1. Introduction
Fibrillary deposits of amyloid beta (Aβ) and tau are characteristic for the pathology of Alzheimer’s disease [
1]. The aggregation of both proteins is thought to be responsible for the neurodegeneration that occurs during the course of Alzheimer’s disease. In its nontoxic monomeric form, tau serves important functions in neurons [
2]. Tau knockout mice show muscle weakness, impairment during fear conditioning, and hyperactivity in a novel environment [
3]. The physiological function of tau is tied to microtubules as tau is a microtubule-associated protein (MAPT). Tau was shown to be an intrinsic component for the building, stabilization, maintenance, and degradation of microtubules [
4]. These neuroprotective features are lost upon the aggregation of tau. The exact initial cause for aggregation is still not clear. During aggregation, the monomeric protein undergoes a structural shift and together with other monomers forms oligomers and later fibrils. These fibrils are then found as deposits in the brain and are most prevalent in Alzheimer patients [
5,
6]. The neurodegeneration present in Alzheimer’s disease is linked to a toxic gain of function of the oligomers and fibrils, in addition to the toxic loss of function due to monomer depletion [
7]. Many therapeutic approaches were developed to target Aβ pathology but did not yield positive results in clinical trials [
8]. Recent studies show that cognitive defects correlate more with tau propagation and hyperphosphorylation [
9,
10], while Aβ is claimed to be responsible for tau hyperphosphorylation via the GSK3 cascade [
11]. Tau hyperphosphorylation is shown to induce the detachment of tau from microtubules and is assumed to precede tau aggregation. This indicates that tau toxicity may play a more active role in neurodegeneration and explains why many tested Aβ-targeting therapeutics fail to deliver their intended effect during clinical trials. Therefore, the tau protein is thought to be an appropriate target for the development of new therapeutic agents. Ideally, new therapeutics should stabilize the monomeric form of the tau protein in order to maintain its function and shift the equilibrium from aggregates towards nontoxic monomers. We decided to use mirror-image phage display to generate all the D-enantiomeric peptides that are able to achieve the desired effect. Mirror image phage display allows the identification of all D-enantiomeric peptides as ligands for a given target, in this case the tau protein. In comparison to the normal, L-enantiomeric peptides, D-peptides are much more resistant to proteolytic degradation and are less immunogenic. These are important properties for potential therapeutics. It has been claimed that the aggregation of tau is dependent on two hexapeptides, namely
306VQIVYK
311 (PHF6) and
275VQIINK
280 (PHF6*) [
12,
13]. PHF6* is located on the second imperfect repeat and only present in tau isoforms that express the second imperfect repeat. The PHF6 region is located in the third imperfect repeat and is present in all tau isoforms. Both peptides are able to induce tau aggregation, but recent studies suggest that the PHF6* region has a stronger effect than the PHF6 region [
14]. Cryo-electron microscopy of AD brains revealed that the core of PHFs contains the residues 306-378 of two identical protofilaments of tau [
15]. Considering this, we decided to choose a 45-amino-acid-long fragment of tau as a target for mirror-image phage display. This target contained the amino acid residues of tau from 271 to 316. Thus, both the PHF6* and the PHF6 region were present in the fragment. We used the mirror image of tau(271-316) as a target for phage display selection and next-generation sequencing (NGS) for the analysis of the selection results. We successfully selected stable all D-enantiomeric peptides that formed large groups of homologous sequences enriched by the selection. Here, we show that the enriched peptides are able to stop tau aggregation in vitro and reduce the amount of tau fibril assembly in HEK293T cells.
3. Discussion
Most of the currently developed therapeutic agents for Alzheimer’s disease and other tauopathies focus on either the elimination of toxic oligomers of aggregated tau or its expression downregulation. Up to now, there is no approved therapeutic agent that effectively targets tau pathology. As recent studies show that the onset of symptoms during Alzheimer’s disease correlates better with tau pathology rather than Aβ, it may be more efficient to target the tau protein [
10]. Therefore, the approach of stabilizing and protecting the monomeric form of tau, while shifting the aggregation equilibrium towards its monomeric conformation and destabilizing aggregates, may prove to be a successful strategy. The use of mirror-image phage display to generate all D-enantiomeric compounds has already been shown to yield all D-peptides which are able to bind tau aggregates [
28,
29,
30]. An all D-enantiomeric peptide that is able to bind and stabilize monomeric tau does not yet exist.
In this study, we demonstrated the successful selection of the all D-enantiomeric compound TF2D-5 and its derivative TF2D-5a, which are able to bind monomeric tau with a nanomolar KD and to inhibit the formation of tau aggregation in vitro and in cells. We were able to identify these compounds by using mirror-image phage display combined with NGS, which allowed for the in-depth analysis of the selections.
A crucial property of selected compounds as potential therapeutic agents is the inhibition of tau aggregation. The ability of TF2D-5 and TF2D-5a to inhibit tau aggregation in vitro was studied with different methods. ThT assays were used to observe changes in the lag time and the fluorescent levels, while gel electrophoresis and AFM measurements were used to visualize the effects of both compounds and to ensure that no amorphous aggregates were formed. All of the obtained results indicate that both compounds have a stabilizing effect on monomeric tau. The ThT assays with the addition of either TF2D-5 or TF2D-5a revealed an increased lag time of tau aggregation, while the addition of TF2D-5 to an ongoing tau aggregation was able to stop its aggregation from the point of introduction. These observations were also consistent with the gel electrophoresis results. The presence of TF2D-5 lowered the amount of tau that sedimented by centrifugation compared to the samples that did not contain TF2D-5. The visualization of the tau aggregation inhibiting effectivity of the investigated compounds by AFM measurements revealed that the addition of TF2D-5 during the lag phase of aggregation led to no observable fibrils on the micas. Similarly, the addition of TF2D-5 during the exponential phase led to a reduction in overall fibril quantity. This might be due to the mode of fibril growth during tau aggregation. Recent studies showed that tau fibril size distribution is dominated by the breaking and linkage of tau fibrils where longer fibrils fracture and act as multiple seeds which elongate by monomer addition and form longer fibrils [
31]. This elongation is disrupted by the TF2D-5 addition as it stabilizes the monomeric tau and prevents its recruitment towards smaller fibrils, resulting in broken fibrils. The sequence specificity of TF2D-5 was verified by the use of a control D-peptide, which features the same amino acid composition as TF2D-5 but in a randomized sequence. This randomized control D-peptide was ineffective for tau aggregation inhibition during the ThT assays.
High affinity towards the target is a key property. We used SPR measurements to determine the K
D of TF2D-5 and TF2D-5a. Both compounds possess high affinities towards the target, resulting in low K
Ds of 1.3 nM and 6.1 nM, respectively. In further experiments, we validated these K
Ds and found that the actual K
D of TF2D-5 towards monomeric tau is even lower, with 308 pM. Recent papers reported on the cryo EM structures of tau fibrils isolated from the PHFs of Alzheimer patients. They revealed that the R3 domain is part of the fibril core [
15], thus binding within this region might destabilize the fibril structure of tau or lead to steric hindrance during the incorporation of monomeric tau with bound TF2D-5 and TF2D-5a. Although TF2D-5 has the higher affinity towards full-length tau (308 pM versus 11.7 nM), we observed that the aggregation inhibition efficiency of TF2D-5a is superior. We assume that the increased inhibition properties of TF2D-5a are due to the proposed binding site of TF2D-5. In
Figure 8C, we show the high affinity of TF2D-5 towards the imperfect repeat R3 of tau. This imperfect repeat was shown to be essential for the aggregation of tau as it is incorporated into the core of the fibril during aggregation [
15]. The binding of TF2D-5 towards this site might lead to steric hindrances during aggregation. The addition of five arginine residues to the peptide results in an elongated peptide that may reach further into the potential fibril core, resulting in a stronger aggregation destabilization. At the same time, this addition is not optimized for tau binding and results in a lower affinity towards the binding site. The expansion of the peptide with five arginine residues was supposed to increase its solubility, and it is suspected that this increases the peptide’s ability to cross the blood–brain barrier [
22]. We believe that the affinity for tau could be increased by replacing the arginine residues with other amino acids.
Considering the data obtained in the monomer quantification, we were able to show that both peptides increase the amount of monomer that is retained during a seeded aggregation. This effect was shown to be present at sub-stoichiometric concentrations, and we hypothesize that this effect is even stronger in a de novo aggregation or with higher concentrations of peptide.
We have shown that the effect of TF2D-5 and TF2D-5a is sub-stoichiometric. This again raises the question of the mechanism by which the aggregation of tau is inhibited. We currently hypothesize that this behavior is related to the binding of monomeric tau by TF2D-5 and TF2D-5a. Considering the SPR results, we assume that the peptides bind in repeat region three of tau. This region was found to be incorporated into the fibril core of aggregated tau [
15]. Binding in this region could destabilize the fibril structure or lead to a change in the energetically most favorable conformation, effectively protecting monomeric tau from conversion into fibrils. The ability of TF2D-5 and TF2D-5a to inhibit tau aggregation at sub-stoichiometric concentrations could come from the amount of “unprotected” tau that is available. A decrease in the amount of tau that can form fibrils may lead to a longer lag phase until this chance-based cascade is triggered. This would fit with the shown data where the addition of very low amounts of TF2D-5 and TF2D-5a increases lag time but does not inhibit fibril formation completely. Another explanation may be found in the structural changes that occur during oligomerization. It was shown that the microtubule binding regions of tau adapt a paperclip-like formation with the N-term and C-term in the monomeric form. This paperclip formation collapses during aggregation, exposing the potential binding site for TF2D-5 and TF2D-5a [
23]. As shown in the SPR experiments, the affinity of the peptides towards this region is in the low picomolar range. TF2D-5 and TF2D-5a could bind to the exposed binding site and stop an ongoing conversion into oligomers due to the steric hindrances introduced due to the binding.
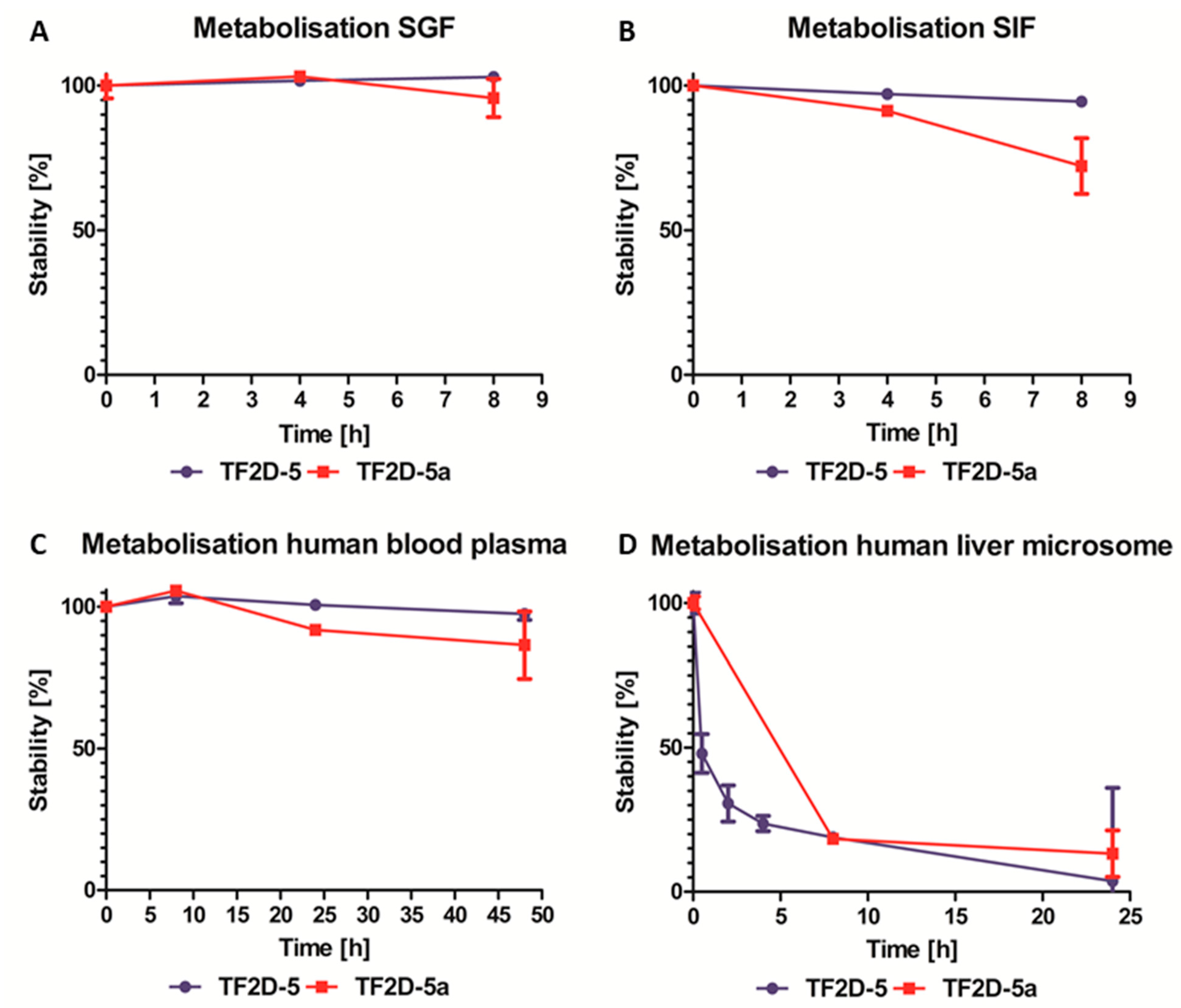
Moreover, as expected for all D-peptides, the metabolic stability of TF2D-5 and TF2D-5a in different simulated environments connected to the uptake and digestion of therapeutic agents was quite favorable. From our experiments, we conclude that both compounds might be stable in SGF, SIF, and human blood plasma. TF2D-5 showed a particularly high stability in SGF, SIF, and human blood plasma. Both compounds are more stable in human blood plasma than RD2, an all D-enantiomeric peptide selected against Aβ that recently successfully finished clinical phase I trials [
32] and was also investigated for its metabolic stability [
33]. TF2D-5 was not very stable in liver microsomes. After 24 h, only 3.7% of TF2D-5 and 13.2% of TF2D-5a were unmetabolized. Compared to other all D-enantiomeric compounds, this metabolization speed is fast [
33]. Moreover, TF2D-5 and TF2D-5a may well cross the blood–brain barrier.
Lastly, TF2D-5a was able to reduce the amount of cells with tau aggregates by about 80% compared to tau aggregation in the absence of TF2D-5a or the presence of a control D-peptide without any affinity for tau.
4. Material and Methods
4.1. Mirror-Image Phage Display
The mirror-image phage display describes a technique to select D-peptide binders of L-target proteins from a library. This is conducted by using a D-target and selecting with the L-peptides presented on the phages. The phages that bind to the D-target are analyzed, and the chosen peptides are synthetically produced as D-peptides. These peptides are then analyzed for their ability to bind the target and are, in the case of successful binding, further developed.
4.2. Target
The target of the phage display selection was a custom designed all D-enantiomeric fragment of tau consisting of the amino acids 271-316 out of the full-length tau 441(D-tau271-316). It encompasses the imperfect repeat 2 and part of the imperfect repeat 3 of the full-length 2N4R tau protein. The hexapeptides PHF6 and PHF6*, which are important for tau aggregation, were present in the imperfect repeat 2 (PHF6*) and 3 (PHF6). Both regions were included in the target design as binding near these regions might stabilize monomeric tau. A biotin tag was added to the N-terminus. The C-terminus was amidated.
4.3. Mass Spectroscopy
Mass spectroscopy was performed at the Central Institute for Engineering, Electronics and Analytics (ZEA-3) within the Forschungszentrum Jülich to verify the identity of the mirror-image D-tau(271-316).
4.4. Selection
4.4.1. Strategy
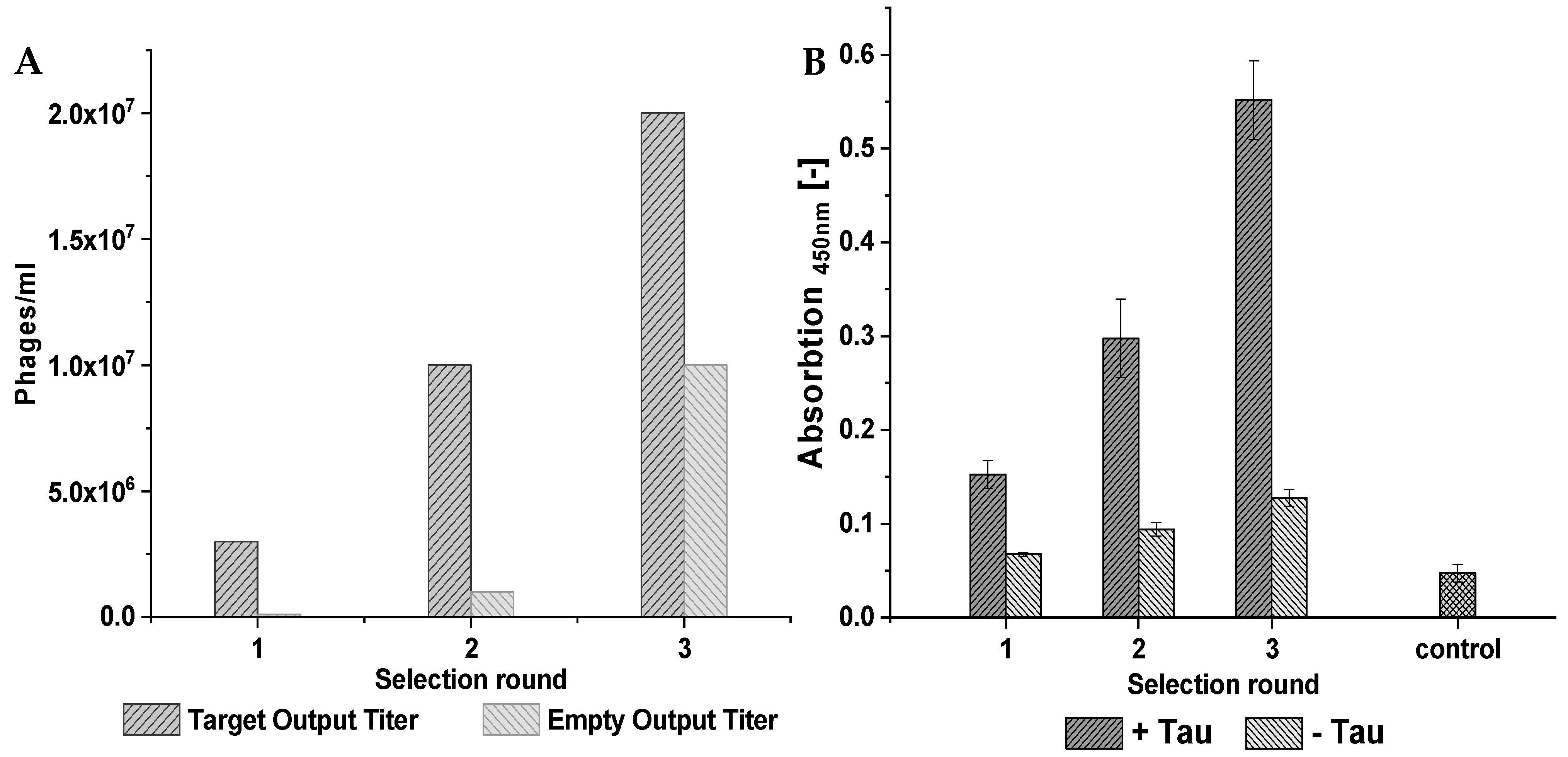
During the phage display selection experiment, multiple control selections were performed to ensure the quality of the selection. From the library, two starting selections were performed. The first, called TS1 for target selection 1, used a surface that contained the target, while the second one, called ES1 for empty selection 1, did not contain the target on the surface and served as a control. The empty selection was performed in parallel to the target selection. The phages derived from TS1 were later applied to the next target selection (TS2) as well as the direct control (DC2), which did not contain the target protein on the surface and served as a negative control for direct comparison. A subsequent third selection round was performed analogously.
Figure 14 illustrates this course of the selection and at which points a control selection had been performed, which subsequently enabled the identification of the target-specific binders via the NGS results. The different selection types were named “target selection” (TS), “direct control” (DC), and “empty selection” (ES).
4.4.2. Implementation
For the target selection rounds, 120 pmol of D-tau(271-316) was immobilized on a Pierce streptavidin plate (ThermoFisher Scientific, Waltham, MA, USA) via biotin-streptavidin binding. The target was solved in phosphate-buffered saline (PBS) buffer, and 100 µL was applied to each used well. For the empty selection and direct control selection, a buffer instead of D-tau(271-316) was used during immobilization. The subsequent procedure was identical for all three selections. The plate was incubated at room temperature for 60 min with shaking. After removing the immobilization solution, a blocking step with PBS and 10 mg/mL milk powder was performed. Two hundred microliters of the blocking buffer was incubated for 30 min under shaking at room temperature. The wells were washed six times with 200 µL washing buffer containing PBS with 2 mg/mL milk powder. The phages, with a total number of 2 × 1011, from a TriCo-16 phage display peptide library (Creative Biolabs, lot number: CBLX090318, Shirley, NY, USA), were solved in 100 µL PBS and added to the prepared wells. The incubation was performed at room temperature for 20 min with shaking. This was followed by 20 washing steps, each with 100 µL washing buffer. The elution of bound phages was performed by incubating the wells with 100 µL 0.2 M glycine HCl (pH 2.2) for 10 min at room temperature with shaking. The eluted phages were transferred into a prepared Eppendorf tube with 25 µL 1M tris-HCl (pH 9.1). Five microliters of the phages was used for output titer determination, and the rest of the phages were amplified in bacteria.
For phage amplification, 20 mL of E.coli K12 ER 2738 in lysogeny broth (LB) medium was added to a 250 mL flask and grown to an OD600 of 0.1. Tetracycline was added to an end concentration of 50 µM. The phage amplification was performed at 37 °C with shaking for 4.5 to 5 h. The 20 mL was harvested into 50 mL falcons and centrifuged at 3000× g at 4 °C for 20 min. The supernatant was mixed with 7 mL of PEG/NaCl (20% (w/v) Polyethylenglycol-8000, 2.5 M NaCl) and incubated overnight on ice. Afterwards, the solution was again centrifuged at 3000× g at 4 °C for 60 min. The supernatant was discarded and the pellet resuspended in 1 mL tris-buffered saline (TBS) and centrifuged for 5 min at 11,000× g. The supernatant was transferred into a new 1.5 mL Eppendorf tube. Two hundred microliters of PEG was added to the Eppendorf tube and incubated on ice for 60 min. After incubation, the mixture was centrifuged for 45 min at 4 °C at 3000× g. Finally, the resulting pellet containing the phages was properly resuspended in 100 µL TBS.
4.4.3. Titer Determination
The output and input titer of each selection round were determined by diluting 1 µL of the output/input samples, taken at the end of each selection round and after amplification, and diluting it in 99 µL LB medium. Five and ten additional 1:10 dilutions were prepared for the output and input titer, respectively. One hundred microliters of E. coli K12 ER2738 with an OD600 of 0.6 was added to each dilution. Eight hundred microliters of warm TOP-Agar were mixed with the 200 µL phage/bacteria mixture and put onto a small LB/IPTG/X-Gal plate. The plates were shaken in a double orbital manner to ensure sufficient distribution of the mixture throughout the plate. After brief drying at room temperature, the plates were flipped and incubated at 37 °C overnight. The respective output or input titer was determined by counting the resulting blue plaques on the plates with different dilution factors.
4.5. ELISA
For the ELISA, a Pierce Streptavidin/Thermo Scientific/15500 plate was used. D-tau(271-316) amounts of 120 pmol, 60 pmol, and 20 pmol were immobilized on the surface of the plate via biotin-streptavidin binding. The immobilization was performed in 100 µL PBS for 60 min at room temperature while shaking. The plate was blocked by incubation with 200 µL PBS containing 10 mg/mL milk powder for 60 min at room temperature and shaking. The wells were washed three times with 200 µL PBS containing 2 mg/mL milk powder and 0.1% Tween20. During the phage incubation, 5 × 1010 phages were used. The binding was performed in 100 µL PBS for 20 min at room temperature with shaking. An additional washing step with five repetitions and a washing volume of 100 µL was performed. The horseradish conjugated anti-M13 antibody from Sino Biological (Eschborn, Germany, product number 11973-MM05T-H) was diluted 1:2500, and 100 µL was incubated for 60 min at room temperature. Afterwards, each well was washed six times. One hundred microliters of 3,3′5,5′-tetramethylbenzidine (TMB) substrate solution was added to each well. After 30 s, the reaction was stopped by the addition of 100 µL 2 M sulfuric acid. The signal intensity was then quantified by detecting the absorption at 450 nm in a CLARIOstar microplate reader (BMG, Offenburg, Germany).
4.6. DNA isolation
For DNA isolation, 10 µL of purified phage suspension was diluted in 580 µL PBS 7.4. Two hundred microliters of PEG/NaCl was added, inverted eight times, and incubated at room temperature for 20 min. After incubation, the phage suspension was centrifuged at 21,000× g at 4 °C for 10 min. The supernatant was discarded, and the remaining pellet was resuspended in 200 µL of a 10:1 mixture of 3 M sodium acetate (pH 5.2) and TE-buffer (10 mM tris-HCl, pH 8, 0.1 mM EDTA). Afterwards, 500 µL of 99% ethanol was added and incubated for 15 min at room temperature, followed by 10 min centrifugation at 21,000× g at 4 °C. The supernatant was discarded, and the pellet was resuspended in 250 µL 70% ethanol. After a final centrifugation of 10 min at 21,000× g at 4 °C the supernatant was discarded, and the resulting pellet was dried in an Eppendorf concentrator 5301 (Eppendorf, Hamburg, Germany). The dried pellet was resuspended in 40 µL 10 mM tris buffer pH 8.
4.7. PCR
The DNA concentration was determined by photometric analysis using a nano photometer (IMPLEN, München, Germany). The PCR was performed with 25 ng of sample (2.5 µL), 0.2 µg of forward primer (5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCGCAATTCCTTTAGTGGTACC-3′, 5 µL), 0.2 µg of reverse primer (5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGCCCTCATAGTTAGCGTAACG-3′, 5 µL), and 12.5 µL of KAPA mix (Kapa Biosystems, Wilmington, USA) for a total volume of 25 µL. The PCR started at 3 min at 95 °C, followed by 25 cycles of 30 s at 95 °C, 30 s at 55 °C, and 30 s at 72 °C. The PCR finished at 5 min at 72 °C and was afterwards on hold at 4 °C. The success of PCR was verified by analysis with a DNA gel.
4.8. Next-Generation Sequencing
Next-generation sequencing (NGS) of the phage DNA was performed by the Genomics & Transcriptomics Labor of the Heinrich-Heine-Universität Düsseldorf. For the NGS, all the target selection inputs, as well as direct control input three and empty selection input three, were prepared and sequenced; this included a sample of the library.
4.9. Data Analysis
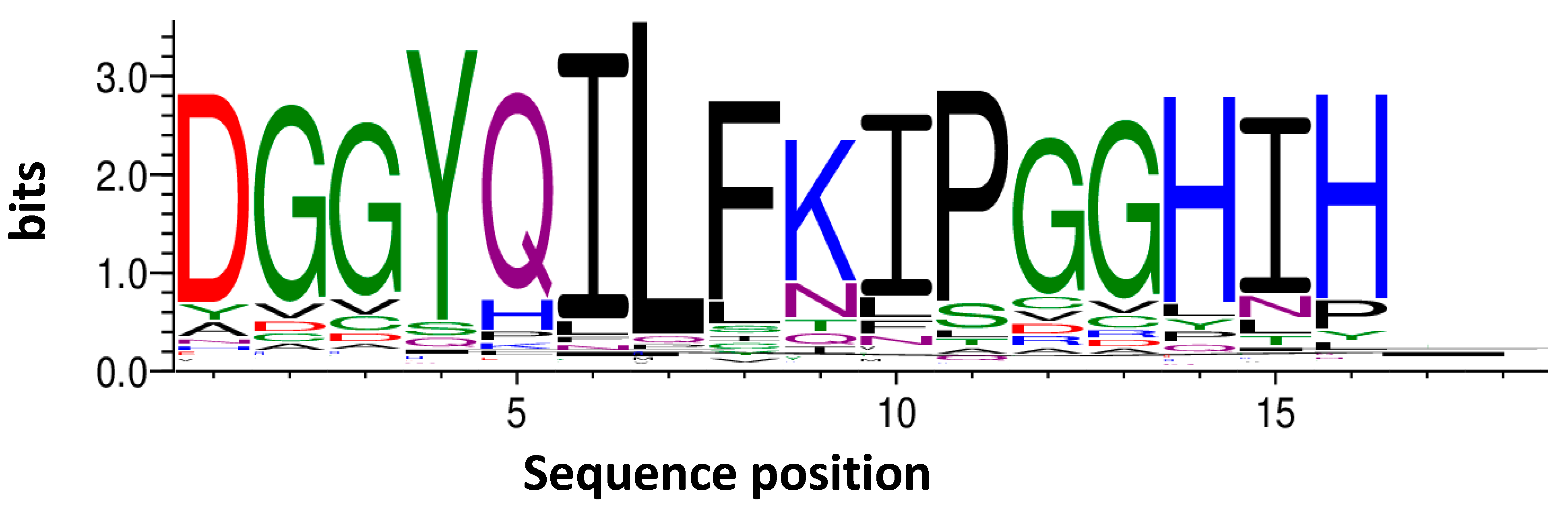
For the initial analysis and the grouping of the NGS data, the program TSAT (Target Sequence Analysis Tool) was used, which was developed in-house. A secondary analysis was performed using the clustering software Hammock.
4.9.1. Target Sequence Analysis Tool (TSAT)
The program TSAT takes raw DNA data in a FASTA or FASTQ format and examines these data for the provided framing regions of the randomized peptide region. If the framing regions are present in the given DNA, the randomized region coding for the peptide is excised, grouped, translated into amino acids, and saved in a database. Once saved in a database, the data are further processed by filtering. During filtering, the sequence frequency of the last target selection round are analyzed for their enrichment from target round to target round as well as compared to the empty selection and direct control. The filtering is conducted according to the following formula:
Only sequence abundancies or frequencies which pass the filter and possess more than eight amino acid residues are added to the new database. Each sequence in the new database is assigned an enrichment score and an empty score. These are based on the ratio between the last target selection round, the library, and the last empty selection round. The scores are calculated according to the following formulas:
If a sequence was not found in the library or the last empty selection, then the lowest value of the corresponding selection is taken and divided by two. Afterwards, this value is taken to calculate the enrichment or empty score. This was decided to first solve the problem of dividing by zero in the cases where the sequence was not found in the empty selection or library and second to highlight these sequences as they either enriched themselves strongly or were very target-specific. All calculations are performed as parts per million (ppm), which means the abundance of the particular sequence within the “read” divided by the total number of sequences obtained by the “read” and multiplied by 1 million, to enable a comparison between the samples. Finally, a document is created where all the sequences that passed the filter are displayed in a descending order according to their empty score and in FASTA format. This final document can be used to run the Hammock software.
4.9.2. Hammock
The program Hammock is software intended for the sequence clustering of large datasets comprising short sequences [
21]. It was used to generate clusters of similar sequences to generate motifs with a high order of similarity. Hammock was performed using the standard parameters of the software.
4.9.3. Peptides TF2D-5, TF2D-5a, and CP1
All the peptides used in this study were ordered from CASLO (Lyngby, Denmark). Both TF2D-5 and CP1 are 16-mers, while TF2D-5a is a 21-mer. The respective sequences are:
| TF2D-5 | dGGyqilfkipGGhih |
| TF2D-5a | dGGyqilfkipGGhihrrrrr |
| CP1 | GiiGpqykdhGhfliG |
All the peptides were synthesized from D-enantiomeric amino acid building blocks (except glycine) with the C-terminus amidated. TF2D-5 was selected during mirror-image phage display selection, while CP1 is a control peptide with the same amino acid residues as in TF2D-5, but in a randomized sequence. TF2D-5a was designed to increase the ability of TF2D-5 to cross membranes.
4.10. Recombinant Full-Length Tau Expression and Purification
The gene for the human full-length tau was prepared on a plasmid called pET28a(+)-tau2N4R. E. coli pRare cells were transformed using the generated plasmid. This was achieved by adding 1 µL (approx. 100 ng) of the plasmid DNA to 100 µL of competent E.coli pRare cells. The mix was incubated for 30 min on ice, followed by heat shock treatment at 42 °C for 45 s. After incubation on ice for 5 min, 900 µL of LB media was added and incubated for 1 h at 37 °C and 130 rpm. Transformation efficiency was controlled by plating out 100 and 800 µL of cell solution onto agar plates containing kanamycin and chloramphenicol. The remaining 100 µL was used to inoculate 100 mL of LBKAN, CHL medium and incubated overnight at 37 °C with shaking at 130 rpm.
For full-length tau expression, a preculture of transformed E.coli pRare cells was grown overnight in LBKAN, CHL at 37 °C with 5% CO2 and shaking at 300 rpm. The antibiotic concentration was adjusted to 1 mM. The overnight culture was transferred to the expression culture and adjusted to an OD600 of 0.1. LBKAN, CHL medium was used as the expression medium. The culture was grown to an OD600 of 0.6 0.7 and expression was induced by adding 1 mM IPTG. Expression was performed at 37 °C at 300 rpm, with shaking for 3 to 4 h. Cells were harvested by centrifugation at 5000× g for 20 min at 4 °C. The resulting pellets were either stored at −20 °C or resuspended directly in lysis buffer containing 500 mM NaCl, 20 mM PIPES, 1 mM EDTA, and 2 mM 2-mercaptoethanol pH 7.0.
The resuspended cell pellets were boiled for 15–20 min to disrupt all the cells and denature most of the proteins. As tau is an intrinsically disordered protein, the target protein is not destroyed by this method. Full-length tau is separated from the denatured proteins and cell debris by centrifugation for 1 h at 11,000× g at 4 °C. In preparation for ion exchange chromatography, the supernatant was dialyzed against 20 mM PIPES, 50 mM NaCl, 2 mM DTT, 0.5 mM EDTA; pH 6.7. Two dialysis steps were performed, with the first at room temperature for one hour and the second overnight at 4 °C. The next day, cation exchange chromatography was performed using a Bio SCX, NP5, SS column from Agilent Technologies (Santa Clara, CA, USA) on a 1200 series HPLC from Agilent Technologies. The elution buffer used was 20 mM PIPES 1 M NaCl, 2 mM DTT, and 0.5 mM EDTA; pH 6.7. The collected fractions were pooled and dialyzed overnight against 20 mM PIPES, 150 mM NaCl, 2 mM DTT; pH 7.4. A size exclusion run on a 1200 series HPLC from Agilent Technologies was performed. For the size exclusion, a Bio SEC 3, 300Å column form Agilent Technologies was used. The concentration of purified tau was measured and afterwards stored at −80 °C.
4.11. Screening for Aggregation Inhibitors via Thioflavin-T Assay
To identify the peptides (compounds) with the highest chance of becoming a successful therapeutic agent, a screening for aggregation inhibitors via ThT assay was performed. All the compounds were only tested in one concentration and added at the start of the experiment. The specifics of the other shown ThT assays are described in the next part. A FLUOstar plate reader (BMG, Offenburg, Germany) was used for the experiment. Fifty micrometers of recombinant full-length tau 441 was mixed with 12.5 µM heparin, 2% DMSO, 20 µM ThT, 500 µM DTT, and 50 µM compound. The total volume was adjusted to 100 µL with PBS pH 7.4, and the plate (Corning, New York, NY, USA) was measured with bottom optics. The experiment ran at 37 °C with measurement intervals of 20 min. Before each measurement, the plate was shaken for 15 s at 300 rpm.
4.12. Thioflavin-T Assay
ThT assays to characterize the aggregation inhibition properties of the selected compounds were conducted as follows. All measurements were performed in a FLUOstar plate reader (BMG, Offenburg, Germany). Recombinant full-length tau 441 (25 µM) was mixed with heparin (12.5 µM), DTT (200 µM), NaN3 (0.05%), and ThT (20 µM). Slight differences in tau concentrations compared to former THT assays are justified by different tau preparations which exhibit different aggregation properties. The compounds were added with a maximum concentration of 25 µM. The total volume was adjusted to 100 µL with PBS pH 7.4, and the plate (Corning, New York, NY, USA) was measured with bottom optics. A 3 mm borosilicate bead (Hilgenberg GmbH, Malsfeld, Germany) was added to each well. The experiment ran at 37 °C, with measurement intervals of 20 min. Before each measurement, the plate was shaken for 15 s at 300 rpm.
4.13. Atomic Force Microscopy (AFM)
All AFM measurements were performed by drying 1 µL of sample on mica followed by two washing steps using 100 µL water. All samples were measured on a JPK Nanowizard3 with an intermittent contact mode on air and an OMCL AC160TS as a cantilever.
4.14. SPR Measurement
4.14.1. Chip Preparation
The chips were prepared using a Biacore T200 (Biacore, GE Healthcare, Uppsala, Sweden) via manual run as well as a Biacore 8K (Biacore, GE Healthcare, Uppsala, Sweden) using an immobilization run. Series S CM5 chips were used, and the flow rate was adjusted to 10 µL/min. A mixture of 50 mM N-hydroxysuccinimide (NHS) and 16.1 mM N-Ethyl-N′-(dimethylaminopropyl)carbodiimide (EDC) (XanTec, Düsseldorf, Germany) was used to activate the chip. The activation duration was 7 min with a flow rate of 10 µL/min. Prior to immobilization, TF2D-5 and TF2D-5a were diluted with 10 mM sodium acetate pH 5 to a concentration of 50 µg/mL and 5 µg/mL, respectively. Each compound was immobilized on separate chips, with the total immobilization level being 130 RU for TF2D-5 and 600 RU for TF2D-5a. Channel one of each chip was used as the reference and only buffer was injected during immobilization for the T200. Flow cell 1 was used as a reference during experiments on the 8K. Thee quenching of both channels (T200) and flow cells (8K) was achieved by injection of 1 M ethanolamine, pH 8.5 (XanTec, Düsseldorf, Germany) for 7 min.
4.14.2. Measurement T200
Measurements of TF2D-5 and TF2D-5a against full-length tau 441 were performed on a Biacore T200 (Biacore, GE Healthcare, Uppsala, Sweden) using multicycle experiments. The flow rate was adjusted to 35 µL/min. Full-length tau 441 with different concentrations was used as an analyte. TBS pH 7.4 with 0.05% Tween20 and 10 µM DTT was used as a running buffer. Before measurement, three startup cycles without analyte were performed to allow for instrument equilibration. The experiment started with two cycles of 0 nM analyte concentration, used to reference the background noise within the experiment. The tau association time was set to ten min and the dissociation time to 60 min. The tau concentrations were 3.1 nM, 6.2 nM, 12.5 nM, and 25 nM for TF2D-5 and 31.2 nM, 62.5 nM, 125 nM, 250 nM, and 500 nM for TF2D-5a. The remaining tau was eluted by three 45 s regeneration cycles with 2 M guanidinium hydrochloride. The data were evaluated using the Biacore T200 Evaluation Software 3.2 using a 1:1 fit (RI = 0).
4.14.3. Measurement 8K
Measurements of TF2D-5a against the R3 domain of tau were performed on a Biacore 8K (GE Healthcare, Uppsala, Sweden) using a single cycle experiment. The all L-enantiomeric R3 domain (304SVQIVYKPVDLSKVTSKCGSLGNIHHKPGGG336) of tau, purchased from CASLO (CASLO ApS, Lyngby, Denmark), was used with concentrations of 3.1 nM, 6.2 nM, 12.5 nM, 25 nM, 50 nM, and 100 nM as the analytes. PBS pH 7.4 with 0.05% Tween20 was used as a running buffer. The flow rate was adjusted to 35 µL/min. Before measurement, three startup cycles without analyte were performed to allow for instrument equilibration. The contact time for each concentration was 3 min. The final dissociation time was set to 60 min. The data were analyzed using the Biacore insight evaluation software 3.0.12.15655 using a 1:1 fit.
4.15. MST Measurements
The affinity of TF2D-5 and TF2D-5a for full-length tau was additionally tested by MST measurements. All peptides were labeled with the fluorophore CF633 kit obtained from Sigma Aldrich (USA) and purified by reverse phase HPLC.
4.15.1. TF2D-5
CF633-labeled TF2D-5 was used at a concentration of 500 nM, and the tau concentration ranged from 2.5 µM to 0.2 pM using a dilution series of 1:2. The full-length tau was centrifuged at 20,000× g at 4 °C for 20 min, after thawing before use. The measurement was performed in 1x PBS pH 7.4 containing 150 mM NaCl.
4.15.2. TF2D-5a
CF633-labeled TF2D-5a was used at a concentration of 62.5 nM, and the tau concentration ranged from 300 nM to 0.5 nM using a 2:1 dilution series. Full-length tau was centrifuged at 20,000× g at 4 °C for 20 min, after thawing before use. Measurements were performed in a 0.1 M sodium phosphate buffer of pH 7.4 containing 75 mM sodium sulfate and 0.05% Tween 20. All measurements were repeated four times. The average of all the measurements was calculated, and a sigmoidal fit (Boltzmann) was used for KD determination.
4.15.3. CP1
CF633-labeled CP1 was used at a concentration of 62.5 nM, and the tau concentration ranged from 2.5 µM to 0.2 pM using a dilution series of 1:2. Full-length tau was centrifuged at 20,000× g at 4 °C for 20 min, after thawing before use. Measurements were performed in 0.1 M sodium phosphate buffer of pH 7.4 containing 75 mM sodium sulfate and 0.05% Tween 20. All measurements were repeated four times. The average of all the measurements was calculated.
4.16. Creation of Soluble Full-Length Tau Aggregates
First, the insoluble PFF were produced by the incubation of 130 µM recombinant tau in a LoBind reaction tube (Eppendorf GmbH, GE) with one borosilicate glass bead (d = 3.0 mm; Hilgenberg, DE) in 1× PBS pH 7.4 with 65 µM Heparin, 1 mM DTT, and 0.05% (w/v) sodium azide for two weeks at 37 °C. The insoluble PFF were harvested by ultracentrifugation at 100.000× g for 30 min at 4 °C, and the pellet was washed several times with 1× PBS pH 7.4. The monomer equivalent concentration was determined by measuring the tau concentration in the supernatant after the first centrifugation and subtracting it from the starting concentration for fibrilization. The insoluble PFF were resuspended in buffer and frozen at −80 °C with liquid N2. PFF oligomers were generated by harsh sonication of 200 µL insoluble Tau with a 13 µM monomer equivalent concentration for 3 × 15 s (1 s. on/off) and 60% amplitude with a tip sonifier (MS 72 micro tip, Sonopolus, Brandelin, GE). The insoluble PFF were separated by centrifugation at 100.000× g for 1 h at 4 °C. The supernatant containing the PFF oligomers was separated, aliquoted, and frozen at −80 °C with liquid N2.
4.17. Monomer Quantification Using HPLC
Quantities equaling 100 nM soluble tau oligomers and 100 nM of monomeric tau were incubated for 96 h at 37 °C at 300 rpm. Sub-stoichiometric concentrations of TF2D-5, TF2D-5a, and CP1 (25 µM, 50 µM) were added. After 96 h, the samples were centrifuged for two min at 11,000× g to sediment the large aggregates. One hundred microliters of each sample was analyzed on an HPLC (Agilent 1200 with quaternary pump, manual injection valve, and variable wavelength detector) with a size exclusion column (Agilent Bio-SEC 3, 300 A, 7.8 × 300 mm, 3 µm). The flow rate was adjusted to 1 mL/min.
4.18. Stability Assay of D-Peptides in Simulated Human Body Fluids
The stability of the compounds was tested according to Elfgen et al. [
33] and Schemmert et al. [
34] Exceptions included the extraction of plasma, simulated gastric fluid (SGF), and simulated intestinal fluid (SIF) samples. These were obtained by the addition of 12.5 µL 0.35 M ZnSO
4, followed by vortexing, the addition of 12.5 µL 0.5 M HCl, additional vortexing, and centrifugation at 14,000×
g for 10 min at 4 °C.
4.19. Cell-Viability Assay
Human bone marrow SH-SY5Y cells and rat pheochromocytoma PC12 cells were cultivated in RPMI medium with 10% fetal bovine serum and 5% horse serum. On a collagen-coated 96-well plate (Gibco, Life Technology, Carlsbad, CA, USA), 10.000 cells were seeded per well. The plate was incubated for 24 h at 37 °C and 5% CO2. TF2D-5 and TF2D-5a were added to the cells at concentrations of 0.2 µM, 1 µM, and 5 µM, respectively. As a positive control, 0.1% Trion X-100 was used. A medium was used as a negative control. The samples were incubated for a further 24 h at 37 °C and 5% CO2. Cell viability was determined by using the Cell Proliferation Kit 1 (MTT) (Roche, Basel, Switzerland) according to the instruction manual. The absorbance of generated formazan products was measured using a CLARIOstar plate reader (BMG, Offenburg, Germany). Absorption at 570 nm was measured and referenced against absorbance at 660 nm.
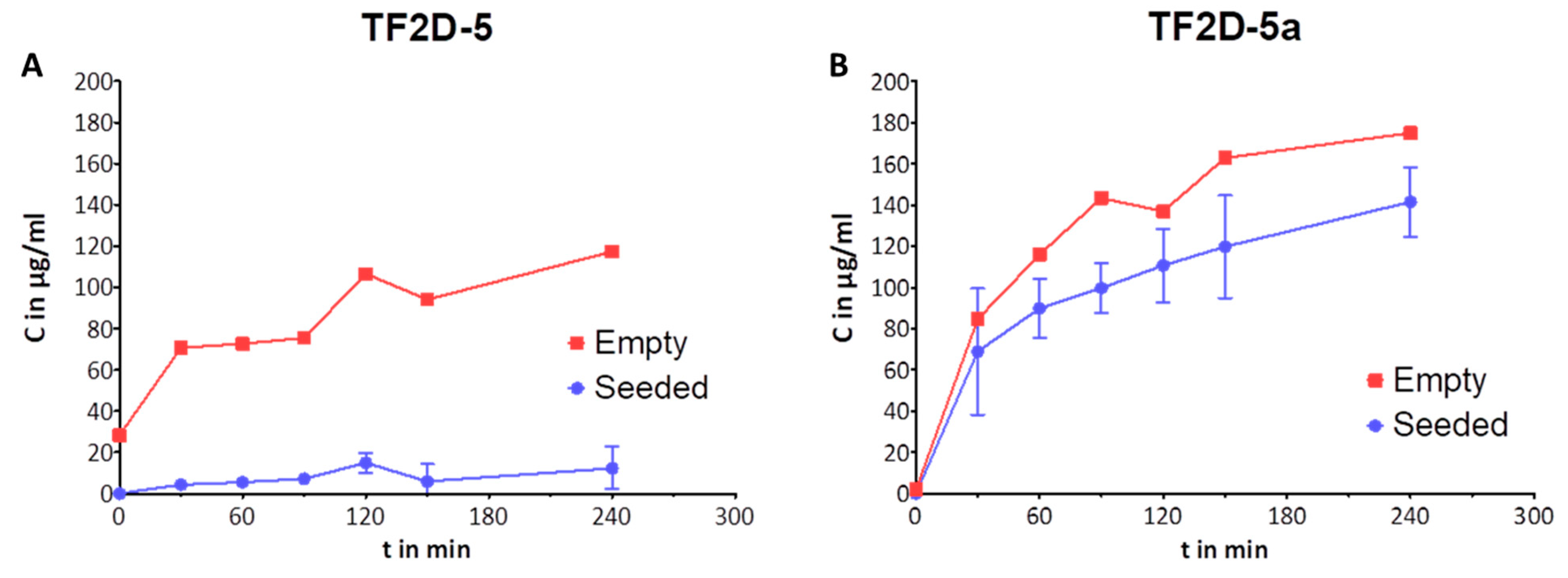
4.20. Blood–Brain Barrier (BBB) Permeability Assay
The preparation of the blood–brain barrier (BBB) model was performed according to Liu et al. [
35], Niego et al. [
36], and Pardridge et al. [
37]. Brain endothelial cells from rats and astroglial cells were used to cultivate the different sides of the porous membrane.
Brain endothelial cells from rats (RBMVEC, Cell Applications Inc., R840-05a, San Diego, CA, USA) were cultivated in Complete Classic medium (Cell Systems, Kirkland, WA, USA, 4Z0-500), and rat astroglial cells (CTX TNA2, ATCC, CRL-2006) were cultivated in Complete Classic medium (Cell Systems, Kirkland, WA, USA, 4Z0-500). The cells were allowed to grow in a humidified incubator with 5% CO2 at 37 °C and a maximum of 20 passages. The cells were passaged every 3 to 5 days, according to their confluence.
In order to set up the in vitro BBB model, the astroglial cells were seeded at a density of 600.000 cells/cm² on the outer (basolateral, “brain”) side of an insert with a porous membrane (Corning Transwell polycarbonate membrane cell culture inserts, CLS3415, 3 µM pore size) and allowed to grow/adhere overnight in the Complete Classic medium. The next day, brain endothelial cells were seeded at a density of 600.000 cells/cm² on the inner side (apical, “blood”) of the corresponding membrane. The seeded inserts were then cultivated to grow to full confluence over a period of 5 to 7 days, with the medium renewed daily. On the day of the assay, the medium was swapped out for Complete Serum-Free medium (Cell Systems, SF-4Z0-500), and the D-peptides TF2D-5 and TF2D-5a were added at a final concentration of 1 mg/mL to the inner chamber. Prior to this, TEER (transendothelial electrical resistance values) were measured in each well to ensure the integrity of the in vitro BBB. At 0, 30, 60, 90, 120, 150, 180, and 240 min, 50 µL of the medium out of the outer chamber was taken and analyzed by HPLC. This 50 µL would then be replaced by the same amount of fresh serum-free medium to keep the fluid levels equal across both chambers. This slight dilution was factored into the following calculations. Furthermore, a dilution series of each reagent was analyzed alongside the samples as a calibration to quantify the analysis. Each substance and time point was tested in triplicate plus a negative control, in which the same set-up was used without cells. The P
app values were calculated as follows (higher is better):
V = Volume of basolateral chamber (mL);
A = Membrane area (cm2);
C = Concentration (µg/µL);
Dc = Measured concentration in basolateral chamber (µg/mL);
Dt = sampling time (s).
4.21. Gel Electrophoresis
Each replicate of the screening via ThT assay was collected and pooled. The samples were centrifuged at 4 °C at 11,000× g for 60 min. The supernatant and pellet were subsequently separated, and the pellet was resuspended in the same volume. The samples were mixed 1:4 with 4× Lämmli and heated to 95 °C. Afterwards, the samples were loaded onto a 15% gel and run at 45 mA per gel for 45 min. Colorization of the gels was achieved by incubation with Coomassie brilliant blue for 30 min. The gels were afterwards decolored by heating them in water with a microwave and subsequent incubation in the heated water with paper tissues. The gels were imaged subsequently on a Gel Doc XR+ Gel Documentation System (Bio-Rad Laboratories, Hercules, CA, USA).
4.22. Cell Assay for Tau Aggregation
A construct encoding the four microtubule binding repeats of the human microtubule-associated protein tau (amino acids 243 to 375, corresponding to 2N4R tau), with the familial mutations P301L and V337M was fused via an 18-amino-acid flexible linker (EFCSRRYRGPGIHRSPTA) to YFP at the C-terminus of tau and introduced into the pMK–RQ expression vector (GeneArt; Thermo Fisher Scientific, Waltham, MA, USA). The tauK18(LM)-YFP construct was subcloned into the pIRESpuro3 vector (Clontech; Takara Bio, JPN) using EcoRI (5′) and NotI (3′) restriction sites. HEK293T cells (American Type Culture Collection) were cultured in high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM; Sigma-Aldrich, St. Louis, MI, USA), supplemented with 10% (v/v) fetal calf serum (Sigma-Aldrich, USA) and 50 units/mL penicillin, as well as 50 μg/mL streptomycin (Sigma-Aldrich, USA). The cells were cultured in a humidified atmosphere of 5% CO2 at 37 °C. The cells plated in DMEM were transfected using lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, USA). The stable cells were selected in DMEM containing 1 μg/mL puromycin (EMD Millipore, Burlington, MA, USA). Monoclonal lines were generated by fluorescence-activated cell sorting of a polyclonal cell population in 96-well plates using a MoFlo XDP cell sorter (Beckman Coulter, USA). Finally, the clonal cell line H7 was selected from among 16 clonal cell lines and is referred to as tauK18(LM)–YFP cells. The compounds were incubated with 1.5% (v/v) lipofectamine 2000 in OptiMEM for 2 h at room temperature. The tauK18(LM)–YFP cells were plated in a 384-well plate with poly-D-lysine coating (Greiner, AT) at a density of 1.000 cells per well with 0.1 μg/mL Hoechst 33342 (Thermo Fisher Scientific, USA), and the previously prepared transfection mix was added directly to the cells in the well. To seed the cellular aggregation of tau in the tauK18(LM)–YFP cells, 25 nM of sonicated tau fibrils (seeds) was incubated with 1.5% (v/v) lipofectamine in OptiMEM for 2 h at room temperature and added to each well 3 h after the first transfection. The plate was then incubated in a humidified atmosphere of 5% CO2 at 37 °C. On day 3, the cells were imaged with an IN Cell Analyzer 6500HS System (Cytiva, SE) using the blue and green fluorescence channels and analyzed using IN Carta Image Analysis Software (Cytiva, SE) after an algorithm was established to identify intracellular aggregates in the living cells. Statistical analysis was performed using one-way ANOVA, followed by Dunnett’s multiple comparisons test (GraphPad Prism 9, GraphPad Software, USA).

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}













