pericoronary Adipose Tissue as Storage and Supply Site for Oxidized Low-Density Lipoprotein in Human Coronary Plaques. PLoS ONE 2016, 11, e0150862. [Google Scholar] [CrossRef] [Green Version]
- Antonopoulos, A.S.; Margaritis, M.; Coutinho, P.; Shirodaria, C.; Psarros, C.; Herdman, L.; Sanna, F.; De Silva, R.; Petrou, M.; Sayeed, R.; et al. Adiponectin as a link between type 2 diabetes and vascular NADPH oxidase activity in the human arterial wall: The regulatory role of perivascular adipose tissue. Diabetes 2015, 64, 2207–2219. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.P.; Hsu, H.L.; Hung, W.C.; Yu, T.H.; Chen, Y.H.; Chiu, C.A.; Lu, L.F.; Chung, F.M.; Shin, S.J.; Lee, Y.J. Increased epicardial adipose tissue (EAT) volume in type 2 diabetes mellitus and association with metabolic syndrome and severity of coronary atherosclerosis. Clin. Endocrinol. 2009, 70, 876–882. [Google Scholar] [CrossRef]
- Ahn, S.G.; Lim, H.S.; Joe, D.Y.; Kang, S.J.; Choi, B.J.; Choi, S.Y.; Yoon, M.H.; Hwang, G.S.; Tahk, S.J.; Shin, J.H. Relationship of epicardial adipose tissue by echocardiography to coronary artery disease. Heart 2008, 94, e7. [Google Scholar] [CrossRef] [Green Version]
- Blumensatt, M.; Greulich, S.; Herzfeld de Wiza, D.; Mueller, H.; Maxhera, B.; Rabelink, M.J.; Hoeben, R.C.; Akhyari, P.; Al-Hasani, H.; Ruige, J.B.; et al. Activin A impairs insulin action in cardiomyocytes via up-regulation of miR-143. Cardiovasc. Res. 2013, 100, 201–210. [Google Scholar] [CrossRef]
- Venteclef, N.; Guglielmi, V.; Balse, E.; Gaborit, B.; Cotillard, A.; Atassi, F.; Amour, J.; Leprince, P.; Dutour, A.; Clement, K.; et al. Human epicardial adipose tissue induces fibrosis of the atrial myocardium through the secretion of adipo-fibrokines. Eur. Heart J. 2015, 36, 795–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baekkevold, E.S.; Roussigne, M.; Yamanaka, T.; Johansen, F.E.; Jahnsen, F.L.; Amalric, F.; Brandtzaeg, P.; Erard, M.; Haraldsen, G.; Girard, J.P. Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. Am. J. Pathol. 2003, 163, 69–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altara, R.; Ghali, R.; Mallat, Z.; Cataliotti, A.; Booz, G.W.; Zouein, F.A. Conflicting vascular and metabolic impact of the IL-33/sST2 axis. Cardiovasc. Res. 2018, 114, 1578–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griesenauer, B.; Paczesny, S. The ST2/IL-33 Axis in Immune Cells during Inflammatory Diseases. Front. Immunol. 2017, 8, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, K.; Sanada, S.; Kudinova, A.Y.; Steinhauser, M.L.; Handa, V.; Gannon, J.; Lee, R.T. Interleukin-33 prevents apoptosis and improves survival after experimental myocardial infarction through ST2 signaling. Circ. Heart Fail. 2009, 2, 684–691. [Google Scholar] [CrossRef] [Green Version]
- Drake, L.Y.; Kita, H. IL-33: Biological properties, functions, and roles in airway disease. Immunol. Rev. 2017, 278, 173–184. [Google Scholar] [CrossRef]
- Gieseck, R.L., 3rd; Wilson, M.S.; Wynn, T.A. Type 2 immunity in tissue repair and fibrosis. Nat. Rev. Immunol. 2018, 18, 62–76. [Google Scholar] [CrossRef]
- Gruzdeva, O.; Uchasova, E.; Dyleva, Y.; Borodkina, D.; Akbasheva, O.; Belik, E.; Karetnikova, V.; Brel, N.; Kokov, A.; Kashtalap, V.; et al. Relationships between epicardial adipose tissue thickness and adipo-fibrokine indicator profiles post-myocardial infarction. Cardiovasc. Diabetol. 2018, 17, 40. [Google Scholar] [CrossRef] [Green Version]
- Vianello, E.; Dozio, E.; Bandera, F.; Schmitz, G.; Nebuloni, M.; Longhi, E.; Tacchini, L.; Guazzi, M.; Corsi Romanelli, M.M. Dysfunctional EAT thickness may promote maladaptive heart remodeling in CVD patients through the ST2-IL33 system, directly related to EPAC protein expression. Sci. Rep. 2019, 9, 10331. [Google Scholar] [CrossRef] [Green Version]
- Dozio, E.; Briganti, S.; Vianello, E.; Dogliotti, G.; Barassi, A.; Malavazos, A.E.; Ermetici, F.; Morricone, L.; Sigruener, A.; Schmitz, G.; et al. Epicardial adipose tissue inflammation is related to vitamin D deficiency in patients affected by coronary artery disease. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 267–273. [Google Scholar] [CrossRef]
- Miller, A.M.; Asquith, D.L.; Hueber, A.J.; Anderson, L.A.; Holmes, W.M.; McKenzie, A.N.; Xu, D.; Sattar, N.; McInnes, I.B.; Liew, F.Y. Interleukin-33 induces protective effects in adipose tissue inflammation during obesity in mice. Circ. Res. 2010, 107, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.; Al-Ghimlas, F.; Warsame, S.; Al-Hubail, A.; Ahmad, R.; Bennakhi, A.; Al-Arouj, M.; Behbehani, K.; Dehbi, M.; Dermime, S. IL-33 is negatively associated with the BMI and confers a protective lipid/metabolic profile in non-diabetic but not diabetic subjects. BMC Immunol. 2014, 15, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Liu, N.; Feng, X.; Yang, Y.; Fang, Y.; Zhuang, S.; Dai, Y.; Liu, M.; Tang, L. Circulating levels of IL-33 are elevated by obesity and positively correlated with metabolic disorders in Chinese adults. J. Transl. Med. 2021, 19, 52. [Google Scholar] [CrossRef] [PubMed]
- Schernthaner, C.; Lichtenauer, M.; Wernly, B.; Paar, V.; Pistulli, R.; Rohm, I.; Jung, C.; Figulla, H.R.; Yilmaz, A.; Cadamuro, J.; et al. Multibiomarker analysis in patients with acute myocardial infarction. Eur. J. Clin. Investig. 2017, 47, 638–648. [Google Scholar] [CrossRef]
- Huang, A.; Glick, S.A. Genetic susceptibility to cutaneous radiation injury. Arch. Dermatol. Res. 2017, 309, 1–10. [Google Scholar] [CrossRef]
- Miller, A.M.; Purves, D.; McConnachie, A.; Asquith, D.L.; Batty, G.D.; Burns, H.; Cavanagh, J.; Ford, I.; McLean, J.S.; Packard, C.J.; et al. Soluble ST2 associates with diabetes but not established cardiovascular risk factors: A new inflammatory pathway of relevance to diabetes? PLoS ONE 2012, 7, e47830. [Google Scholar] [CrossRef] [Green Version]
- Weir, R.A.; Miller, A.M.; Murphy, G.E.; Clements, S.; Steedman, T.; Connell, J.M.; McInnes, I.B.; Dargie, H.J.; McMurray, J.J. Serum soluble ST2: A potential novel mediator in left ventricular and infarct remodeling after acute myocardial infarction. J. Am. Coll. Cardiol. 2010, 55, 243–250. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Chandrasekera, P.C.; Pippin, J.J. Leptin- and leptin receptor-deficient rodent models: Relevance for human type 2 diabetes. Curr. Diabetes Rev. 2014, 10, 131–145. [Google Scholar] [CrossRef] [Green Version]
- Conrotto, P.; Yakymovych, I.; Yakymovych, M.; Souchelnytskyi, S. Interactome of transforming growth factor-beta type I receptor (TbetaRI): Inhibition of TGFbeta signaling by Epac1. J. Proteome Res. 2007, 6, 287–297. [Google Scholar] [CrossRef]
- Tan, Y.Q.; Li, J.; Chen, H.W. Epac, a positive or negative signaling molecule in cardiovascular diseases. Biomed. Pharmacother. Biomed. Pharmacother. 2022, 148, 112726. [Google Scholar] [CrossRef]
- Demyanets, S.; Kaun, C.; Pentz, R.; Krychtiuk, K.A.; Rauscher, S.; Pfaffenberger, S.; Zuckermann, A.; Aliabadi, A.; Groger, M.; Maurer, G.; et al. Components of the interleukin-33/ST2 system are differentially expressed and regulated in human cardiac cells and in cells of the cardiac vasculature. J. Mol. Cell. Cardiol. 2013, 60, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, N.; Lu, Y.; Gu, M.; Li, N.; Liu, M.; Jiao, J.; Zhu, Z.; Li, J.; Li, D.; Tang, T.; et al. A Unique Population of Regulatory T Cells in Heart Potentiates Cardiac Protection From Myocardial Infarction. Circulation 2020, 142, 1956–1973. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.J.; Seto, E. Unlocking the mechanisms of transcription factor YY1: Are chromatin modifying enzymes the key? Gene 1999, 236, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Cornwell, J.D.; McDermott, J.C. MEF2 in cardiac hypertrophy in response to hypertension. Trends Cardiovasc. Med. 2022, 13, 1. [Google Scholar] [CrossRef]
- Iglesias, M.J.; Pineiro, R.; Blanco, M.; Gallego, R.; Dieguez, C.; Gualillo, O.; Gonzalez-Juanatey, J.R.; Lago, F. Growth hormone releasing peptide (ghrelin) is synthesized and secreted by cardiomyocytes. Cardiovasc. Res. 2004, 62, 481–488. [Google Scholar] [CrossRef] [PubMed]
- de Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Rui, T.; Tang, Q. IL-33 attenuates anoxia/reoxygenation-induced cardiomyocyte apoptosis by inhibition of PKCbeta/JNK pathway. PLoS ONE 2013, 8, e56089. [Google Scholar] [CrossRef]
- Zeyda, M.; Wernly, B.; Demyanets, S.; Kaun, C.; Hammerle, M.; Hantusch, B.; Schranz, M.; Neuhofer, A.; Itariu, B.K.; Keck, M.; et al. Severe obesity increases adipose tissue expression of interleukin-33 and its receptor ST2, both predominantly detectable in endothelial cells of human adipose tissue. Int. J. Obes. 2013, 37, 658–665. [Google Scholar] [CrossRef] [Green Version]
- Baron, M.A.; Ferreira, L.R.P.; Teixeira, P.C.; Moretti, A.I.S.; Santos, R.H.B.; Frade, A.F.; Kuramoto, A.; Debbas, V.; Benvenuti, L.A.; Gaiotto, F.A.; et al. Matrix Metalloproteinase 2 and 9 Enzymatic Activities are Selectively Increased in the Myocardium of Chronic Chagas Disease Cardiomyopathy Patients: Role of TIMPs. Front. Cell. Infect. Microbiol. 2022, 12, 836242. [Google Scholar] [CrossRef]
- Robichaux, W.G., 3rd; Cheng, X. Intracellular cAMP Sensor EPAC: Physiology, Pathophysiology, and Therapeutics Development. Physiol. Rev. 2018, 98, 919–1053. [Google Scholar] [CrossRef]
- Wu, Q.Q.; Xiao, Y.; Yuan, Y.; Ma, Z.G.; Liao, H.H.; Liu, C.; Zhu, J.X.; Yang, Z.; Deng, W.; Tang, Q.Z. Mechanisms contributing to cardiac remodelling. Clin. Sci. 2017, 131, 2319–2345. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Umemura, M.; Yokoyama, U.; Okumura, S.; Ishikawa, Y. The role of Epac in the heart. Cell. Mol. Life Sci. 2017, 74, 591–606. [Google Scholar] [CrossRef] [PubMed]
- Metrich, M.; Laurent, A.C.; Breckler, M.; Duquesnes, N.; Hmitou, I.; Courillau, D.; Blondeau, J.P.; Crozatier, B.; Lezoualc’h, F.; Morel, E. Epac activation induces histone deacetylase nuclear export via a Ras-dependent signalling pathway. Cell. Signal. 2010, 22, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Asensio-Lopez, M.C.; Lax, A.; Fernandez Del Palacio, M.J.; Sassi, Y.; Hajjar, R.J.; Januzzi, J.L.; Bayes-Genis, A.; Pascual-Figal, D.A. Yin-Yang 1 transcription factor modulates ST2 expression during adverse cardiac remodeling post-myocardial infarction. J. Mol. Cell. Cardiol. 2019, 130, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Ma, Z.; Zhang, Z.; Xiong, X.; Wang, X.; Zhang, H.; Shi, G.; Xia, X.; Ning, G.; Li, X. Yin Yang 1 promotes hepatic steatosis through repression of farnesoid X receptor in obese mice. Gut 2014, 63, 170–178. [Google Scholar] [CrossRef]
- Moran, O.; Phillip, M. Leptin: Obesity, diabetes and other peripheral effects—A review. Pediatr. Diabetes 2003, 4, 101–109. [Google Scholar] [CrossRef]
- Takaya, K.; Ogawa, Y.; Isse, N.; Okazaki, T.; Satoh, N.; Masuzaki, H.; Mori, K.; Tamura, N.; Hosoda, K.; Nakao, K. Molecular cloning of rat leptin receptor isoform complementary DNAs—Identification of a missense mutation in Zucker fatty (fa/fa) rats. Biochem. Biophys. Res. Commun. 1996, 225, 75–83. [Google Scholar] [CrossRef]
- Kai, Y.; Gao, J.; Liu, H.; Wang, Y.; Tian, C.; Guo, S.; He, L.; Li, M.; Tian, Z.; Song, X. Effects of IL-33 on 3T3-L1 cells and obese mice models induced by a high-fat diet. Int. Immunopharmacol. 2021, 101, 108209. [Google Scholar] [CrossRef]
- Abston, E.D.; Barin, J.G.; Cihakova, D.; Bucek, A.; Coronado, M.J.; Brandt, J.E.; Bedja, D.; Kim, J.B.; Georgakopoulos, D.; Gabrielson, K.L.; et al. IL-33 independently induces eosinophilic pericarditis and cardiac dilation: ST2 improves cardiac function. Circ. Heart Fail. 2012, 5, 366–375. [Google Scholar] [CrossRef] [Green Version]
- Albarran-Zeckler, R.G.; Smith, R.G. The ghrelin receptors (GHS-R1a and GHS-R1b). Endocr. Dev. 2013, 25, 5–15. [Google Scholar] [CrossRef]
- McHedlidze, T.; Waldner, M.; Zopf, S.; Walker, J.; Rankin, A.L.; Schuchmann, M.; Voehringer, D.; McKenzie, A.N.; Neurath, M.F.; Pflanz, S.; et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity 2013, 39, 357–371. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prime Name | Primer Code | Amplicon Length |
|---|---|---|
| Rat Yy1 | PPR53391A | 99 bp |
| Rat Socs3 | PPR06602A | 145 bp |
| Rat Rapgef3 (Epac1) | PPR49530A | 131 bp |
| Rat Rapgef4 (Epac2) | PPR49522A | 119 bp |
| Rat Pparα | PPR44459A | 110 bp |
| Rat Mef2A | PPR62504B | 100 bp |
| Rat IL33 | PPR564110A | 97 bp |
| Rar Hdac4 | PPR47615A | 89 bp |
| Rat Ghsr | PPR51984A | 121 bp |
| Rat Ghrl | PPR49492A | 107 bp |
| Rat Gapdh | PPR06557B | 200 bp |
| Prime Name | Primer Code | Amplicon Length |
|---|---|---|
| Forward Rat St2L | AGTTGTGCATTTACGGGAGAG | 68 bp |
| Reverse Rat St2L | GGATACTGCTTTCCACCACAG | |
| Forward Rat St2s | GGTGTGACCGACAAGGACT | 119 bp |
| Reverse Rat St2s | TTGTGAGAGACACTCCTTAC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sitzia, C.; Vianello, E.; Dozio, E.; Kalousová, M.; Zima, T.; Brizzola, S.; Roccabianca, P.; Tedeschi, G.; Lamont, J.; Tacchini, L.; et al. Unveiling IL-33/ST2 Pathway Unbalance in Cardiac Remodeling Due to Obesity in Zucker Fatty Rats. Int. J. Mol. Sci. 2023, 24, 1991. https://doi.org/10.3390/ijms24031991
Sitzia C, Vianello E, Dozio E, Kalousová M, Zima T, Brizzola S, Roccabianca P, Tedeschi G, Lamont J, Tacchini L, et al. Unveiling IL-33/ST2 Pathway Unbalance in Cardiac Remodeling Due to Obesity in Zucker Fatty Rats. International Journal of Molecular Sciences. 2023; 24(3):1991. https://doi.org/10.3390/ijms24031991
Chicago/Turabian StyleSitzia, Clementina, Elena Vianello, Elena Dozio, Marta Kalousová, Tomáš Zima, Stefano Brizzola, Paola Roccabianca, Gabriella Tedeschi, John Lamont, Lorenza Tacchini, and et al. 2023. "Unveiling IL-33/ST2 Pathway Unbalance in Cardiac Remodeling Due to Obesity in Zucker Fatty Rats" International Journal of Molecular Sciences 24, no. 3: 1991. https://doi.org/10.3390/ijms24031991
APA StyleSitzia, C., Vianello, E., Dozio, E., Kalousová, M., Zima, T., Brizzola, S., Roccabianca, P., Tedeschi, G., Lamont, J., Tacchini, L., & Corsi-Romanelli, M. M. (2023). Unveiling IL-33/ST2 Pathway Unbalance in Cardiac Remodeling Due to Obesity in Zucker Fatty Rats. International Journal of Molecular Sciences, 24(3), 1991. https://doi.org/10.3390/ijms24031991