1. Introduction
Prostate cancer (PCa) is one of the most common cancers in men worldwide; more than one million new cases are diagnosed annually [
1]. Prostate cancer is characterized by high clinical heterogeneity, which is manifested by a different propensity for recurrence of the disease, as well as the onset of progression after surgical treatment. The clinical heterogeneity of PCa, in turn, is due to molecular heterogeneity—in the tumors of patients, there are disorders that are accompanied by various changes in signaling pathways and metabolic processes [
2,
3]. At the same time, for the development of aggressive forms of PCa, the occurrence of only a few driver disorders is sufficient [
4,
5,
6].
Despite a wide range of therapeutic approaches, the problem of choosing the tactics for treating a patient after radical prostatectomy is quite acute, especially for the category of patients with locally advanced PCa (LAPC). In addition to invasion of the prostatic capsule, metastasis to regional lymph nodes is often observed in LAPC. In this case, as a rule, the choice is between the immediate start of adjuvant hormone therapy or active monitoring of the level of prostate-specific antigen (PSA) in the blood.
The choice of a therapeutic concept is based on determining the prognosis for the patient, which, in turn, is based on such clinical and pathological parameters as the PSA level, the size of the primary tumor (stage T), and the Gleason score. Indeed, it is worth noting the emergence of an improved classification of cancer cell differentiation based on the Gleason score, a system developed by the ISUP (International Society of Uropathologists) [
7]. The division into groups occurs between 1 and 5, depending on the Gleason score, the method providing the most accurate stratification of tumors.
However, these indicators are not informative enough to identify a group of patients with tumors that have high potential for progression, the development of an aggressive phenotype, and metastasis [
8,
9]. Thus, reliable prognostic markers are needed, the use of which, in combination with clinicopathological parameters, will help to reliably identify an aggressive tumor phenotype and thus choose the best therapeutic approach for the patient. One of the promising approaches in the search for prognostic markers may be the analysis of the transcriptomic data of tumors [
10,
11,
12]. Currently, several prognostic expression panels of PCa markers based on tissue analysis have been identified, such as Decipher and Oncotype DX Genomic Prostate Score (GPS).
The Decipher test measures the RNA expression levels of 22 different genes, selected based on unique differential expression patterns in early metastasis [
13]. The Decipher test has shown high discrimination in the prediction of clinical metastases (AUC = 0.75–0.83) and mortality from PCa (AUC = 0.78) in validation studies, significantly exceeding the available clinicopathological characteristics (AUC = 0.69) [
14]. The Oncotype DX panel is a tissue biopsy-based genomic assay that measures the mRNA expression of 17 genes responsible for tumor cell growth and survival [
15]. At the development stage of this test, we used the results of quantitative PCR, based on the archival material of tissue samples obtained after the surgical treatment of patients with PCa in low- and intermediate-risk groups. Based on validation studies, the Oncotype DX panel has been shown to be highly correlated with the biochemical recurrence of PCa and a poor prognosis, highlighting its predictive value for patients with PCa in low- and intermediate-risk groups.
Previously, we conducted a study that included miRNA-Seq analysis of 44 PCa tissue samples with and without lymphatic dissemination (N1 group = 20 samples; N0 group = 24 samples), as a result of which we identified a number of miRNAs, the expression of which could potentially be associated with lymphatic dissemination [
16]. In the present study, we performed RNA-Seq profiling of an expanded sample of 73 PCa tissue samples from Russian patients and validated the obtained results by quantitative PCR (qPCR). This study also included an independent sample of 37 PCa tissue samples based on archival material. The obtained qPCR data for both PCa samples were used to analyze the predictive value of models based on combinations of candidate markers and clinicopathological parameters, using various machine learning algorithms (Logistic Regression (LR; scikit-learn ver.1.1.3), Light Gradient Boosting Machine (LGBM; ver.3.3.2), CatBoost (ver.1.0.6), Random Forest (scikit-learn ver.1.1.3) and XGBoost (ver.1.6.2)). The results of the study can be used to develop an expression panel for assessing the metastatic potential of high-risk PCa when choosing a therapeutic concept for a patient.
3. Discussion
In the present study, a comprehensive transcriptomic analysis of LAPC tissue samples in a sample of Russian patients was carried out. This procedure aimed at identifying promising candidate markers of early metastasis. Based on the obtained transcriptomic profile, the biological pathways associated with lymphatic dissemination were first considered. The results of the biological pathways analysis with high enrichment predominantly demonstrated the activation of translational processes in tumor cells. This translational activity is likely to be closely related to increased metabolism, directed at obtaining energy from various sources to promote the epithelial–mesenchymal transition of tumor cells and metastasis.
Among all of the pathways identified, it is worth noting the decreased activation of the “Fatty Acid Metabolism” pathway. It is known that malignant transformation of a tumor depends on complex intercellular interactions, supported by a wide network of physical and chemical mediators that make up the tumor microenvironment [
17]. Recently, various researchers have emphasized the key role of adipose tissue as a key component in the progression of solid tumors [
18]. The prostate gland is surrounded by periprostatic adipose tissue, and extraprostatic expansion to adipose tissue is a widely recognized poor prognostic factor in PCa and an important predictor of recurrence after treatment [
19]. The positive relationship between obesity and aggressive PCa, determined by an increase in local and distant spread, also supports the role of adipose tissue in tumor progression [
20].
Interactions between adipocytes and tumor cells in the tumor microenvironment can create a metabolic symbiosis, leading to growth and metastasis. In combination with glucose, fatty acids are also vital for the synthesis of membrane lipids in tumor cells, energy production, and the synthesis of carcinogenesis-associated lipid-signaling molecules such as lysophosphatidic acids [
21,
22,
23]. As a result, tumor cells activate de novo fatty acid synthesis, and elevated levels of fatty acid synthase are negatively correlated with prognosis [
24]. Thus, in addition to synthesis, tumor cells can also use exogenous fatty acids as a source of nutrition. Thus, our results also highlight the importance of lipid metabolism in the progression of PCa.
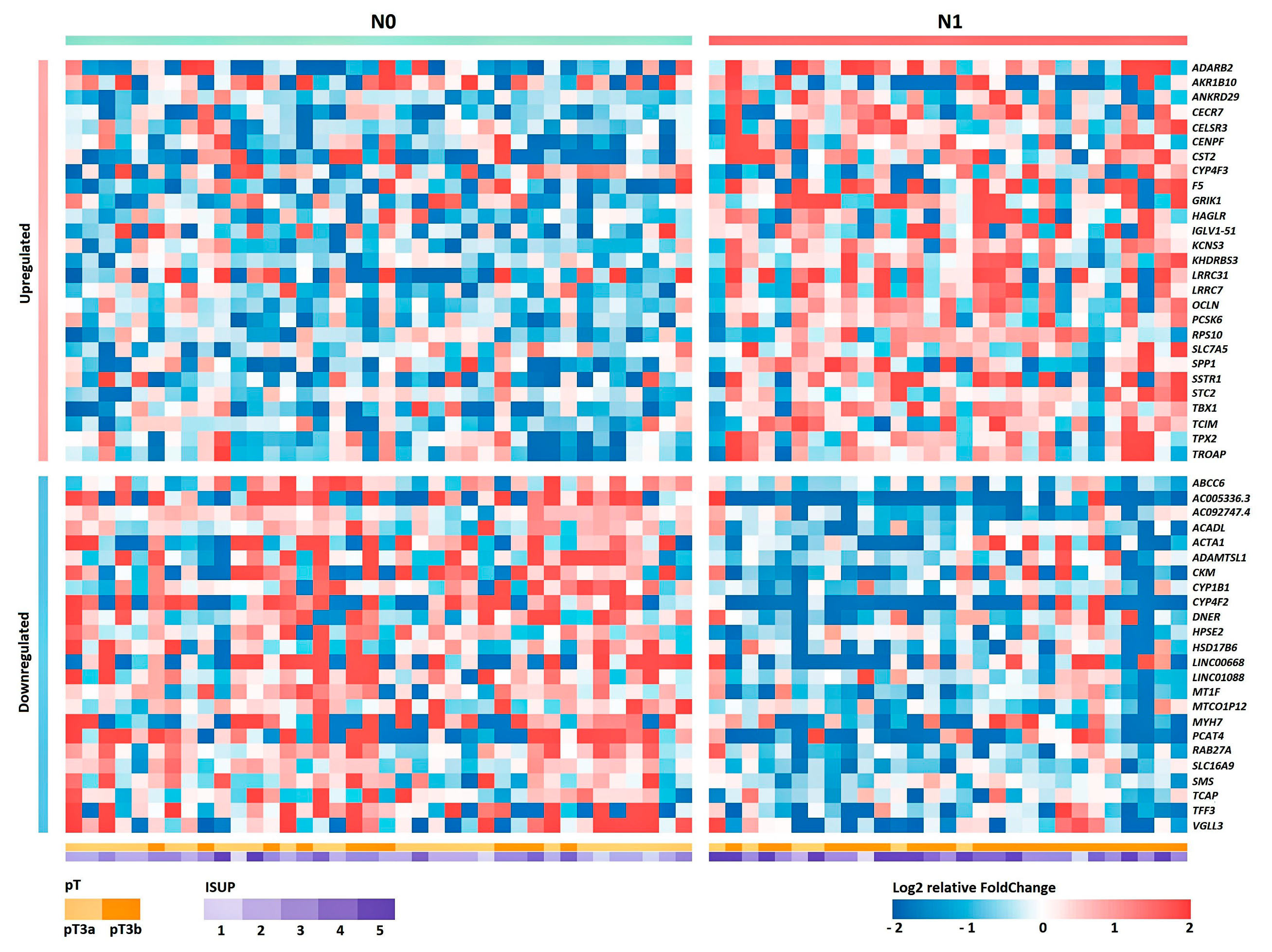
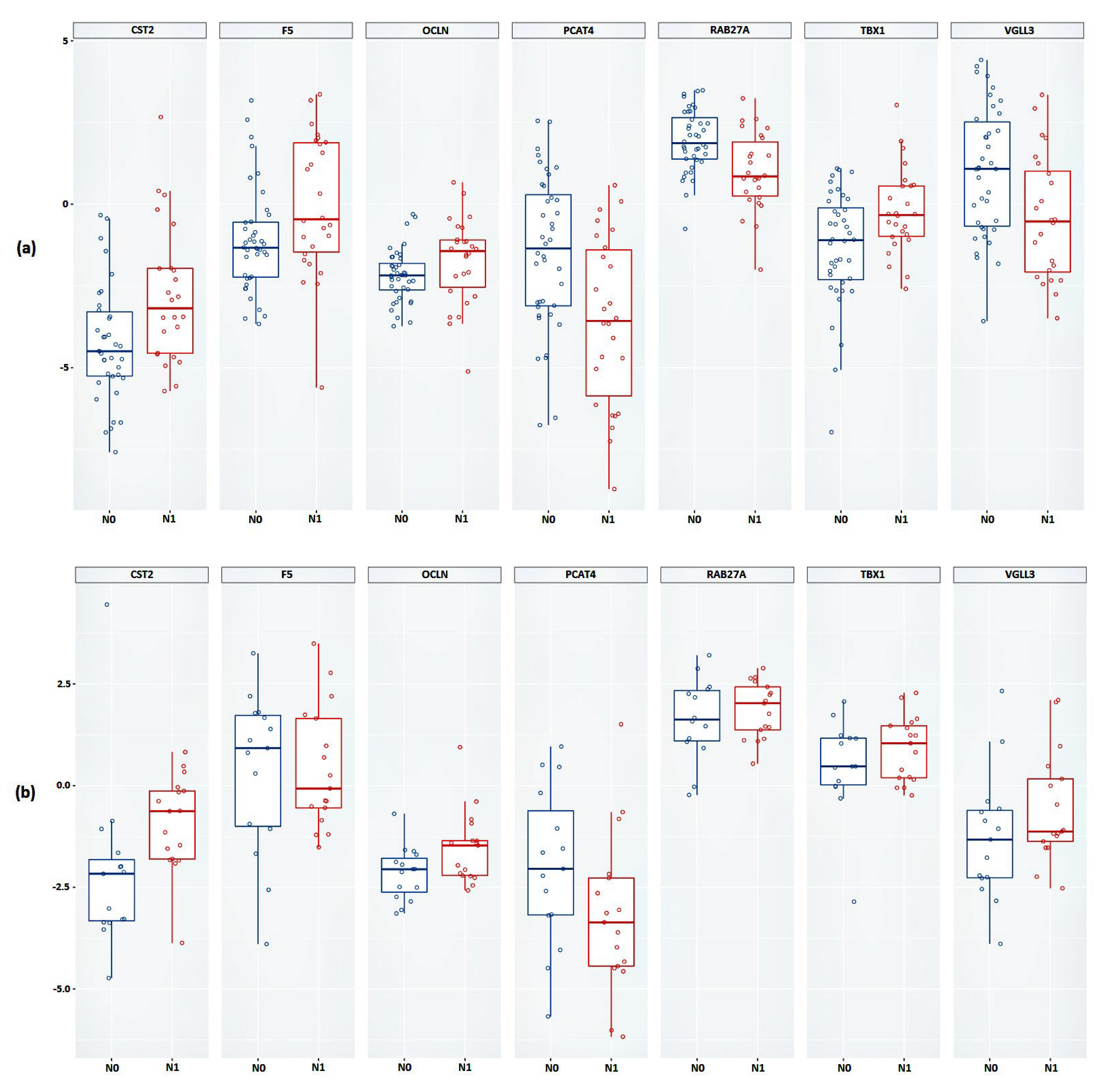
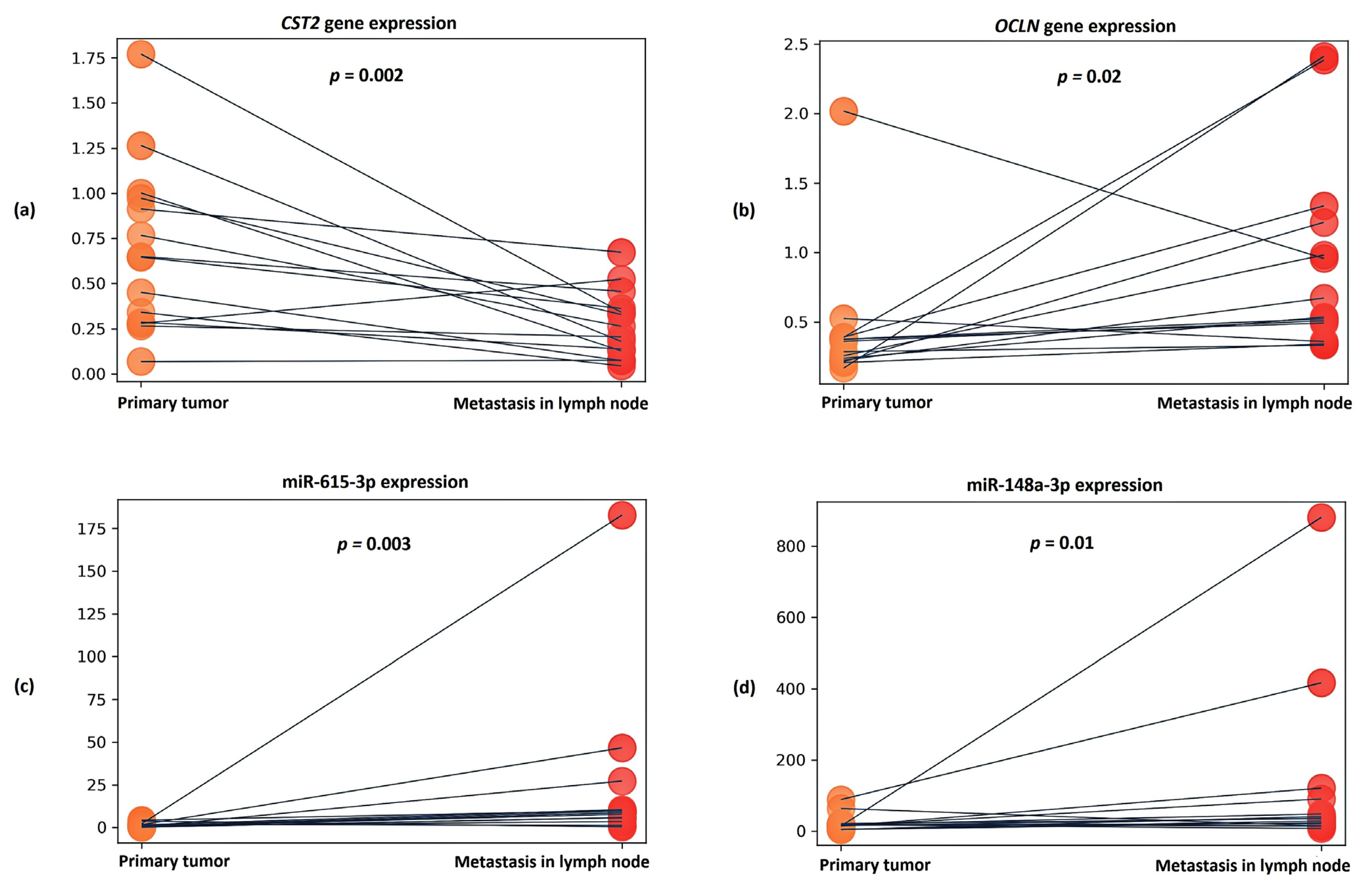
Furthermore, based on the obtained transcriptomic profile, we searched for promising candidate markers based on gene expression. As a result of the validation, we confirmed the statistical significance of the expression of the PCAT4, OCLN and CST2 genes in lymphatic dissemination in an independent sample.
The PCAT4 (prostate cancer associated transcript 4; PCAN1; GDEP) gene is characterized by high tissue specificity for prostate tissue, but there are no published data on the biological function of this gene. According to the data obtained, we saw a significant decrease in the expression of this gene in the N1 group.
The
OCLN (occludin) gene encodes the occludin protein, which belongs to tight junction proteins. Tight junctions are one of the key components in tumor metastasis, as tumor cells must pass through a series of barriers to successfully metastasize to secondary lesions [
25]. OCLN is widely expressed in tissues and cells with tight junctions and is a membrane protein with four trans-membrane domains [
26]. According to the literature, high expression of OCLN has been found in lung cancer, and when
OCLN was knocked down in cell lines of lung cancer (A549, NCL-H1650, SPC-A1, HCC827, NCI-H1299, and MSTO-211H), inhibition of cell proliferation was observed in vitro and in vivo. In addition,
OCLN knockdown promoted apoptosis of lung cancer cell lines and reduced their ability to invade, on the basis of which the role of
OCLN as a tumor promoter and prometastatic factor was shown for the first time [
27]. Based on our data, for the first time, an association between increased expression of the
OCLN gene and the presence of lymphatic dissemination in LAPC was shown.
The
CST2 (cystatin SA) gene is a member of the cystatin family. Based on several studies, it has been shown that high expression of this gene is associated with the development of carcinogenesis. In breast cancer, increased expression of the
CST2 gene has been shown to be associated with tumor cell proliferation, movement, and adhesion [
28]. Based on our data, we observed an association between increased expression of the
CST2 gene and lymphatic dissemination in LAPC.
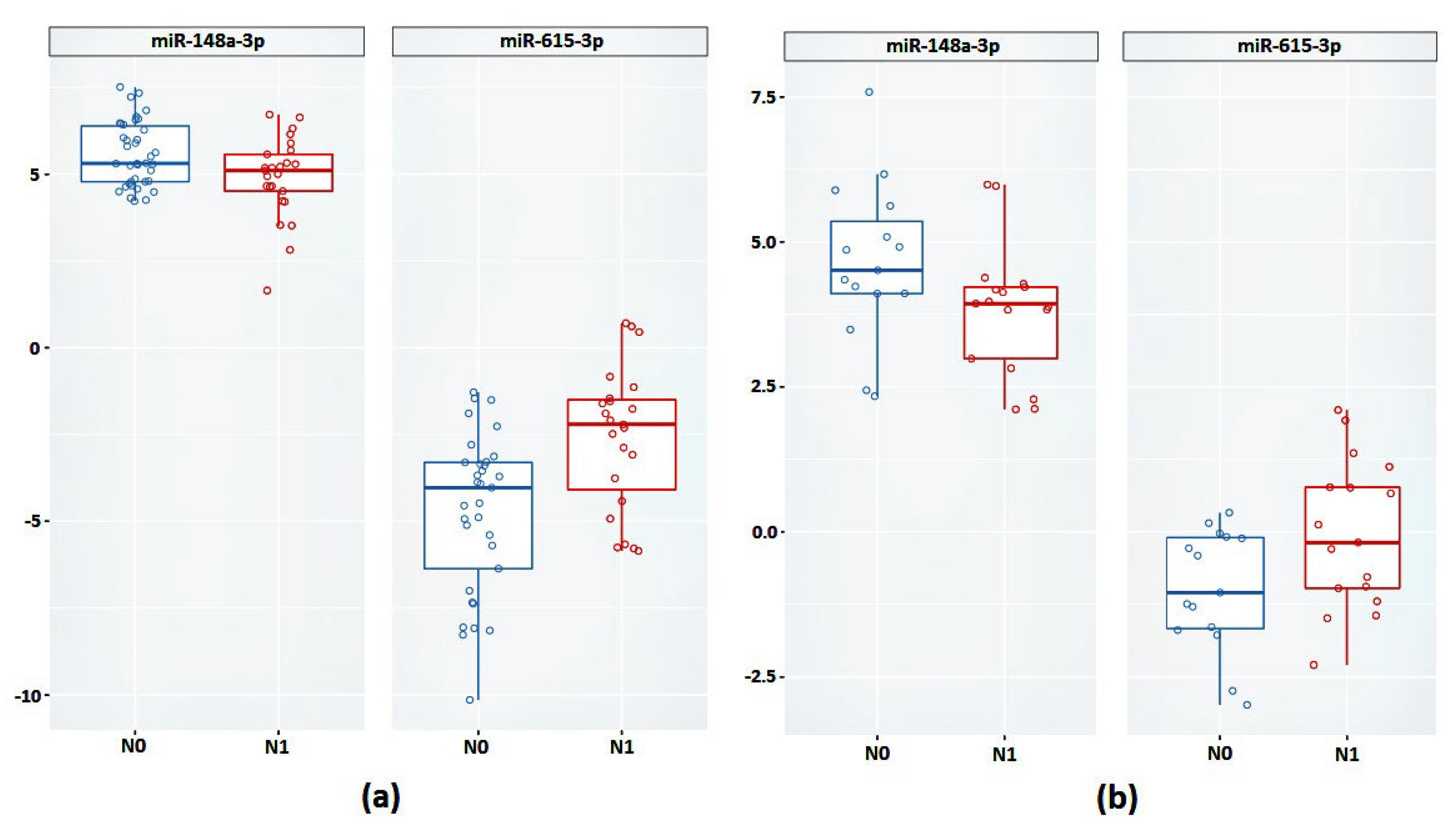
In addition, we also validated previously identified promising markers based on miRNA expression, specifically miR-615-3p and miR-148a-3p, which also confirmed their association with lymphatic dissemination in the case of an independent sample of LAPC.
Aberrant expression of miR-615-3p has been described in many forms of cancer, including PCa, where overexpression of miR-615-3p has been observed in the most aggressive forms [
29,
30,
31]. Experiments on cell lines of various types of cancer have shown that miR-615-3p overexpression supports cell proliferation and migration [
29,
30]. Functional studies performed on PCa cell lines have shown that miR-615-3p promotes proliferation, apoptosis, and migration of the PC3M cell line in vitro, indicating that miR-615-3p is an important oncogenic microRNA in PCa [
31].
miR-148a-3p is one of the most highly expressed miRNAs in PCa tissues, as well as the most dominant in PCa metastasis [
32]. High-grade tumors have been shown to exhibit reduced levels of miR-148a-3 expression. miR-148a expression has also been shown to be downregulated in docetaxel-resistant variants of PCa cell lines, including PC-3 and DU145, and downregulation of miR-148a has been observed in PCa with a risk of biochemical recurrence [
33].
Evaluation of the expression of these candidate markers in samples of affected lymph nodes showed a further linear increase in the expression of the OCLN gene and miR-615-3p, as well as increased expression of miR-148a-3p. It can be assumed that the increased expression of these markers is not only associated with lymphatic dissemination in LAPC, but also supports the formation of secondary tumor foci.
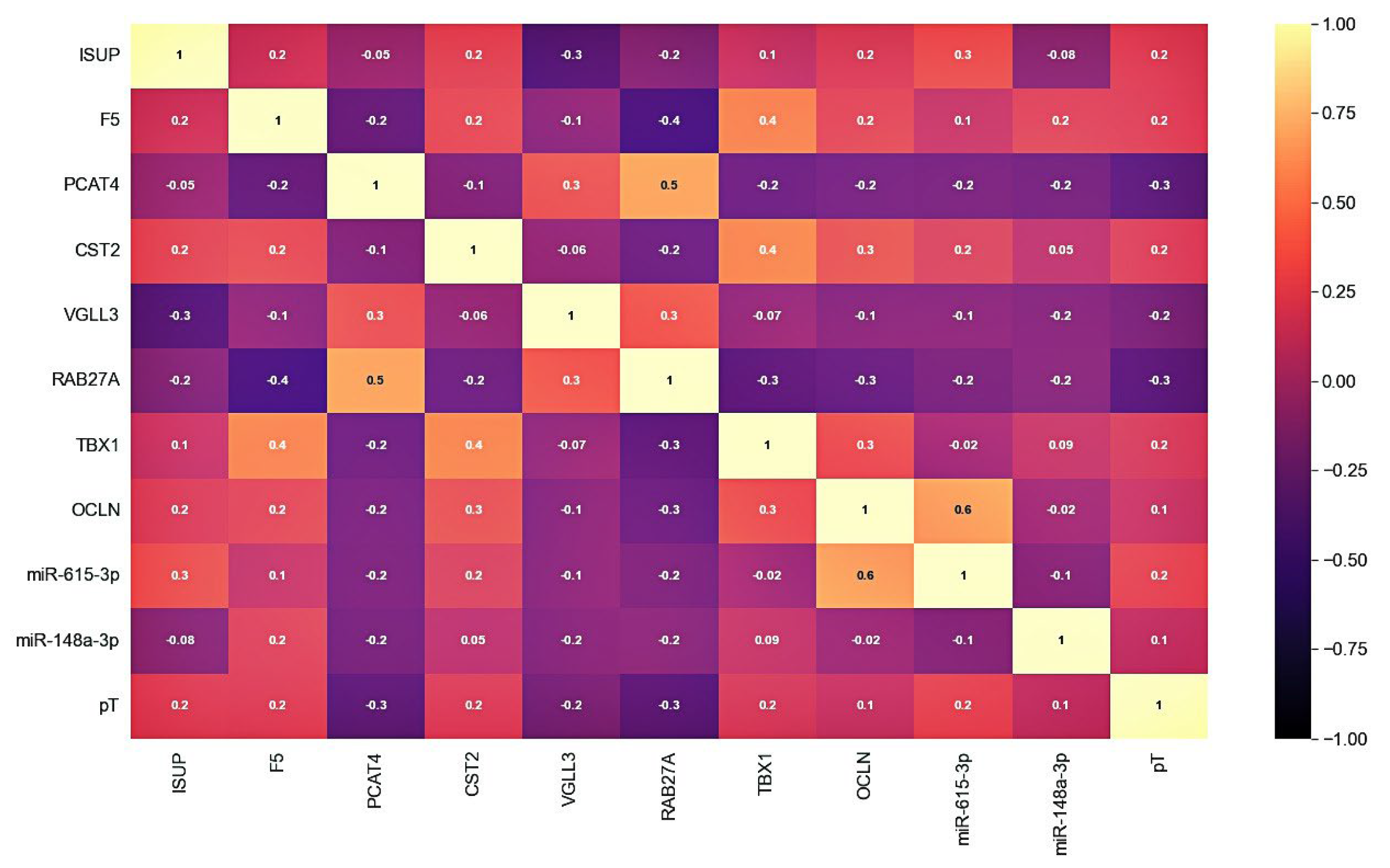
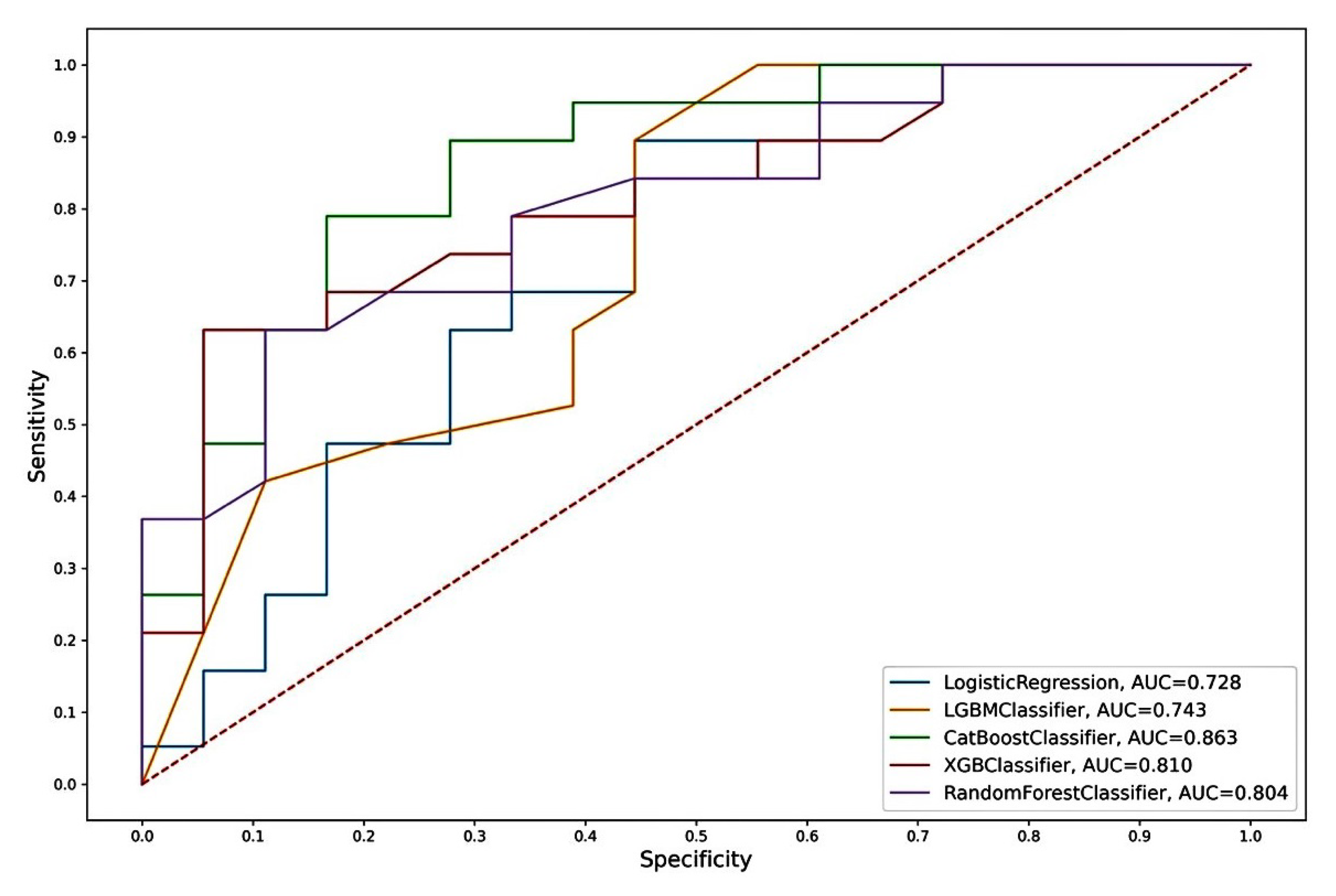
We assessed the prognostic significance of PCAT4, OCLN, CST2, miR-615-3p, and miR-148a-3p for various combinations of predictors, both with each other and with such clinicopathological parameters as ISUP and pT. We considered the main metrics for models based on five machine learning algorithms for a classification problem.
The “CST2 + OCLN + pT” model, based on the CatBoost algorithm, had the highest metrics. In addition to the highest AUC (0.863), this model also had the highest sensitivity (SE = 0.83), specificity (SP = 0.79), and accuracy (AC = 0.81) of an independent sample of archival material. The parameters of this model based on other algorithms also differed in the highest rates (AUC = 0.73–0.81; SE = 0.56–0.78; SP = 0.68–0.84; AC = 0.62–0.73). Thus, the model we identified based on the predictor combination of «CST2 + OCLN + pT» had the highest prognostic potential for determining lymphatic dissemination in LAPC, both based on freshly frozen surgical material and, in the case of archival material, based on FFPE blocks.

 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}