
Liaisons dangereuses: Intrinsic Disorder in Cellular Proteins Recruited to Viral Infection-Related Biocondensates





Abstract
:1. Introduction
2. Results
2.1. Data Set Generation and Global Disorder Analysis of the Selected Eukaryotic Proteins

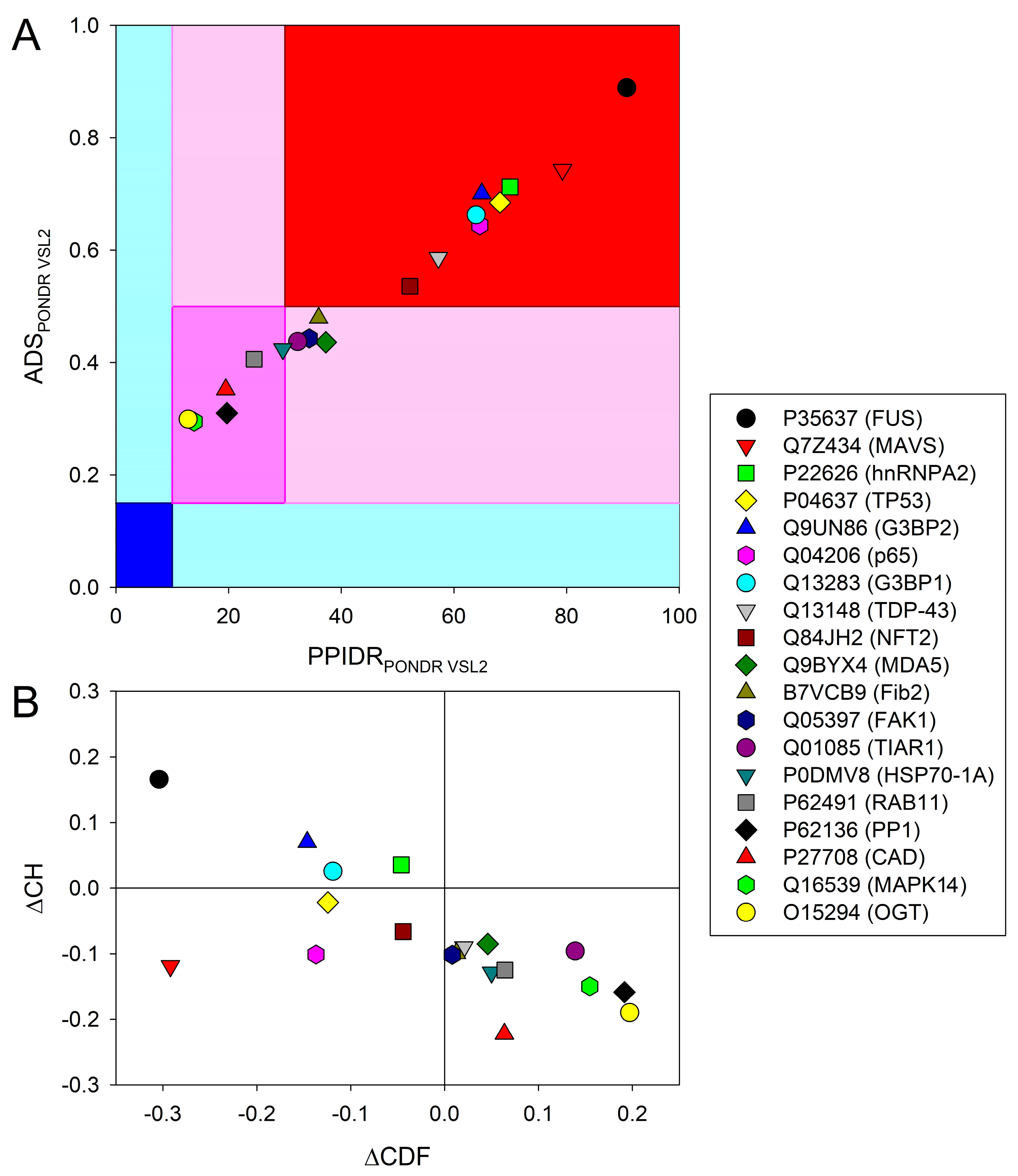{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cellular Protein | Organism UniProt ID | Type of Condensate | Virus | Known to Phase Separate | PPIDR (PONDR® VSL2) | IDRs (PONDR® VSL2) | MoRFs | FuzDrop pLLPS Status DPRs | PSP Score | DisProt Entry # (IDRs) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| FUS 1 | Homo sapiens P35637 | Cellular MLO | SARS-CoV-2 | YES | 90.68 | 1–286 314–315 330–347 356–526 | 1–19 33–61 75–83 111–165 175–196 205–212 231–240 257–268 285–312 347–375 423–428 432–446 478–486 489–512 | 0.9999 Driver 1–294 360–437 443–526 | 0.99 | DP01102 (1–507) | [66] |
| MAVS 2 | Homo sapiens Q7Z434 | Viral IBs | RSV | NO | 79.26 | 1–6 91–508 537–540 | 123–148 156–268 277–290 294–325 338–377 397–410 416–449 463–479 | 0.9996 Driver 81–456 470–513 | 0.58 | none | [68] |
| hnRNPA2 3 | Homo sapiens P22626 | Cellular MLO | SARS-CoV-2 | YES | 69.97 | 1–21 57–62 72–108 120–126 148–152 183–353 | 65–71 155–160 168–177 205–212 238–244 | 0.9808 Driver 1–12 187–353 | 0.99 | DP01109 (190–341) | [66] |
| p53 4 | Homo sapiens P04637 | Viral replication foci | HPV (human papilloma virus) | YES | 68.19 | 1–107 165–166 168–189 222–224 260–393 | 11–57 106–115 132–141 232–239 251–258 265–277 322–355 363–387 | 0.9848 Driver 1–24 28–108 277–337 341–393 | 0.94 | DP00086 (1–93, 291–312, 361–393) | [69] |
| G3BP2 5 | Homo sapiens Q9UN86 | Cellular MLO | SARS-CoV-2 | YES | 64.94 | 1–7 43–53 100–106 127–331 400–482 | 90–97 109–115 119–145 168–192 207–224 232–281 323–338 348–358 371–379 387–399 436–448 456–482 | 0.9976 Driver 130–325 399–482 | 0.94 | none | [70] |
| p65 6 | Homo sapiens Q04206 | Viral IBs | RSV | NO | 64.61 | 1–4 14–92 169–175 257–457 487–551 | 1–11 31–41 62–76 98–103 110–118 285–290 305–317 350–380 398–414 433–483 492–504 523–551 | 0.9487 Driver 37–71 77–96 309–355 367–441 503–526 | 0.83 | DP00085 (428–551) | [71] |
| G3BP1 7 | Homo sapiens Q13283 | Cellular MLO | SARS-CoV-2 | YES | 63.95 | 1–7 37–52 139–342 352–356 401–466 | 123–143 165–193 206–280 304–311 337–348 355–369 376–386 395–406 435–466 | 0.9937 Driver 135–339 405–466 | 0.49 | none | [70] |
| TDP-43 8 | Homo sapiens Q13148 | Cellular MLO | SARS-CoV-2 | YES | 57.25 | 1–23 79–98 137–143 176–195 197–197 199–207 258–414 | 28–35 245–255 311–342 380–387 397–402 | 0.8981 Driver 251–414 | 0.98 | DP01108 (263–414) | [66] |
| NTF2 9 | Nicotiana benthamiana Q84JH2 This is a homolog from Arabidopsis thaliana | Cellular MLO | Pea enation mosaic virus 2 (PEMV2) | YES | 52.18 | 1–8 82–87 141–144 160–174 177–315 392–458 | 173–179 197–202 233–257 276–284 292–299 318–334 360–365 424–429 450–458 | 0.7408 Driver 184–200 213–312 396–451 | 0.96 | none | [72] |
| MDA5 10 | Homo sapiens Q9BYX4 | Viral IBs | RSV | NO | 37.27 | 1–8 98–110 153–164 192–311 347–356 425–430 466–477 493–500 524–553 568–568 570–576 585–598 631–718 757–774 824–825 865–896 995–999 1022–1025 | 233–239 243–276 324–329 503–515 | 0.6164 Driver 239–308 489–499 566–595 641–661 | 0.11 | none | [68] |
| Fib2 11 | Nicotiana benthamiana B7VCB9 | Cellular MLO | PEMV2 | YES | 35.99 | 1–83 116–119 277–292 305–314 | 1–16 29–44 72–90 | 0.3248 Client 1–86 | 0.99 | none | [72] |
| FAK1 12 | Homo sapiens Q05397 | Negri bodies (viral IBs) | RABV | YES | 34.32 | 1–32 107–113 188–194 306–313 363–418 576–580 638–647 660–751 771 785–926 941–949 1014–1018 1046–1052 | 1–7 36–42 341–359 652–660 672–681 698–705 726–769 792–808 830–845 848–867 882–905 922–938 958–965 | 0.6417 Driver 1–35 683–736 743–767 812–922 | 0.07 | DP03144 (565–583) | [31,73] |
| TIAR1 13 | Homo sapiens Q01085 | Viral IBs | Ebola Virus (EBOV) | YES | 32.27 | 1–8 85–94 132–138 174–201 280–284 303–309 320–375 | 140–148 205–213 339–345 | 0.5857 Client 1–16 174–185 311–375 | 0.36 | none | [74] |
| HSP70-1A 14 | Homo sapiens P0DMV8 | Negri bodies (viral IBs) | RABV | NO | 29.64 | 1–5 100–106 153–158 230–230 243–286 361–363 491–572 588–598 611–641 | 476–486 541–550 573–584 602–614 | 0.3828 Client 548–569 606–641 | 0.12 | DP02353 (229–306) | [31] |
| RAB11 15 | Homo sapiens P62491 | Viral assembly compartment | IAV | NO | 24.54 | 1–6 24–25 35–40 178–216 | 166–176 210–216 | 0.1679 Client 176–209 | 0.02 | none | [58] |
| PP1 16 | Homo sapiens P62136 | Viral IBs | RSV | NO | 19.70 | 1–10 14–14 18–30 179–183 213–216 299–330 | Not found | 0.1692 Client 300–330 | 0.04 | none | [75] |
| CAD 17 | Homo sapiens P27708 | IBs (viral factories) | EBOV | NO | 19.51 | 1–5 118–155 222–232 239–243 319–320 338–343 360–402 405–406 408–412 525–544 567–573 689–696 797–804 852–860 1041–1042 1117–1119 1156–1159 1287–1289 1325–1326 1401–1403 1540–1545 1649–1665 1690–1708 1711–1711 1713–1727 1807–1923 1972–1986 1988–2009 2122–2135 2177–2177 2190–2196 2223–2225 | 327–333 346–357 1675–1684 1709–1718 1768–1805 1841–1864 1874–1891 1903–1930 2161–2166 | 0.2011 Client 379–392 1812–1923 2043–2053 | 0.11 | DP01024 (1822–1846) | [76] |
| MAPK14 18 | Homo sapiens Q16539 | Viral IBs | RSV | NO | 13.89 | 1–7 176–178 247–256 313–327 342–345 350–360 | Not found | 0.1119 Not related to LLPS | 0.00 | None | [77] |
| OGT 19 | Homo sapiens O15294 | Viral IBs | RSV | NO | 12.83 | 1–17 34–36 106–106 305–308 377–377 405–405 435–445 499–512 541–553 564–579 683–693 759–770 814–819 908–911 1031–1034 1039–1046 | Not found | 0.1567 Client 1–15 758–772 | 0.10 | None | [77] |
2.2. Per-Residue Disorder Predictions and Interactivity Analysis of the Eukaryotic Proteins Recruited to Vir-MLOs
2.3. LLPS Propensities of the Eukaryotic Proteins Recruited to Vir-MLOs
2.4. Relationships between Disorder Content and Role in LLPS
2.5. Structural Organization and Biological Functions of the Eukaryotic Proteins Recruited to Vir-MLOs
2.5.1. Human hnRNPs: FUS, TDP-43, and hnRNPA2
2.5.2. MAVS/IPS1 and MDA5/IFIH1
2.5.3. Ras-GAP SH3 Domain-Binding Proteins 1 and 2 (G3BP1 and G3BP2)
2.5.4. p53
2.5.5. p65
2.5.6. Fibrillarin-2 and G3BP-like SG Nucleator from N. benthamiana
2.5.7. p38MAPKα
2.5.8. FAK1
2.5.9. TIAR1
2.5.10. HSP70-1
2.5.11. RAB11
2.5.12. PP1
2.5.13. CAD
2.5.14. OGT
3. Discussion
4. Materials and Methods
4.1. Data Set Generation
4.2. Predictions of LLPS Propensity
4.3. Disorder Predictions
4.4. Generation of Functional Disorder Profiles
4.5. Analysis of Protein Interactivity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Flory, P.J. Fundamental principles of condensation polymerization. Chem. Rev. 1946, 39, 137–197. [Google Scholar] [CrossRef] [PubMed]
- Antifeeva, I.A.; Fonin, A.V.; Fefilova, A.S.; Stepanenko, O.V.; Povarova, O.I.; Silonov, S.A.; Kuznetsova, I.M.; Uversky, V.N.; Turoverov, K.K. Liquid-liquid phase separation as an organizing principle of intracellular space: Overview of the evolution of the cell compartmentalization concept. Cell. Mol. Life Sci. 2022, 79, 251. [Google Scholar] [CrossRef] [PubMed]
- Nesterov, S.V.; Ilyinsky, N.S.; Uversky, V.N. Liquid-liquid phase separation as a common organizing principle of intracellular space and biomembranes providing dynamic adaptive responses. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119102. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Hyman, A.A. Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 196–213. [Google Scholar] [CrossRef] [PubMed]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Van Den Bosch, L.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, eaaf4382. [Google Scholar] [CrossRef] [Green Version]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Darling, A.L.; Uversky, V.N. Known types of membrane-less organelles and biomolecular condensates. In Droplets of Life: Membrane-Less Organelles, Biomolecular Condensates, and Biological Liquid-Liquid Phase Separation; Uversky, V.N., Ed.; Elsevier: Amsterdam, The Netherlands, 2023; pp. 271–335. [Google Scholar]
- Li, J.; Zhang, M.; Ma, W.; Yang, B.; Lu, H.; Zhou, F.; Zhang, L. Post-translational modifications in liquid-liquid phase separation: A comprehensive review. Mol. Biomed. 2022, 3, 13. [Google Scholar] [CrossRef]
- Riback, J.A.; Brangwynne, C.P. Can phase separation buffer cellular noise? Science 2020, 367, 364–365. [Google Scholar] [CrossRef]
- Banani, S.F.; Rice, A.M.; Peeples, W.B.; Lin, Y.; Jain, S.; Parker, R.; Rosen, M.K. Compositional Control of Phase-Separated Cellular Bodies. Cell 2016, 166, 651–663. [Google Scholar] [CrossRef]
- Ditlev, J.A.; Case, L.B.; Rosen, M.K. Who’s In and Who’s Out-Compositional Control of Biomolecular Condensates. J. Mol. Biol. 2018, 430, 4666–4684. [Google Scholar] [CrossRef]
- Farahi, N.; Lazar, T.; Wodak, S.J.; Tompa, P.; Pancsa, R. Integration of Data from Liquid-Liquid Phase Separation Databases Highlights Concentration and Dosage Sensitivity of LLPS Drivers. Int. J. Mol. Sci. 2021, 22, 3017. [Google Scholar] [CrossRef]
- Uversky, V.N. Recent Developments in the Field of Intrinsically Disordered Proteins: Intrinsic Disorder-Based Emergence in Cellular Biology in Light of the Physiological and Pathological Liquid-Liquid Phase Transitions. Annu. Rev. Biophys. 2021, 50, 135–156. [Google Scholar] [CrossRef]
- Gomes, E.; Shorter, J. The molecular language of membraneless organelles. J. Biol. Chem. 2019, 294, 7115–7127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, M.; Li, T.; Zhou, S.; Song, J. Arg/Lys-containing IDRs are cryptic binding domains for ATP and nucleic acids that interplay to modulate LLPS. Commun. Biol. 2022, 5, 1315. [Google Scholar] [CrossRef]
- Schuster, B.S.; Reed, E.H.; Parthasarathy, R.; Jahnke, C.N.; Caldwell, R.M.; Bermudez, J.G.; Ramage, H.; Good, M.C.; Hammer, D.A. Controllable protein phase separation and modular recruitment to form responsive membraneless organelles. Nat. Commun. 2018, 9, 2985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badaczewska-Dawid, A.E.; Uversky, V.N.; Potoyan, D.A. BIAPSS: A Comprehensive Physicochemical Analyzer of Proteins Undergoing Liquid-Liquid Phase Separation. Int. J. Mol. Sci. 2022, 23, 6204. [Google Scholar] [CrossRef] [PubMed]
- Garaizar, A.; Sanchez-Burgos, I.; Collepardo-Guevara, R.; Espinosa, J.R. Expansion of Intrinsically Disordered Proteins Increases the Range of Stability of Liquid-Liquid Phase Separation. Molecules 2020, 25, 4705. [Google Scholar] [CrossRef]
- Darling, A.L.; Uversky, V.N. Intrinsic Disorder and Posttranslational Modifications: The Darker Side of the Biological Dark Matter. Front. Genet. 2018, 9, 158. [Google Scholar] [CrossRef]
- Martin, E.W.; Holehouse, A.S.; Peran, I.; Farag, M.; Incicco, J.J.; Bremer, A.; Grace, C.R.; Soranno, A.; Pappu, R.V.; Mittag, T. Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science 2020, 367, 694–699. [Google Scholar] [CrossRef]
- Jo, Y.; Jang, J.; Song, D.; Park, H.; Jung, Y. Determinants for intrinsically disordered protein recruitment into phase-separated protein condensates. Chem. Sci. 2022, 13, 522–530. [Google Scholar] [CrossRef]
- Wang, S.; Dai, T.; Qin, Z.; Pan, T.; Chu, F.; Lou, L.; Zhang, L.; Yang, B.; Huang, H.; Lu, H.; et al. Targeting liquid-liquid phase separation of SARS-CoV-2 nucleocapsid protein promotes innate antiviral immunity by elevating MAVS activity. Nat. Cell Biol. 2021, 23, 718–732. [Google Scholar] [CrossRef]
- Risso-Ballester, J.; Galloux, M.; Cao, J.; Le Goffic, R.; Hontonnou, F.; Jobart-Malfait, A.; Desquesnes, A.; Sake, S.M.; Haid, S.; Du, M.; et al. A condensate-hardening drug blocks RSV replication in vivo. Nature 2021, 595, 596–599. [Google Scholar] [CrossRef]
- Wei, W.; Bai, L.; Yan, B.; Meng, W.; Wang, H.; Zhai, J.; Si, F.; Zheng, C. When liquid-liquid phase separation meets viral infections. Front. Immunol. 2022, 13, 985622. [Google Scholar] [CrossRef]
- Williams, A.W.; Lowden, M.M. The etiology and diagnosis of hydrophobia. J. Infect. Dis. 1906, 3, 452–483. [Google Scholar] [CrossRef]
- Negri, A. Contributo allo studio dell’eziologia della rabbia. Boll. Della Soc. Med. Chir. Di Pavia 1904, 2, 88–115. [Google Scholar]
- Lahaye, X.; Vidy, A.; Pomier, C.; Obiang, L.; Harper, F.; Gaudin, Y.; Blondel, D. Functional characterization of Negri bodies (NBs) in rabies virus-infected cells: Evidence that NBs are sites of viral transcription and replication. J. Virol. 2009, 83, 7948–7958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristensson, K.; Dastur, D.K.; Manghani, D.K.; Tsiang, H.; Bentivoglio, M. Rabies: Interactions between neurons and viruses. A review of the history of Negri inclusion bodies. Neuropathol. Appl. Neurobiol. 1996, 22, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, J.; Lagaudriere-Gesbert, C.; Scrima, N.; Blondel, D.; Gaudin, Y. Structure and Function of Negri Bodies. Adv. Exp. Med. Biol. 2019, 1215, 111–127. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, J.; Le Bars, R.; Lama, Z.; Scrima, N.; Lagaudriere-Gesbert, C.; Gaudin, Y.; Blondel, D. Negri bodies are viral factories with properties of liquid organelles. Nat. Commun. 2017, 8, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevers, Q.; Albertini, A.A.; Lagaudriere-Gesbert, C.; Gaudin, Y. Negri bodies and other virus membrane-less replication compartments. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118831. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Shofa, M.; Ode, H.; Yumiya, M.; Hirano, J.; Okamoto, T.; Yoshimura, S.H. How Do Flaviviruses Hijack Host Cell Functions by Phase Separation? Viruses 2021, 13, 1479. [Google Scholar] [CrossRef] [PubMed]
- Etibor, T.A.; Yamauchi, Y.; Amorim, M.J. Liquid Biomolecular Condensates and Viral Lifecycles: Review and Perspectives. Viruses 2021, 13, 366. [Google Scholar] [CrossRef] [PubMed]
- Brocca, S.; Grandori, R.; Longhi, S.; Uversky, V. Liquid-Liquid Phase Separation by Intrinsically Disordered Protein Regions of Viruses: Roles in Viral Life Cycle and Control of Virus-Host Interactions. Int. J. Mol. Sci. 2020, 21, 9045. [Google Scholar] [CrossRef]
- Caragliano, E.; Brune, W.; Bosse, J.B. Herpesvirus Replication Compartments: Dynamic Biomolecular Condensates? Viruses 2022, 14, 960. [Google Scholar] [CrossRef]
- Papa, G.; Borodavka, A.; Desselberger, U. Viroplasms: Assembly and Functions of Rotavirus Replication Factories. Viruses 2021, 13, 1349. [Google Scholar] [CrossRef]
- Dolnik, O.; Gerresheim, G.K.; Biedenkopf, N. New Perspectives on the Biogenesis of Viral Inclusion Bodies in Negative-Sense RNA Virus Infections. Cells 2021, 10, 1460. [Google Scholar] [CrossRef]
- Scoca, V.; Di Nunzio, F. Membraneless organelles restructured and built by pandemic viruses: HIV-1 and SARS-CoV-2. J. Mol. Cell Biol. 2021, 13, 259–268. [Google Scholar] [CrossRef]
- Su, J.M.; Wilson, M.Z.; Samuel, C.E.; Ma, D. Formation and Function of Liquid-Like Viral Factories in Negative-Sense Single-Stranded RNA Virus Infections. Viruses 2021, 13, 126. [Google Scholar] [CrossRef]
- Hidalgo, P.; Gonzalez, R.A. Formation of adenovirus DNA replication compartments. FEBS Lett. 2019, 593, 3518–3530. [Google Scholar] [CrossRef] [Green Version]
- Guseva, S.; Milles, S.; Jensen, M.R.; Schoehn, G.; Ruigrok, R.W.; Blackledge, M. Structure, dynamics and phase separation of measles virus RNA replication machinery. Curr. Opin. Virol. 2020, 41, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Pesce, G.; Brocca, S.; Grandori, R.; Longhi, S.; Uversky, V.N. Droplets of life: Role of phase separation in virus replication and compartmentalization. In Droplets of Life: Membrane-Less Organelles, Biomolecular Condensates, and Biological Liquid-Liquid Phase Separation; Academic Press, Elsevier: Amsterdam, The Netherlands, 2023; pp. 567–615. [Google Scholar]
- Lopez, N.; Camporeale, G.; Salgueiro, M.; Borkosky, S.S.; Visentin, A.; Peralta-Martinez, R.; Loureiro, M.E.; de Prat-Gay, G. Deconstructing virus condensation. PLoS Pathog. 2021, 17, e1009926. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Holehouse, A.S.; Leung, D.W.; Amarasinghe, G.K.; Dutch, R.E. Liquid Phase Partitioning in Virus Replication: Observations and Opportunities. Annu. Rev. Virol. 2022, 9, 285–306. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ernst, C.; Kolonko-Adamska, M.; Greb-Markiewicz, B.; Man, J.; Parissi, V.; Ng, B.W. Phase separation in viral infections. Trends Microbiol. 2022, 30, 1217–1231. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, B.S.; Maliga, Z.; Stein, D.A.; Hyman, A.A.; Whelan, S.P.J. Phase Transitions Drive the Formation of Vesicular Stomatitis Virus Replication Compartments. mBio 2018, 9, e02290-17. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Su, J.M.; Samuel, C.E.; Ma, D. Measles Virus Forms Inclusion Bodies with Properties of Liquid Organelles. J. Virol. 2019, 93, e00948-19. [Google Scholar] [CrossRef]
- Guseva, S.; Milles, S.; Jensen, M.R.; Salvi, N.; Kleman, J.P.; Maurin, D.; Ruigrok, R.W.H.; Blackledge, M. Measles virus nucleo- and phosphoproteins form liquid-like phase-separated compartments that promote nucleocapsid assembly. Sci. Adv. 2020, 6, eaaz7095. [Google Scholar] [CrossRef] [Green Version]
- Galloux, M.; Risso-Ballester, J.; Richard, C.A.; Fix, J.; Rameix-Welti, M.A.; Eleouet, J.F. Minimal Elements Required for the Formation of Respiratory Syncytial Virus Cytoplasmic Inclusion Bodies In Vivo and In Vitro. mBio 2020, 11, e01202-20. [Google Scholar] [CrossRef]
- Boggs, K.B.; Edmonds, K.; Cifuentes-Munoz, N.; El Najjar, F.; Ossandón, C.; Roe, M.; Wu, C.; Moncman, C.L.; Creamer, T.P.; Amarasinghe, G.K.; et al. Human Metapneumovirus Phosphoprotein Independently Drives Phase Separation and Recruits Nucleoprotein to Liquid-Like Bodies. mBio 2022, 13, e0109922. [Google Scholar] [CrossRef]
- Hirai, Y.; Tomonaga, K.; Horie, M. Borna disease virus phosphoprotein triggers the organization of viral inclusion bodies by liquid-liquid phase separation. Int. J. Biol. Macromol. 2021, 192, 55–63. [Google Scholar] [CrossRef]
- Ambroggio, E.E.; Costa Navarro, G.S.; Perez Socas, L.B.; Bagatolli, L.A.; Gamarnik, A.V. Dengue and Zika virus capsid proteins bind to membranes and self-assemble into liquid droplets with nucleic acids. J. Biol. Chem. 2021, 297, 101059. [Google Scholar] [CrossRef] [PubMed]
- Savastano, A.; Ibanez de Opakua, A.; Rankovic, M.; Zweckstetter, M. Nucleocapsid protein of SARS-CoV-2 phase separates into RNA-rich polymerase-containing condensates. Nat. Commun. 2020, 11, 6041. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Cui, Y.; Han, X.; Hu, W.; Sun, M.; Zhang, Y.; Wang, P.H.; Song, G.; Chen, W.; Lou, J. Liquid-liquid phase separation by SARS-CoV-2 nucleocapsid protein and RNA. Cell Res. 2020, 30, 1143–1145. [Google Scholar] [CrossRef] [PubMed]
- Geiger, F.; Acker, J.; Papa, G.; Wang, X.; Arter, W.E.; Saar, K.L.; Erkamp, N.A.; Qi, R.; Bravo, J.P.; Strauss, S.; et al. Liquid-liquid phase separation underpins the formation of replication factories in rotaviruses. EMBO J. 2021, 40, e107711. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.; Campbell, E.A.; Wells, J.; Simpson, J.; Nazki, S.; Hawes, P.C.; Broadbent, A.J. Birnaviridae Virus Factories Show Features of Liquid-Liquid Phase Separation and Are Distinct from Paracrystalline Arrays of Virions Observed by Electron Microscopy. J. Virol. 2022, 96, e0202421. [Google Scholar] [CrossRef]
- Alenquer, M.; Vale-Costa, S.; Etibor, T.A.; Ferreira, F.; Sousa, A.L.; Amorim, M.J. Influenza A virus ribonucleoproteins form liquid organelles at endoplasmic reticulum exit sites. Nat. Commun. 2019, 10, 1629. [Google Scholar] [CrossRef] [Green Version]
- Lyonnais, S.; Sadiq, S.K.; Lorca-Oro, C.; Dufau, L.; Nieto-Marquez, S.; Escriba, T.; Gabrielli, N.; Tan, X.; Ouizougun-Oubari, M.; Okoronkwo, J.; et al. The HIV-1 Nucleocapsid Regulates Its Own Condensation by Phase-Separated Activity-Enhancing Sequestration of the Viral Protease during Maturation. Viruses 2021, 13, 2312. [Google Scholar] [CrossRef]
- Monette, A.; Niu, M.; Nijhoff Asser, M.; Gorelick, R.J.; Mouland, A.J. Scaffolding viral protein NC nucleates phase separation of the HIV-1 biomolecular condensate. Cell Rep. 2022, 40, 111251. [Google Scholar] [CrossRef]
- Seyffert, M.; Georgi, F.; Tobler, K.; Bourqui, L.; Anfossi, M.; Michaelsen, K.; Vogt, B.; Greber, U.F.; Fraefel, C. The HSV-1 Transcription Factor ICP4 Confers Liquid-Like Properties to Viral Replication Compartments. Int. J. Mol. Sci. 2021, 22, 4447. [Google Scholar] [CrossRef]
- Zhou, S.; Fu, Z.; Zhang, Z.; Jia, X.; Xu, G.; Sun, L.; Sun, F.; Gao, P.; Xu, P.; Deng, H. Liquid-liquid phase separation mediates the formation of herpesvirus assembly compartments. J. Cell Biol. 2023, 222, e202201088. [Google Scholar] [CrossRef]
- Cubuk, J.; Alston, J.J.; Incicco, J.J.; Singh, S.; Stuchell-Brereton, M.D.; Ward, M.D.; Zimmerman, M.I.; Vithani, N.; Griffith, D.; Wagoner, J.A.; et al. The SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA. Nat. Commun. 2021, 12, 1936. [Google Scholar] [CrossRef] [PubMed]
- Gaete-Argel, A.; Marquez, C.L.; Barriga, G.P.; Soto-Rifo, R.; Valiente-Echeverria, F. Strategies for Success. Viral Infections and Membraneless Organelles. Front. Cell Infect. Microbiol. 2019, 9, 336. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ma, L.; Cai, S.; Zhuang, Z.; Zhao, Z.; Jin, S.; Xie, W.; Zhou, L.; Zhang, L.; Zhao, J.; et al. RNA-induced liquid phase separation of SARS-CoV-2 nucleocapsid protein facilitates NF-kappaB hyper-activation and inflammation. Signal Transduct. Target 2021, 6, 167. [Google Scholar] [CrossRef] [PubMed]
- Perdikari, T.M.; Murthy, A.C.; Ryan, V.H.; Watters, S.; Naik, M.T.; Fawzi, N.L. SARS-CoV-2 nucleocapsid protein phase-separates with RNA and with human hnRNPs. EMBO J. 2020, 39, e106478. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.R.; Asfaha, J.B.; Ghent, C.M.; Howard, C.J.; Hartooni, N.; Safari, M.; Frankel, A.D.; Morgan, D.O. Phosphoregulation of Phase Separation by the SARS-CoV-2 N Protein Suggests a Biophysical Basis for its Dual Functions. Mol. Cell 2020, 80, 1092–1103.e1094. [Google Scholar] [CrossRef] [PubMed]
- Lifland, A.W.; Jung, J.; Alonas, E.; Zurla, C.; Crowe, J.E., Jr.; Santangelo, P.J. Human respiratory syncytial virus nucleoprotein and inclusion bodies antagonize the innate immune response mediated by MDA5 and MAVS. J. Virol. 2012, 86, 8245–8258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borkosky, S.S.; Fassolari, M.; Campos-León, K.; Rossi, A.H.; Salgueiro, M.; Pascuale, C.A.; Martínez, R.P.; Gaston, K.; de Prat Gay, G. Biomolecular Condensation of the Human Papillomavirus E2 Master Regulator with p53: Implications in Viral Replication. J. Mol. Biol. 2022, 167889. [Google Scholar] [CrossRef]
- Nabeel-Shah, S.; Lee, H.; Ahmed, N.; Burke, G.L.; Farhangmehr, S.; Ashraf, K.; Pu, S.; Braunschweig, U.; Zhong, G.; Wei, H.; et al. SARS-CoV-2 nucleocapsid protein binds host mRNAs and attenuates stress granules to impair host stress response. iScience 2022, 25, 103562. [Google Scholar] [CrossRef]
- Jobe, F.; Simpson, J.; Hawes, P.; Guzman, E.; Bailey, D. Respiratory Syncytial Virus Sequesters NF-kappaB Subunit p65 to Cytoplasmic Inclusion Bodies To Inhibit Innate Immune Signaling. J. Virol. 2020, 94, e01380-20. [Google Scholar] [CrossRef]
- Brown, S.L.; Garrison, D.J.; May, J.P. Phase separation of a plant virus movement protein and cellular factors support virus-host interactions. PLoS Pathog. 2021, 17, e1009622. [Google Scholar] [CrossRef]
- Fouquet, B.; Nikolic, J.; Larrous, F.; Bourhy, H.; Wirblich, C.; Lagaudriere-Gesbert, C.; Blondel, D. Focal adhesion kinase is involved in rabies virus infection through its interaction with viral phosphoprotein P. J. Virol. 2015, 89, 1640–1651. [Google Scholar] [CrossRef] [PubMed]
- Le Sage, V.; Cinti, A.; McCarthy, S.; Amorim, R.; Rao, S.; Daino, G.L.; Tramontano, E.; Branch, D.R.; Mouland, A.J. Ebola virus VP35 blocks stress granule assembly. Virology 2017, 502, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, C.A.; Rincheval, V.; Lassoued, S.; Fix, J.; Cardone, C.; Esneau, C.; Nekhai, S.; Galloux, M.; Rameix-Welti, M.A.; Sizun, C.; et al. RSV hijacks cellular protein phosphatase 1 to regulate M2-1 phosphorylation and viral transcription. PLoS Pathog. 2018, 14, e1006920. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.; Wendt, L.; Bodmer, B.S.; Mettenleiter, T.C.; Hoenen, T. The Cellular Protein CAD is Recruited into Ebola Virus Inclusion Bodies by the Nucleoprotein NP to Facilitate Genome Replication and Transcription. Cells 2020, 9, 1126. [Google Scholar] [CrossRef]
- Fricke, J.; Koo, L.Y.; Brown, C.R.; Collins, P.L. p38 and OGT sequestration into viral inclusion bodies in cells infected with human respiratory syncytial virus suppresses MK2 activities and stress granule assembly. J. Virol. 2013, 87, 1333–1347. [Google Scholar] [CrossRef] [Green Version]
- Obradovic, Z.; Peng, K.; Vucetic, S.; Radivojac, P.; Dunker, A.K. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins: Struct. Funct. Bioinform. 2005, 61, 176–182. [Google Scholar] [CrossRef]
- Necci, M.; Piovesan, D.; Predictors, C.; DisProt, C.; Tosatto, S.C.E. Critical assessment of protein intrinsic disorder prediction. Nat. Methods 2021, 18, 472–481. [Google Scholar] [CrossRef]
- Hatos, A.; Hajdu-Soltesz, B.; Monzon, A.M.; Palopoli, N.; Alvarez, L.; Aykac-Fas, B.; Bassot, C.; Benitez, G.I.; Bevilacqua, M.; Chasapi, A.; et al. DisProt: Intrinsic protein disorder annotation in 2020. Nucleic Acids Res. 2020, 48, D269–D276. [Google Scholar] [CrossRef] [Green Version]
- Quaglia, F.; Meszaros, B.; Salladini, E.; Hatos, A.; Pancsa, R.; Chemes, L.B.; Pajkos, M.; Lazar, T.; Pena-Diaz, S.; Santos, J.; et al. DisProt in 2022: Improved quality and accessibility of protein intrinsic disorder annotation. Nucleic Acids Res 2022, 50, D480–D487. [Google Scholar] [CrossRef]
- Sickmeier, M.; Hamilton, J.A.; LeGall, T.; Vacic, V.; Cortese, M.S.; Tantos, A.; Szabo, B.; Tompa, P.; Chen, J.; Uversky, V.N.; et al. DisProt: The Database of Disordered Proteins. Nucleic Acids Res. 2007, 35, D786–D793. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, K.; Mooney, S.M.; Parekh, N.; Getzenberg, R.H.; Kulkarni, P. A majority of the cancer/testis antigens are intrinsically disordered proteins. J. Cell Biochem. 2011, 112, 3256–3267. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Sullivan, W.J., Jr.; Radivojac, P.; Dunker, A.K.; Uversky, V.N. Intrinsic disorder in pathogenic and non-pathogenic microbes: Discovering and analyzing the unfoldomes of early-branching eukaryotes. Mol. Biosyst. 2008, 4, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Ruff, K.M.; Pappu, R.V. AlphaFold and Implications for Intrinsically Disordered Proteins. J. Mol. Biol. 2021, 433, 167208. [Google Scholar] [CrossRef] [PubMed]
- Piovesan, D.; Monzon, A.M.; Tosatto, S.C.E. Intrinsic protein disorder and conditional folding in AlphaFoldDB. Protein Sci. 2022, 31, e4466. [Google Scholar] [CrossRef] [PubMed]
- Lieutaud, P.; Canard, B.; Longhi, S. MeDor: A metaserver for predicting protein disorder. BMC Genom. 2008, 9 (Suppl. 2), S25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinform. 2006, 7, 208. [Google Scholar] [CrossRef] [Green Version]
- Peng, K.; Vucetic, S.; Radivojac, P.; Brown, C.J.; Dunker, A.K.; Obradovic, Z. Optimizing long intrinsic disorder predictors with protein evolutionary information. J. Bioinform. Comput. Biol. 2005, 3, 35–60. [Google Scholar] [CrossRef]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dosztányi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [Green Version]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J. Mol. Biol. 2005, 347, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Oates, M.E.; Romero, P.; Ishida, T.; Ghalwash, M.; Mizianty, M.J.; Xue, B.; Dosztanyi, Z.; Uversky, V.N.; Obradovic, Z.; Kurgan, L.; et al. D(2)P(2): Database of disordered protein predictions. Nucleic Acids Res. 2013, 41, D508–D516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Oldfield, C.J.; Meng, J.; Romero, P.; Uversky, V.N.; Dunker, A.K. Mining alpha-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry 2007, 46, 13468–13477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vacic, V.; Oldfield, C.J.; Mohan, A.; Radivojac, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Characterization of molecular recognition features, MoRFs, and their binding partners. J. Proteome Res. 2007, 6, 2351–2366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meszaros, B.; Erdos, G.; Dosztanyi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orti, F.; Navarro, A.M.; Rabinovich, A.; Wodak, S.J.; Marino-Buslje, C. Insight into membraneless organelles and their associated proteins: Drivers, Clients and Regulators. Comput. Struct. Biotechnol. J. 2021, 19, 3964–3977. [Google Scholar] [CrossRef]
- Mészáros, B.; Erdős, G.; Szabó, B.; Schád, É.; Tantos, Á.; Abukhairan, R.; Horváth, T.; Murvai, N.; Kovács, O.P.; Kovács, M.; et al. PhaSePro: The database of proteins driving liquid-liquid phase separation. Nucleic Acids Res. 2020, 48, D360–D367. [Google Scholar] [CrossRef] [PubMed]
- You, K.; Huang, Q.; Yu, C.; Shen, B.; Sevilla, C.; Shi, M.; Hermjakob, H.; Chen, Y.; Li, T. PhaSepDB: A database of liquid-liquid phase separation related proteins. Nucleic Acids Res. 2020, 48, D354–D359. [Google Scholar] [CrossRef] [PubMed]
- Ning, W.; Guo, Y.; Lin, S.; Mei, B.; Wu, Y.; Jiang, P.; Tan, X.; Zhang, W.; Chen, G.; Peng, D.; et al. DrLLPS: A data resource of liquid-liquid phase separation in eukaryotes. Nucleic Acids Res. 2020, 48, D288–D295. [Google Scholar] [CrossRef] [PubMed]
- Geuens, T.; Bouhy, D.; Timmerman, V. The hnRNP family: Insights into their role in health and disease. Hum. Genet. 2016, 135, 851–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.C.; Liang, T.Y.; Mazur, C.; et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.Y.; Riley, T.R.; Coady, T.; Bussemaker, H.J.; Manley, J.L. TLS/FUS (translocated in liposarcoma/fused in sarcoma) regulates target gene transcription via single-stranded DNA response elements. Proc. Natl. Acad. Sci. USA 2012, 109, 6030–6035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrocola, A.S.; Kim, S.H.; Trinh, A.T.; Rodenkirch, L.A.; Tibbetts, R.S. The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J. Biol. Chem. 2013, 288, 24731–24741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, H.; Gao, K.; Jankovic, J. The role of FUS gene variants in neurodegenerative diseases. Nat. Rev. Neurol. 2014, 10, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Dormann, D.; Rodde, R.; Edbauer, D.; Bentmann, E.; Fischer, I.; Hruscha, A.; Than, M.E.; Mackenzie, I.R.; Capell, A.; Schmid, B.; et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010, 29, 2841–2857. [Google Scholar] [CrossRef] [Green Version]
- Coady, T.H.; Manley, J.L. ALS mutations in TLS/FUS disrupt target gene expression. Genes Dev. 2015, 29, 1696–1706. [Google Scholar] [CrossRef] [Green Version]
- Qiu, H.; Lee, S.; Shang, Y.; Wang, W.Y.; Au, K.F.; Kamiya, S.; Barmada, S.J.; Finkbeiner, S.; Lui, H.; Carlton, C.E.; et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Investig. 2014, 124, 981–999. [Google Scholar] [CrossRef]
- Shang, Y.; Huang, E.J. Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res. 2016, 1647, 65–78. [Google Scholar] [CrossRef] [Green Version]
- Daigle, J.G.; Lanson, N.A., Jr.; Smith, R.B.; Casci, I.; Maltare, A.; Monaghan, J.; Nichols, C.D.; Kryndushkin, D.; Shewmaker, F.; Pandey, U.B. RNA-binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS-linked mutations. Hum. Mol. Genet. 2013, 22, 1193–1205. [Google Scholar] [CrossRef] [Green Version]
- Han, T.W.; Kato, M.; Xie, S.; Wu, L.C.; Mirzaei, H.; Pei, J.; Chen, M.; Xie, Y.; Allen, J.; Xiao, G.; et al. Cell-free formation of RNA granules: Bound RNAs identify features and components of cellular assemblies. Cell 2012, 149, 768–779. [Google Scholar] [CrossRef] [Green Version]
- Freibaum, B.D.; Chitta, R.K.; High, A.A.; Taylor, J.P. Global Analysis of TDP-43 Interacting Proteins Reveals Strong Association with RNA Splicing and Translation Machinery. J. Proteome Res. 2010, 9, 1104–1120. [Google Scholar] [CrossRef]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; Konig, J.; Hortobagyi, T.; Nishimura, A.L.; Zupunski, V.; et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buratti, E.; Brindisi, A.; Giombi, M.; Tisminetzky, S.; Ayala, Y.M.; Baralle, F.E. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: An important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J. Biol. Chem. 2005, 280, 37572–37584. [Google Scholar] [CrossRef] [Green Version]
- Baralle, M.; Buratti, E.; Baralle, F.E. The role of TDP-43 in the pathogenesis of ALS and FTLD. Biochem. Soc. Trans. 2013, 41, 1536–1540. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, H.; Sawada, J.; Hideyama, T.; Yamashita, T.; Katayama, T.; Hasebe, N.; Kimura, T.; Yahara, O.; Kwak, S. TDP-43 pathology in sporadic ALS occurs in motor neurons lacking the RNA editing enzyme ADAR2. Acta Neuropathol. 2010, 120, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Gal, J.; Kuang, L.; Barnett, K.R.; Zhu, B.Z.; Shissler, S.C.; Korotkov, K.V.; Hayward, L.J.; Kasarskis, E.J.; Zhu, H. ALS mutant SOD1 interacts with G3BP1 and affects stress granule dynamics. Acta Neuropathol. 2016, 132, 563–576. [Google Scholar] [CrossRef] [Green Version]
- Liu-Yesucevitz, L.; Bilgutay, A.; Zhang, Y.J.; Vanderwyde, T.; Citro, A.; Mehta, T.; Zaarur, N.; McKee, A.; Bowser, R.; Sherman, M.; et al. Tar DNA Binding Protein-43 (TDP-43) Associates with Stress Granules: Analysis of Cultured Cells and Pathological Brain Tissue. PLoS ONE 2010, 5, e13250. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Chakrabartty, A. Phase to Phase with TDP-43. Biochemistry 2017, 56, 809–823. [Google Scholar] [CrossRef]
- Santamaria, N.; Alhothali, M.; Harreguy Alfonso, M.; Breydo, L.; Uversky, V.N. Intrinsic disorder in proteins involved in amyotrophic lateral sclerosis. Cell. Mol. Life Sci. 2017, 74, 1297–1318. [Google Scholar] [CrossRef]
- He, Y.; Smith, R. Nuclear functions of heterogeneous nuclear ribonucleoproteins A/B. Cell. Mol. Life Sci. 2009, 66, 1239–1256. [Google Scholar] [CrossRef]
- Shorter, J.; Taylor, J.P. Disease mutations in the prion-like domains of hnRNPA1 and hnRNPA2/B1 introduce potent steric zippers that drive excess RNP granule assembly. Rare Dis. 2013, 1, e25200. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, N.C.; Wang, Y.D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Tong, X.; Ye, X. Ndfip1 negatively regulates RIG-I-dependent immune signaling by enhancing E3 ligase Smurf1-mediated MAVS degradation. J. Immunol. 2012, 189, 5304–5313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshiumi, H.; Sakai, K.; Matsumoto, M.; Seya, T. DEAD/H BOX 3 (DDX3) helicase binds the RIG-I adaptor IPS-1 to up-regulate IFN-beta-inducing potential. Eur. J. Immunol. 2010, 40, 940–948. [Google Scholar] [CrossRef]
- Dixit, E.; Boulant, S.; Zhang, Y.; Lee, A.S.; Odendall, C.; Shum, B.; Hacohen, N.; Chen, Z.J.; Whelan, S.P.; Fransen, M.; et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 2010, 141, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Oshiumi, H.; Ikeda, M.; Matsumoto, M.; Watanabe, A.; Takeuchi, O.; Akira, S.; Kato, N.; Shimotohno, K.; Seya, T. Hepatitis C virus core protein abrogates the DDX3 function that enhances IPS-1-mediated IFN-beta induction. PLoS ONE 2010, 5, e14258. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.H.; Macmillan, J.B.; Chen, Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [Green Version]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273, Table of Contents. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, H.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Uematsu, S.; Matsui, K.; Tsujimura, T.; Takeda, K.; Fujita, T.; Takeuchi, O.; et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity 2005, 23, 19–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Lee, J.H.; Parker, Z.M.; Acharya, D.; Chiang, J.J.; van Gent, M.; Riedl, W.; Davis-Gardner, M.E.; Wies, E.; Chiang, C.; et al. ISG15-dependent activation of the sensor MDA5 is antagonized by the SARS-CoV-2 papain-like protease to evade host innate immunity. Nat. Microbiol. 2021, 6, 467–478. [Google Scholar] [CrossRef]
- Wu, X.M.; Zhang, J.; Li, P.W.; Hu, Y.W.; Cao, L.; Ouyang, S.; Bi, Y.H.; Nie, P.; Chang, M.X. NOD1 Promotes Antiviral Signaling by Binding Viral RNA and Regulating the Interaction of MDA5 and MAVS. J. Immunol. 2020, 204, 2216–2231. [Google Scholar] [CrossRef]
- Zhao, J.; Qin, C.; Liu, Y.; Rao, Y.; Feng, P. Herpes Simplex Virus and Pattern Recognition Receptors: An Arms Race. Front. Immunol. 2020, 11, 613799. [Google Scholar] [CrossRef]
- Rebendenne, A.; Valadao, A.L.C.; Tauziet, M.; Maarifi, G.; Bonaventure, B.; McKellar, J.; Planes, R.; Nisole, S.; Arnaud-Arnould, M.; Moncorge, O.; et al. SARS-CoV-2 triggers an MDA-5-dependent interferon response which is unable to control replication in lung epithelial cells. J. Virol. 2021, 95, e02415-20. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Riva, L.; Pu, Y.; Martin-Sancho, L.; Kanamune, J.; Yamamoto, Y.; Sakai, K.; Gotoh, S.; Miorin, L.; De Jesus, P.D.; et al. MDA5 Governs the Innate Immune Response to SARS-CoV-2 in Lung Epithelial Cells. Cell Rep. 2021, 34, 108628. [Google Scholar] [CrossRef] [PubMed]
- Urcuqui-Inchima, S.; Cabrera, J.; Haenni, A.L. Interplay between dengue virus and Toll-like receptors, RIG-I/MDA5 and microRNAs: Implications for pathogenesis. Antivir. Res. 2017, 147, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Wehbe, M.; Leveque, N.; Bodet, C. Skin innate immune response to flaviviral infection. Eur. Cytokine Netw. 2017, 28, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Sherry, B. Rotavirus and reovirus modulation of the interferon response. J. Interferon Cytokine Res. 2009, 29, 559–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahun, A.S.; Goodfellow, I.G. Interferon responses to norovirus infections: Current and future perspectives. J. Gen. Virol. 2021, 102, 001660. [Google Scholar] [CrossRef]
- Unterholzner, L. The interferon response to intracellular DNA: Why so many receptors? Immunobiology 2013, 218, 1312–1321. [Google Scholar] [CrossRef]
- Nikolic, J.; Civas, A.; Lama, Z.; Lagaudriere-Gesbert, C.; Blondel, D. Rabies Virus Infection Induces the Formation of Stress Granules Closely Connected to the Viral Factories. PLoS Pathog. 2016, 12, e1005942. [Google Scholar] [CrossRef] [Green Version]
- Cifuentes-Munoz, N.; Branttie, J.; Slaughter, K.B.; Dutch, R.E. Human Metapneumovirus Induces Formation of Inclusion Bodies for Efficient Genome Replication and Transcription. J. Virol. 2017, 91, e01282-17. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.; Garcia-Barreno, B.; Vivo, A.; Melero, J.A. Cytoplasmic inclusions of respiratory syncytial virus-infected cells: Formation of inclusion bodies in transfected cells that coexpress the nucleoprotein, the phosphoprotein, and the 22K protein. Virology 1993, 195, 243–247. [Google Scholar] [CrossRef]
- Rincheval, V.; Lelek, M.; Gault, E.; Bouillier, C.; Sitterlin, D.; Blouquit-Laye, S.; Galloux, M.; Zimmer, C.; Eleouet, J.F.; Rameix-Welti, M.A. Functional organization of cytoplasmic inclusion bodies in cells infected by respiratory syncytial virus. Nat. Commun. 2017, 8, 563. [Google Scholar] [CrossRef] [PubMed]
- Parker, F.; Maurier, F.; Delumeau, I.; Duchesne, M.; Faucher, D.; Debussche, L.; Dugue, A.; Schweighoffer, F.; Tocque, B. A Ras-GTPase-activating protein SH3-domain-binding protein. Mol. Cell. Biol. 1996, 16, 2561–2569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.H.; Wang, J.X.; Cai, M.L.; Shao, R.; Liu, H.; Zhao, W.L. The roles and mechanisms of G3BP1 in tumour promotion. J. Drug Target 2019, 27, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Jin, J.; Li, J.; Ye, M.; Jin, X. The roles of G3BP1 in human diseases (review). Gene 2022, 821, 146294. [Google Scholar] [CrossRef] [PubMed]
- Gallouzi, I.E.; Parker, F.; Chebli, K.; Maurier, F.; Labourier, E.; Barlat, I.; Capony, J.P.; Tocque, B.; Tazi, J. A novel phosphorylation-dependent RNase activity of GAP-SH3 binding protein: A potential link between signal transduction and RNA stability. Mol. Cell Biol. 1998, 18, 3956–3965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.S.; Cai, H.; Xue, W.; Wang, M.; Xia, T.; Li, W.J.; Xing, J.Q.; Zhao, M.; Huang, Y.J.; Chen, S.; et al. G3BP1 promotes DNA binding and activation of cGAS. Nat. Immunol. 2019, 20, 18–28. [Google Scholar] [CrossRef]
- Matsuki, H.; Takahashi, M.; Higuchi, M.; Makokha, G.N.; Oie, M.; Fujii, M. Both G3BP1 and G3BP2 contribute to stress granule formation. Genes Cells 2013, 18, 135–146. [Google Scholar] [CrossRef]
- French, J.; Stirling, R.; Walsh, M.; Kennedy, H.D. The expression of Ras-GTPase activating protein SH3 domain-binding proteins, G3BPs, in human breast cancers. Histochem. J. 2002, 34, 223–231. [Google Scholar] [CrossRef]
- Kang, W.; Wang, Y.; Yang, W.; Zhang, J.; Zheng, H.; Li, D. Research Progress on the Structure and Function of G3BP. Front. Immunol. 2021, 12, 718548. [Google Scholar] [CrossRef]
- Brady, C.A.; Jiang, D.; Mello, S.S.; Johnson, T.M.; Jarvis, L.A.; Kozak, M.M.; Kenzelmann Broz, D.; Basak, S.; Park, E.J.; McLaughlin, M.E.; et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 2011, 145, 571–583. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J. P53 and The Immune Response: 40 Years of Exploration-A Plan for the Future. Int. J. Mol. Sci. 2020, 21, 541. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. Structural biology of the tumor suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.D.; Galbraith, M.D.; Andrysik, Z.; Espinosa, J.M. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018, 25, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure-Function Continuum Concept. Int. J. Mol. Sci. 2016, 17, 1874. [Google Scholar] [CrossRef] [Green Version]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, E.A.; Spector, D.H. p53 and RPA are sequestered in viral replication centers in the nuclei of cells infected with human cytomegalovirus. J. Virol. 1998, 72, 2033–2039. [Google Scholar] [CrossRef] [Green Version]
- Gannon, J.V.; Lane, D.P. p53 and DNA polymerase alpha compete for binding to SV40 T antigen. Nature 1987, 329, 456–458. [Google Scholar] [CrossRef]
- König, C.; Roth, J.; Dobbelstein, M. Adenovirus type 5 E4orf3 protein relieves p53 inhibition by E1B-55-kilodalton protein. J. Virol. 1999, 73, 2253–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcock, D.; Lane, D.P. Localization of p53, retinoblastoma and host replication proteins at sites of viral replication in herpes-infected cells. Nature 1991, 349, 429–431. [Google Scholar] [CrossRef]
- Lecoq, L.; Raiola, L.; Chabot, P.R.; Cyr, N.; Arseneault, G.; Legault, P.; Omichinski, J.G. Structural characterization of interactions between transactivation domain 1 of the p65 subunit of NF-kappaB and transcription regulatory factors. Nucleic Acids Res. 2017, 45, 5564–5576. [Google Scholar] [CrossRef]
- Joyce, D.; Albanese, C.; Steer, J.; Fu, M.; Bouzahzah, B.; Pestell, R.G. NF-kappaB and cell-cycle regulation: The cyclin connection. Cytokine Growth Factor Rev. 2001, 12, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Verma, I.M.; Stevenson, J.K.; Schwarz, E.M.; Van Antwerp, D.; Miyamoto, S. Rel/NF-kappa B/I kappa B family: Intimate tales of association and dissociation. Genes Dev. 1995, 9, 2723–2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baeuerle, P.A.; Baltimore, D. NF-kappa B: Ten years after. Cell 1996, 87, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunto, S.T.; Shin, K.K.; Kim, H.G.; Park, S.H.; Oh, J.; Sung, G.H.; Hossain, M.A.; Rho, H.S.; Lee, J.; Kim, J.H.; et al. Phosphatidylinositide 3-Kinase Contributes to the Anti-Inflammatory Effect of Abutilon crispum L. Medik Methanol Extract. Evid. Based Complement. Altern. Med. 2018, 2018, 1935902. [Google Scholar] [CrossRef] [Green Version]
- Yoboua, F.; Martel, A.; Duval, A.; Mukawera, E.; Grandvaux, N. Respiratory syncytial virus-mediated NF-kappa B p65 phosphorylation at serine 536 is dependent on RIG-I, TRAF6, and IKK beta. J. Virol. 2010, 84, 7267–7277. [Google Scholar] [CrossRef] [Green Version]
- Baum, A.; Garcia-Sastre, A. Induction of type I interferon by RNA viruses: Cellular receptors and their substrates. Amino Acids 2010, 38, 1283–1299. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Jamaluddin, M.; Li, K.; Garofalo, R.P.; Casola, A.; Brasier, A.R. Retinoic acid-inducible gene I mediates early antiviral response and Toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J. Virol. 2007, 81, 1401–1411. [Google Scholar] [CrossRef] [Green Version]
- Pollock, N.; Taylor, G.; Jobe, F.; Guzman, E. Modulation of the transcription factor NF-kappaB in antigen-presenting cells by bovine respiratory syncytial virus small hydrophobic protein. J. Gen. Virol. 2017, 98, 1587–1599. [Google Scholar] [CrossRef] [Green Version]
- Taylor, G.; Wyld, S.; Valarcher, J.F.; Guzman, E.; Thom, M.; Widdison, S.; Buchholz, U.J. Recombinant bovine respiratory syncytial virus with deletion of the SH gene induces increased apoptosis and pro-inflammatory cytokines in vitro, and is attenuated and induces protective immunity in calves. J. Gen. Virol. 2014, 95, 1244–1254. [Google Scholar] [CrossRef]
- Liu, J.; Perumal, N.B.; Oldfield, C.J.; Su, E.W.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder in transcription factors. Biochemistry 2006, 45, 6873–6888. [Google Scholar] [CrossRef] [PubMed]
- Ryabov, E.V.; Robinson, D.J.; Taliansky, M. Umbravirus-encoded proteins both stabilize heterologous viral RNA and mediate its systemic movement in some plant species. Virology 2001, 288, 391–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canetta, E.; Kim, S.H.; Kalinina, N.O.; Shaw, J.; Adya, A.K.; Gillespie, T.; Brown, J.W.; Taliansky, M. A plant virus movement protein forms ringlike complexes with the major nucleolar protein, fibrillarin, in vitro. J. Mol. Biol. 2008, 376, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Macfarlane, S.; Kalinina, N.O.; Rakitina, D.V.; Ryabov, E.V.; Gillespie, T.; Haupt, S.; Brown, J.W.; Taliansky, M. Interaction of a plant virus-encoded protein with the major nucleolar protein fibrillarin is required for systemic virus infection. Proc. Natl. Acad. Sci. USA 2007, 104, 11115–11120. [Google Scholar] [CrossRef] [Green Version]
- Frottin, F.; Schueder, F.; Tiwary, S.; Gupta, R.; Korner, R.; Schlichthaerle, T.; Cox, J.; Jungmann, R.; Hartl, F.U.; Hipp, M.S. The nucleolus functions as a phase-separated protein quality control compartment. Science 2019, 365, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Krapp, S.; Greiner, E.; Amin, B.; Sonnewald, U.; Krenz, B. The stress granule component G3BP is a novel interaction partner for the nuclear shuttle proteins of the nanovirus pea necrotic yellow dwarf virus and geminivirus abutilon mosaic virus. Virus Res. 2017, 227, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Soni, S.; Anand, P.; Padwad, Y.S. MAPKAPK2: The master regulator of RNA-binding proteins modulates transcript stability and tumor progression. J. Exp. Clin. Cancer Res. 2019, 38, 121. [Google Scholar] [CrossRef] [Green Version]
- Arthur, J.S.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef]
- Golubovskaya, V.M. Targeting FAK in human cancer: From finding to first clinical trials. Front. Biosci. (Landmark Ed.) 2014, 19, 687–706. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Wang, J.; Liang, Q.; Tong, R.; Huang, J.; Yang, X.; Xu, Y.; Wang, W.; Sun, M.; Shi, J. Recent progress on FAK inhibitors with dual targeting capabilities for cancer treatment. Biomed. Pharm. 2022, 151, 113116. [Google Scholar] [CrossRef]
- Schaller, M.D. Cellular functions of FAK kinases: Insight into molecular mechanisms and novel functions. J. Cell Sci. 2010, 123, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.L.; Gupta, M.; Li, W.; Miller, I.; Anderson, P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J. Cell Biol. 1999, 147, 1431–1442. [Google Scholar] [CrossRef]
- Nelson, E.V.; Schmidt, K.M.; Deflube, L.R.; Doganay, S.; Banadyga, L.; Olejnik, J.; Hume, A.J.; Ryabchikova, E.; Ebihara, H.; Kedersha, N.; et al. Ebola Virus Does Not Induce Stress Granule Formation during Infection and Sequesters Stress Granule Proteins within Viral Inclusions. J. Virol. 2016, 90, 7268–7284. [Google Scholar] [CrossRef] [Green Version]
- Reineke, L.C.; Lloyd, R.E. Diversion of stress granules and P-bodies during viral infection. Virology 2013, 436, 255–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dember, L.M.; Kim, N.D.; Liu, K.Q.; Anderson, P. Individual RNA recognition motifs of TIA-1 and TIAR have different RNA binding specificities. J. Biol. Chem. 1996, 271, 2783–2788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartl, F.U.; Hayer-Hartl, M. Molecular chaperones in the cytosol: From nascent chain to folded protein. Science 2002, 295, 1852–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, M.P. Gymnastics of molecular chaperones. Mol. Cell 2010, 39, 321–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005, 62, 670–684. [Google Scholar] [CrossRef] [Green Version]
- Nagy, P.D.; Wang, R.Y.; Pogany, J.; Hafren, A.; Makinen, K. Emerging picture of host chaperone and cyclophilin roles in RNA virus replication. Virology 2011, 411, 374–382. [Google Scholar] [CrossRef] [Green Version]
- Mayer, M.P. Recruitment of Hsp70 chaperones: A crucial part of viral survival strategies. Rev. Physiol. Biochem. Pharm. 2005, 153, 1–46. [Google Scholar] [CrossRef]
- Zhang, X.; Bourhis, J.M.; Longhi, S.; Carsillo, T.; Buccellato, M.; Morin, B.; Canard, B.; Oglesbee, M. Hsp72 recognizes a P binding motif in the measles virus N protein C-terminus. Virology 2005, 337, 162–174. [Google Scholar] [CrossRef]
- Zhang, X.; Glendening, C.; Linke, H.; Parks, C.L.; Brooks, C.; Udem, S.A.; Oglesbee, M. Identification and characterization of a regulatory domain on the carboxyl terminus of the measles virus nucleocapsid protein. J. Virol. 2002, 76, 8737–8746. [Google Scholar] [CrossRef] [Green Version]
- Phillips, B.; Abravaya, K.; Morimoto, R.I. Analysis of the specificity and mechanism of transcriptional activation of the human hsp70 gene during infection by DNA viruses. J. Virol. 1991, 65, 5680–5692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Zhang, J.; Tong, X.; Liu, W.; Ye, X. Heat shock protein 70 inhibits the activity of Influenza A virus ribonucleoprotein and blocks the replication of virus in vitro and in vivo. PLoS ONE 2011, 6, e16546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, R.; Zhang, Z.; Mei, X.; Gong, W.; Wei, L. Protective effect of a RSV subunit vaccine candidate G1F/M2 was enhanced by a HSP70-Like protein in mice. Biochem. Biophys. Res. Commun. 2008, 377, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Broquet, A.H.; Lenoir, C.; Gardet, A.; Sapin, C.; Chwetzoff, S.; Jouniaux, A.M.; Lopez, S.; Trugnan, G.; Bachelet, M.; Thomas, G. Hsp70 negatively controls rotavirus protein bioavailability in caco-2 cells infected by the rotavirus RF strain. J. Virol. 2007, 81, 1297–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirayama, E.; Atagi, H.; Hiraki, A.; Kim, J. Heat shock protein 70 is related to thermal inhibition of nuclear export of the influenza virus ribonucleoprotein complex. J. Virol. 2004, 78, 1263–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Marco, A.; Santoro, M.G. Antiviral effect of short hyperthermic treatment at specific stages of vesicular stomatitis virus replication cycle. J. Gen. Virol. 1993, 74 Pt 8, 1685–1690. [Google Scholar] [CrossRef]
- Weeks, S.A.; Shield, W.P.; Sahi, C.; Craig, E.A.; Rospert, S.; Miller, D.J. A targeted analysis of cellular chaperones reveals contrasting roles for heat shock protein 70 in flock house virus RNA replication. J. Virol. 2010, 84, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Lahaye, X.; Vidy, A.; Fouquet, B.; Blondel, D. Hsp70 protein positively regulates rabies virus infection. J. Virol. 2012, 86, 4743–4751. [Google Scholar] [CrossRef] [Green Version]
- Oglesbee, M.; Ringler, S.; Krakowka, S. Interaction of canine distemper virus nucleocapsid variants with 70K heat-shock proteins. J. Gen. Virol. 1990, 71 Pt 7, 1585–1590. [Google Scholar] [CrossRef]
- Diekmann, Y.; Seixas, E.; Gouw, M.; Tavares-Cadete, F.; Seabra, M.C.; Pereira-Leal, J.B. Thousands of rab GTPases for the cell biologist. PLoS Comput. Biol. 2011, 7, e1002217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amorim, M.J. A Comprehensive Review on the Interaction Between the Host GTPase Rab11 and Influenza A Virus. Front. Cell Dev. Biol. 2018, 6, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Castro Martin, I.F.; Fournier, G.; Sachse, M.; Pizarro-Cerda, J.; Risco, C.; Naffakh, N. Influenza virus genome reaches the plasma membrane via a modified endoplasmic reticulum and Rab11-dependent vesicles. Nat. Commun. 2017, 8, 1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceulemans, H.; Bollen, M. Functional diversity of protein phosphatase-1, a cellular economizer and reset button. Physiol. Rev. 2004, 84, 1–39. [Google Scholar] [CrossRef] [Green Version]
- Bollen, M.; Peti, W.; Ragusa, M.J.; Beullens, M. The extended PP1 toolkit: Designed to create specificity. Trends Biochem. Sci. 2010, 35, 450–458. [Google Scholar] [CrossRef] [Green Version]
- Choy, M.S.; Page, R.; Peti, W. Regulation of protein phosphatase 1 by intrinsically disordered proteins. Biochem. Soc. Trans. 2012, 40, 969–974. [Google Scholar] [CrossRef]
- Del Cano-Ochoa, F.; Ramon-Maiques, S. Deciphering CAD: Structure and function of a mega-enzymatic pyrimidine factory in health and disease. Protein Sci. 2021, 30, 1995–2008. [Google Scholar] [CrossRef]
- Luthra, P.; Naidoo, J.; Pietzsch, C.A.; De, S.; Khadka, S.; Anantpadma, M.; Williams, C.G.; Edwards, M.R.; Davey, R.A.; Bukreyev, A.; et al. Inhibiting pyrimidine biosynthesis impairs Ebola virus replication through depletion of nucleoside pools and activation of innate immune responses. Antivir. Res. 2018, 158, 288–302. [Google Scholar] [CrossRef]
- Martin, S.; Chiramel, A.I.; Schmidt, M.L.; Chen, Y.C.; Whitt, N.; Watt, A.; Dunham, E.C.; Shifflett, K.; Traeger, S.; Leske, A.; et al. A genome-wide siRNA screen identifies a druggable host pathway essential for the Ebola virus life cycle. Genome Med. 2018, 10, 58. [Google Scholar] [CrossRef]
- Lloyd, R.E. How do viruses interact with stress-associated RNA granules? PLoS Pathog. 2012, 8, e1002741. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Lloyd, R.E. Regulation of stress granules in virus systems. Trends Microbiol. 2012, 20, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Ohn, T.; Anderson, P. The role of posttranslational modifications in the assembly of stress granules. Wiley Interdiscip. Rev. RNA 2010, 1, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Ohn, T.; Kedersha, N.; Hickman, T.; Tisdale, S.; Anderson, P. A functional RNAi screen links O-GlcNAc modification of ribosomal proteins to stress granule and processing body assembly. Nat. Cell Biol. 2008, 10, 1224–1231. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, S.; Kedersha, N.; Anderson, P.; Ivanov, P. Molecular mechanisms of stress granule assembly and disassembly. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118876. [Google Scholar] [CrossRef]
- Corbet, G.A.; Parker, R. RNP Granule Formation: Lessons from P-Bodies and Stress Granules. Cold Spring Harb Symp. Quant. Biol. 2019, 84, 203–215. [Google Scholar] [CrossRef]
- Sanders, D.W.; Kedersha, N.; Lee, D.S.W.; Strom, A.R.; Drake, V.; Riback, J.A.; Bracha, D.; Eeftens, J.M.; Iwanicki, A.; Wang, A.; et al. Competing Protein-RNA Interaction Networks Control Multiphase Intracellular Organization. Cell 2020, 181, 306–324 e328. [Google Scholar] [CrossRef]
- Costa-Mattioli, M.; Walter, P. The integrated stress response: From mechanism to disease. Science 2020, 368, eaat5314. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Hardenberg, M.; Horvath, A.; Ambrus, V.; Fuxreiter, M.; Vendruscolo, M. Widespread occurrence of the droplet state of proteins in the human proteome. Proc. Natl. Acad. Sci. USA 2020, 117, 33254–33262. [Google Scholar] [CrossRef]
- Hatos, A.; Tosatto, S.C.E.; Vendruscolo, M.; Fuxreiter, M. FuzDrop on AlphaFold: Visualizing the sequence-dependent propensity of liquid-liquid phase separation and aggregation of proteins. Nucleic Acids Res. 2022, 50, W337–W344. [Google Scholar] [CrossRef] [PubMed]
- Vendruscolo, M.; Fuxreiter, M. Sequence Determinants of the Aggregation of Proteins Within Condensates Generated by Liquid-liquid Phase Separation. J. Mol. Biol. 2022, 434, 167201. [Google Scholar] [CrossRef] [PubMed]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodova, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koca, J.; Rose, A.S. Mol* Viewer: Modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Chu, X.; Sun, T.; Li, Q.; Xu, Y.; Zhang, Z.; Lai, L.; Pei, J. Prediction of liquid-liquid phase separating proteins using machine learning. BMC Bioinform. 2022, 23, 72. [Google Scholar] [CrossRef]
- Necci, M.; Piovesan, D.; Clementel, D.; Dosztanyi, Z.; Tosatto, S.C.E. MobiDB-lite 3.0: Fast consensus annotation of intrinsic disorder flavours in proteins. Bioinformatics 2020, 36, 5533–5534. [Google Scholar] [CrossRef]
- Linding, R.; Russell, R.B.; Neduva, V.; Gibson, T.J. GlobPlot: Exploring protein sequences for globularity and disorder. Nucleic Acids Res 2003, 31, 3701–3708. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.R.; Thomson, R.; McNeil, P.; Esnouf, R.M. RONN: The bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics 2005, 21, 3369–3376. [Google Scholar] [CrossRef] [Green Version]
- Prilusky, J.; Felder, C.E.; Zeev-Ben-Mordehai, T.; Rydberg, E.H.; Man, O.; Beckmann, J.S.; Silman, I.; Sussman, J.L. FoldIndex: A simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 2005, 21, 3435–3438. [Google Scholar] [CrossRef]
- Galzitskaya, O.V.; Garbuzynskiy, S.O.; Lobanov, M.Y. FoldUnfold: Web server for the prediction of disordered regions in protein chain. Bioinformatics 2006, 22, 2948–2949. [Google Scholar] [CrossRef] [PubMed]
- Piovesan, D.; Tabaro, F.; Micetic, I.; Necci, M.; Quaglia, F.; Oldfield, C.J.; Aspromonte, M.C.; Davey, N.E.; Davidovic, R.; Dosztanyi, Z.; et al. DisProt 7.0: A major update of the database of disordered proteins. Nucleic Acids Res 2017, 45, D219–D227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, A.J.; Walsh, I.; Tosatto, S.C. MOBI: A web server to define and visualize structural mobility in NMR protein ensembles. Bioinformatics 2010, 26, 2916–2917. [Google Scholar] [CrossRef] [Green Version]
- Walsh, I.; Martin, A.J.; Di Domenico, T.; Tosatto, S.C. ESpritz: Accurate and fast prediction of protein disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linding, R.; Jensen, L.J.; Diella, F.; Bork, P.; Gibson, T.J.; Russell, R.B. Protein disorder prediction: Implications for structural proteomics. Structure 2003, 11, 1453–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callebaut, I.; Courvalin, J.C.; Worman, H.J.; Mornon, J.P. Hydrophobic cluster analysis reveals a third chromodomain in the Tetrahymena Pdd1p protein of the chromo superfamily. Biochem. Biophys. Res. Commun. 1997, 235, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Callebaut, I.; Labesse, G.; Durand, P.; Poupon, A.; Canard, L.; Chomilier, J.; Henrissat, B.; Mornon, J.P. Deciphering protein sequence information through hydrophobic cluster analysis (HCA): Current status and perspectives. Cell. Mol. Life Sci. 1997, 53, 621–645. [Google Scholar] [CrossRef]
- Ramraj, V. Exploiting Whole-PDB Analysis in Novel Bioinformatics Applications. Doctoral Dissertation, University of Oxford, Oxford, UK, 2014. [Google Scholar]
- Wootton, J.C. Non-globular domains in protein sequences: Automated segmentation using complexity measures. Comput. Chem. 1994, 18, 269–285. [Google Scholar] [CrossRef]
- Chandonia, J.M. StrBioLib: A Java library for development of custom computational structural biology applications. Bioinformatics 2007, 23, 2018–2020. [Google Scholar] [CrossRef] [Green Version]
- Dayhoff, G.W., 2nd; Uversky, V.N. Rapid prediction and analysis of protein intrinsic disorder. Protein Sci. 2022, 31, e4496. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Romero, P.; Uversky, V.N.; Dunker, A.K. Coupled Folding and Binding with alpha-Helix-Forming Molecular Recognition Elements. Biochemistry 2005, 44, 12454–12470. [Google Scholar] [CrossRef] [PubMed]
- Campen, A.; Williams, R.M.; Brown, C.J.; Meng, J.; Uversky, V.N.; Dunker, A.K. TOP-IDP-scale: A new amino acid scale measuring propensity for intrinsic disorder. Protein Pept. Lett. 2008, 15, 956–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dosztanyi, Z.; Meszaros, B.; Simon, I. ANCHOR: Web server for predicting protein binding regions in disordered proteins. Bioinformatics 2009, 25, 2745–2746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, B.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N. CDF it all: Consensus prediction of intrinsically disordered proteins based on various cumulative distribution functions. FEBS Lett. 2009, 583, 1469–1474. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Oldfield, C.; Meng, J.; Hsu, W.L.; Xue, B.; Uversky, V.N.; Romero, P.; Dunker, A.K. Subclassifying disordered proteins by the CH-CDF plot method. Biocomputing 2012 2012, 128–139. [Google Scholar]
- Huang, F.; Oldfield, C.J.; Xue, B.; Hsu, W.L.; Meng, J.; Liu, X.; Shen, L.; Romero, P.; Uversky, V.N.; Dunker, A. Improving protein order-disorder classification using charge-hydropathy plots. BMC Bioinform. 2014, 15 (Suppl. 17), S4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Brown, C.J.; Uversky, V.N.; Dunker, A.K. Comparing and combining predictors of mostly disordered proteins. Biochemistry 2005, 44, 1989–2000. [Google Scholar] [CrossRef]
- He, B.; Wang, K.; Liu, Y.; Xue, B.; Uversky, V.N.; Dunker, A.K. Predicting intrinsic disorder in proteins: An overview. Cell Res. 2009, 19, 929–949. [Google Scholar] [CrossRef] [Green Version]
- Ishida, T.; Kinoshita, K. PrDOS: Prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007, 35, W460–W464. [Google Scholar] [CrossRef] [Green Version]
- Andreeva, A.; Howorth, D.; Brenner, S.E.; Hubbard, T.J.; Chothia, C.; Murzin, A.G. SCOP database in 2004: Refinements integrate structure and sequence family data. Nucleic Acids Res. 2004, 32, D226–D229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murzin, A.G.; Brenner, S.E.; Hubbard, T.; Chothia, C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995, 247, 536–540. [Google Scholar] [CrossRef] [PubMed]
- de Lima Morais, D.A.; Fang, H.; Rackham, O.J.; Wilson, D.; Pethica, R.; Chothia, C.; Gough, J. SUPERFAMILY 1.75 including a domain-centric gene ontology method. Nucleic Acids Res. 2011, 39, D427–D434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meszaros, B.; Simon, I.; Dosztanyi, Z. Prediction of protein binding regions in disordered proteins. PLoS Comput. Biol. 2009, 5, e1000376. [Google Scholar] [CrossRef] [Green Version]
- Hornbeck, P.V.; Kornhauser, J.M.; Tkachev, S.; Zhang, B.; Skrzypek, E.; Murray, B.; Latham, V.; Sullivan, M. PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012, 40, D261–D270. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–D568. [Google Scholar] [CrossRef]







| GO Terms | GO ID | p Value |
|---|---|---|
| Biological processes (five most enriched) | ||
| Symbiotic process | 0044403 | 1.68 × 10−7 |
| Viral process | 0016032 | 1.17 × 10−7 |
| Interspecies interaction between organisms | 0044419 | 1.20 × 10−5 |
| Negative regulation of catabolic process | 0009895 | 1.69 × 10−5 |
| Cellular response to stimulus | 0051716 | 1.85 × 10−5 |
| Molecular functions (five most enriched) | ||
| mRNA binding | 0003729 | 9.29 × 10−6 |
| mRNA 3-UTR binding | 0003730 | 1.86 × 10−5 |
| RNA binding | 0003723 | 0.0031 |
| Protein phosphatase binding | 0019903 | 0.0064 |
| Organic cyclic compound binding | 0097159 | 0.0073 |
| Cellular components (five most enriched) | ||
| Cytoplasmic stress granule | 0010494 | 0.00086 |
| Ribonucleoprotein complex | 1990904 | 0.00086 |
| Cell junction | 0030054 | 0.0107 |
| Protein-containing complex | 0032991 | 0.0107 |
| Nuclear matrix | 0016363 | 0.0193 |
| GO Terms | GO ID | p Value |
|---|---|---|
| Biological processes (five most enriched) | ||
| Positive regulation of nitrogen compound metabolic process | 0051173 | 1.34 × 10−122 |
| Positive regulation of cellular metabolic process | 0031325 | 3.13 × 10−119 |
| Positive regulation of cellular process | 0048522 | 1.80 × 10−116 |
| Positive regulation of macromolecule metabolic process | 0010604 | 1.19 × 10−115 |
| Regulation of cellular metabolic process | 0031323 | 1.87 × 10−115 |
| Molecular functions (five most enriched) | ||
| Enzyme binding | 0019899 | 1.02 × 10−93 |
| Protein binding | 0005515 | 1.58 × 10−92 |
| Binding | 0005488 | 1.39 × 10−60 |
| Transcription factor binding | 0008134 | 8.70 × 10−51 |
| Kinase binding | 0019900 | 2.08 × 10−59 |
| Cellular components (five most enriched) | ||
| Nucleoplasm | 0005654 | 1.67 × 10−95 |
| Nuclear lumen | 0031981 | 5.16 × 10−83 |
| Nucleus | 0005634 | 4.98 × 10−82 |
| Intracellular organelle lumen | 0070013 | 1.14 × 10−73 |
| Cytosol | 0005829 | 3.48 × 10−65 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bianchi, G.; Brocca, S.; Longhi, S.; Uversky, V.N. Liaisons dangereuses: Intrinsic Disorder in Cellular Proteins Recruited to Viral Infection-Related Biocondensates. Int. J. Mol. Sci. 2023, 24, 2151. https://doi.org/10.3390/ijms24032151
Bianchi G, Brocca S, Longhi S, Uversky VN. Liaisons dangereuses: Intrinsic Disorder in Cellular Proteins Recruited to Viral Infection-Related Biocondensates. International Journal of Molecular Sciences. 2023; 24(3):2151. https://doi.org/10.3390/ijms24032151
Chicago/Turabian StyleBianchi, Greta, Stefania Brocca, Sonia Longhi, and Vladimir N. Uversky. 2023. "Liaisons dangereuses: Intrinsic Disorder in Cellular Proteins Recruited to Viral Infection-Related Biocondensates" International Journal of Molecular Sciences 24, no. 3: 2151. https://doi.org/10.3390/ijms24032151
APA StyleBianchi, G., Brocca, S., Longhi, S., & Uversky, V. N. (2023). Liaisons dangereuses: Intrinsic Disorder in Cellular Proteins Recruited to Viral Infection-Related Biocondensates. International Journal of Molecular Sciences, 24(3), 2151. https://doi.org/10.3390/ijms24032151