
Genetic Diversity Analysis and Core Germplasm Collection Construction of Radish Cultivars Based on Structure Variation Markers


Abstract
:1. Introduction
2. Results
2.1. Construction of DNA Fingerprint Profiles by Structure Variation Markers
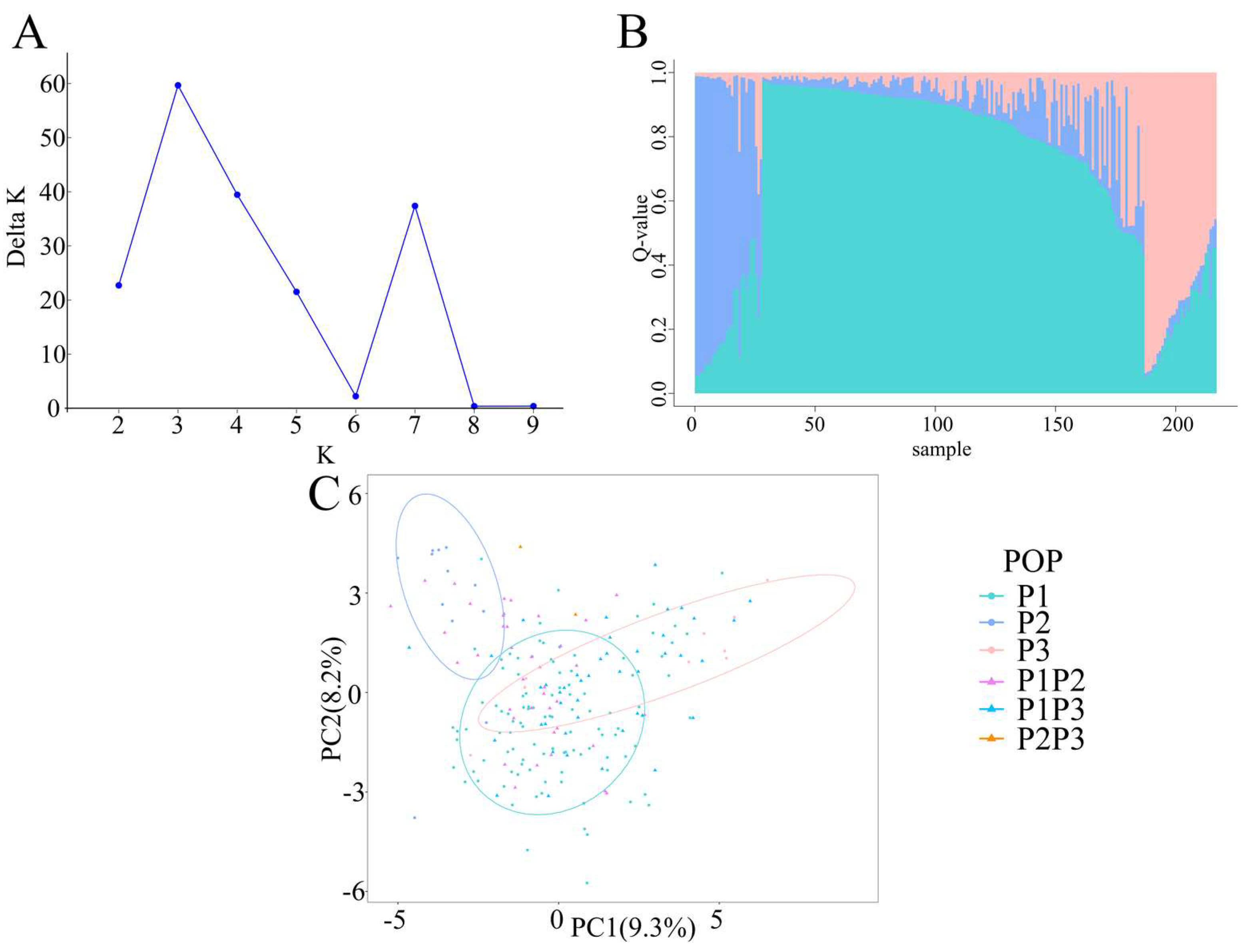
2.2. Genetic Structure of Radish Accessions
2.3. Genetic Diversity of 217 Radish Accessions
2.4. Geographical Distribution of Radish Germplasms
2.5. Phenotypic Analysis of Subpopulations
2.6. Collection of Core Germplasms
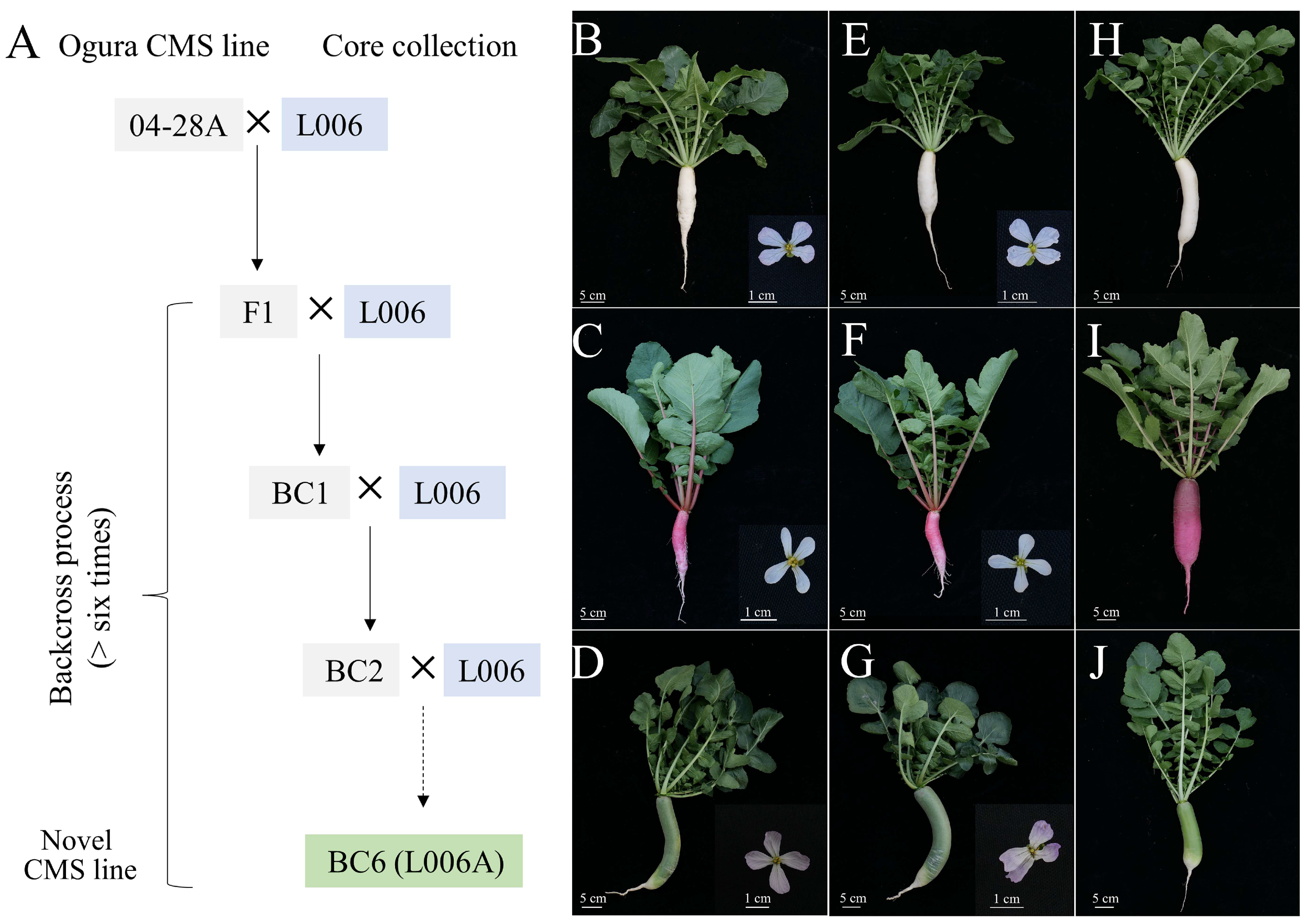
2.7. Utilization of Core Germplasms in Radish
3. Discussion
3.1. Genetic Diversity in Radish Accessions
3.2. Powerful Application of SV Markers
3.3. Application of Core Germplasms to Breed Improvement
4. Materials and Methods
4.1. Plant Materials
4.2. Phenotypic Analysis
4.3. Development of Structure Variation Markers
4.4. PCR Amplification
4.5. Principal Component Analysis
4.6. Genetic Structure Analysis
4.7. Screening of Core Germplasm and Their Utilization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nishio, T. Economic and academic importance of radish. In The Radish Genome; Nishio, T., Kitashiba, H., Eds.; Compendium of Plant Genomes; Springer International Publishing: Cham, Switzerland, 2017; pp. 1–10. [Google Scholar]
- Sahli, H.F.; Conner, J.K.; Shaw, F.H.; Howe, S.; Lale, A. Adaptive differentiation of quantitative traits in the globally distributed weed, wild radish (Raphanus raphanistrum). Genetics 2008, 180, 945–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitashiba, H.; Akanuma, T.; Hirakawa, H.; Fukino, N.; Shirasawa, K.; Kobayashi, H. Identification of genome-wide single-nucleotide polymorphisms among geographically diverse radish accessions. DNA Res. 2020, 27, 1–8. [Google Scholar]
- Campbell, L.G.; Snow, A.A.; Sweeney, P.M. When divergent life histories hybridize: Insights into adaptive life-history traits in an annual weed. New Phytol. 2009, 184, 806–818. [Google Scholar] [CrossRef]
- Yamagishi, H.; Terachi, T. Molecular and biological studies on male-sterile cytoplasm in the Cruciferae. IV. Ogura-type cytoplasm found in the wild radish, Raphanus raphanistrum. Theor. Appl. Genet. 1996, 93, 325–332. [Google Scholar] [CrossRef]
- Jin, L.; Lu, Y.; Xiao, P.; Sun, M.; Corke, H.; Bao, J. Genetic diversity and population structure of a diverse set of rice germplasm for association mapping. Theor. Appl. Genet. 2010, 121, 475–487. [Google Scholar] [CrossRef]
- Mondini, L.; Noorani, A.; Pagnotta, M. Assessing plant genetic diversity by molecular tools. Diversity 2009, 1, 19–35. [Google Scholar] [CrossRef] [Green Version]
- Sultan, S.E. Phenotypic plasticity for plant development, function and life history. Trends Plant Sci. 2000, 5, 537–542. [Google Scholar] [CrossRef]
- Benor, S.; Zhang, M.; Wang, Z.; Zhang, H. Assessment of genetic variation in tomato (Solanum lycopersicum L.) inbred lines using SSR molecular markers. J. Genet. Genom. 2008, 35, 373–379. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, H.; Li, L.; Lan, H.; Ren, Z.; Liu, D.; Wu, L.; Liu, H.; Jaqueth, J.; Li, B.; et al. Characterizing the population structure and genetic diversity of maize breeding germplasm in Southwest China using genome-wide SNP markers. BMC Genom. 2016, 17, 697. [Google Scholar] [CrossRef] [Green Version]
- Dar, A.A.; Mahajan, R.; Lay, P.; Sharma, S. Genetic diversity and population structure of Cucumis sativus L. by using SSR markers. 3 Biotech 2017, 7, 307. [Google Scholar] [CrossRef]
- Huang, J.; Liu, Y.; Han, F.; Fang, Z.; Yang, L.; Zhuang, M.; Zhang, Y.; Lv, H.; Wang, Y.; Ji, J.; et al. Genetic diversity and population structure analysis of 161 broccoli cultivars based on SNP markers. Hortic. Plant J. 2021, 7, 423–433. [Google Scholar] [CrossRef]
- Garrido-Cardenas, J.A.; Mesa-Valle, C.; Manzano-Agugliaro, F. Trends in plant research using molecular markers. Planta 2018, 247, 543–557. [Google Scholar] [CrossRef]
- Adal, A.M.; Demissie, Z.A.; Mahmoud, S.S. Identification, validation and cross-species transferability of novel Lavandula EST-SSRs. Planta 2015, 241, 987–1004. [Google Scholar] [CrossRef]
- King, D.J.; Noss, R.R. Toxicity of polyacrylamide and acrylamide monomer. Rev. Environ. Health 1989, 8, 3–16. [Google Scholar] [CrossRef]
- Thomson, M.J. High-Throughput SNP genotyping to accelerate crop improvement. Plant Breed. Biotechnol. 2014, 2, 195–212. [Google Scholar] [CrossRef]
- Yan, C.; Cui, L.; Ren, Z.; Huang, Y.; Jiao, Z.; Zhu, F.; Zhang, Z.; Gan, C.; Deng, X.; Zhengming, Q. Establishment of molecular identify card for radish germplasm resources based on genome structural variation. China Veg. 2022, 1, 49–57. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Wang, R.; Yu, Y.; Zhao, J.; Shi, Y.; Song, Y.; Wang, T.; Li, Y. Population structure and linkage disequilibrium of a mini core set of maize inbred lines in China. Theor. Appl. Genet. 2008, 117, 1141–1153. [Google Scholar] [CrossRef] [PubMed]
- Beukelaer, H.D.; Smýkal, P.; Davenport, G.F.; Fack, V. Core Hunter II: Fast core subset selection based on multiple genetic diversity measures using Mixed Replica search. BMC Bioinform. 2012, 13, 312. [Google Scholar] [CrossRef] [Green Version]
- Thachuk, C.; Crossa, J.; Franco, J.; Dreisigacker, S.; Warburton, M.; Davenport, G.F. Core Hunter: An algorithm for sampling genetic resources based on multiple genetic measures. BMC Bioinform. 2009, 10, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Liu, T.; Wang, J.; Wang, P.; Qiu, Y.; Zhao, W.; Pang, S.; Li, X.; Wang, H.; Song, J.; et al. Pan-genome of Raphanus highlights genetic variation and introgression among domesticated, wild, and weedy radishes. Mol. Plant 2021, 14, 2032–2055. [Google Scholar] [CrossRef] [PubMed]
- Vejchasarn, P.; Shearman, J.R.; Chaiprom, U.; Phansenee, Y.; Suthanthangjai, A.; Jairin, J.; Chamarerk, V.; Tulyananda, T.; Amornbunchornvej, C. Population structure of nation-wide rice in Thailand. Rice 2021, 14, 88. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yu, H.; Li, Z.; Liu, X.; Fang, Z.; Liu, Y.; Yang, L.; Zhuang, M.; Lv, H.; Zhang, Y. Heterotic group classification of 63 inbred lines and hybrid purity identification by using SSR markers in winter cabbage (Brassica oleracea L. var. capitata). Hortic. Plant J. 2018, 4, 158–164. [Google Scholar] [CrossRef]
- Lv, J.; Qi, J.; Shi, Q.; Shen, D.; Zhang, S.; Shao, G.; Li, H.; Sun, Z.; Weng, Y.; Shang, Y.; et al. Genetic diversity and population structure of cucumber (Cucumis sativus L.). PLoS ONE 2012, 7, e46919. [Google Scholar] [CrossRef]
- Wang, W.; Chen, B.; Zhang, L.; Yan, J.; Lu, Y.; Zhang, X.; Jiang, Y.; Wu, T.; van Peer, A.F.; Li, S.; et al. Structural variation (SV) markers in the basidiomycete Volvariella volvacea and their application in theconstruction of a genetic map. Int. J. Mol. Sci. 2015, 16, 16669–16682. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Yang, J.; Chen, B.; Zhang, X.; Zhang, J.; Yang, K.; Geng, S.; Wen, C. Target sequencing reveals genetic diversity, population structure, core-SNP markers, and fruit shape-associated loci in pepper varieties. BMC Plant Biol. 2019, 19, 578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhang, J.; Han, R.; Zhang, F.; Mao, A.; Luo, J.; Dong, B.; Liu, H.; Tang, H.; Zhang, J.; et al. Target SSR-Seq: A novel SSR genotyping technology associate with perfect SSRs in genetic analysis of cucumber varieties. Front. Plant Sci. 2019, 10, 531. [Google Scholar] [CrossRef] [Green Version]
- Rauf, S.; Al-Khayri, J.M.; Zaharieva, M.; Monneveux, P.; Khalil, F. Breeding strategies to enhance drought tolerance in crops. In Advances in Plant Breeding Strategies: Agronomic, Abiotic and Biotic Stress Traits; Al-Khayri, J.M., Jain, S.M., Johnson, D.V., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 397–445. [Google Scholar]
- Chai, A.L.; Xie, X.W.; Shi, Y.X.; Li, B.J. Research status of clubroot (Plasmodiophora brassicae) on cruciferous crops in China. Can. J. Plant Pathol. 2014, 36, 142–153. [Google Scholar] [CrossRef]
- Gan, C.; Yan, C.; Pang, W.; Cui, L.; Fu, P.; Yu, X.; Qiu, Z.; Zhu, M.; Piao, Z.; Deng, X. Identification of novel locus RsCr6 related to clubroot resistance in radish (Raphanus sativus L.). Front. Plant Sci. 2022, 13, 866211. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Webb, D.M.; Knapp, S.J. DNA extraction from a previously recalcitrant plant genus. Plant Mol. Biol. Report. 1990, 8, 180–185. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Tian, M.; An, G.; Zhang, W.; Chen, J.; Yan, C. Rapid identification of the purple stem (Ps) gene of Chinese kale (Brassica oleracea var. alboglabra) in a segregation distortion population by bulked segregant analysis and RNA sequencing. Mol. Breed. 2017, 37, 153. [Google Scholar] [CrossRef]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R package for multivariate analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.Q.; Ge, H.M.; Sun, X.; Yang, A.G.; Zhang, Z.F.; Ren, M. Data Formater, a software for SSR data formatting to develop population genetics analysis. Mol. Plant Breed. 2016, 14, 265–270. [Google Scholar]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [Green Version]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2011, 4, 359–361. [Google Scholar] [CrossRef]
- Peakall, R.O.D.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Slatkin, M.; Barton, N.H. A comparison of three indirect methods for estimating average levels of gene flow. Evolution 1989, 43, 1349–1368. [Google Scholar] [CrossRef]
- Gouesnard, B.; Bataillon, T.M.; Decoux, G.; Rozale, C.; Schoen, D.J.; David, J.L. MSTRAT: An algorithm for building germplasm core collections by maximizing allelic or phenotypic richness. J. Hered. 2001, 92, 93–94. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | Chromosome | Position | Na | Ne | Ho | He | I | PIC |
|---|---|---|---|---|---|---|---|---|
| HY002 | Rs1 | 10,851,495 | 2 | 1.6030 | 0.1860 | 0.3770 | 0.5636 | 0.3807 |
| HY004 | Rs1 | 16,735,725 | 2 | 1.9348 | 0.2271 | 0.4843 | 0.6762 | 0.4839 |
| HY009 | Rs2 | 23,055,813 | 2 | 1.9258 | 0.2243 | 0.4819 | 0.6738 | 0.4807 |
| HY012 | Rs2 | 30,122,826 | 2 | 1.4973 | 0.1121 | 0.3329 | 0.5143 | 0.3321 |
| HY013 | Rs2 | 33,833,149 | 2 | 1.7481 | 0.2685 | 0.4289 | 0.6192 | 0.4279 |
| HY018 | Rs3 | 6,705,102 | 2 | 1.3301 | 0.0783 | 0.2488 | 0.4142 | 0.2482 |
| HY022 | Rs3 | 31,947,431 | 2 | 1.9832 | 0.0783 | 0.4969 | 0.6889 | 0.4958 |
| HY024 | Rs4 | 2,191,687 | 2 | 1.9966 | 0.1567 | 0.5003 | 0.6923 | 0.4991 |
| HY027 | Rs4 | 7,584,415 | 2 | 1.9625 | 0.0415 | 0.4916 | 0.6836 | 0.4904 |
| HY033 | Rs4 | 30,474,763 | 2 | 1.2064 | 0.0876 | 0.1715 | 0.3128 | 0.1711 |
| HY038 | Rs5 | 454,972 | 2 | 1.6381 | 0.1705 | 0.3904 | 0.5782 | 0.3895 |
| HY040 | Rs5 | 7,436,512 | 2 | 1.8864 | 0.2176 | 0.4710 | 0.6627 | 0.4699 |
| HY042 | Rs5 | 21,313,041 | 2 | 1.6647 | 0.1512 | 0.4003 | 0.5888 | 0.3993 |
| HY050 | Rs6 | 27,231 | 2 | 1.8895 | 0.1349 | 0.4718 | 0.6636 | 0.4708 |
| HY054 | Rs6 | 17,841,141 | 2 | 1.9892 | 0.1982 | 0.4984 | 0.6904 | 0.4973 |
| HY055 | Rs6 | 19,819,455 | 2 | 1.2173 | 0.0876 | 0.1789 | 0.3231 | 0.1785 |
| HY060 | Rs6 | 42,550,591 | 2 | 1.6381 | 0.0968 | 0.3904 | 0.5782 | 0.3895 |
| HY062 | Rs6 | 46,581,963 | 2 | 1.9892 | 0.1982 | 0.4984 | 0.6904 | 0.4973 |
| HY068 | Rs7 | 5,086,840 | 2 | 1.4181 | 0.0461 | 0.2955 | 0.4710 | 0.2948 |
| HY073 | Rs7 | 16,823,631 | 2 | 1.9621 | 0.1852 | 0.4915 | 0.6835 | 0.4904 |
| HY076 | Rs8 | 3,031,541 | 2 | 1.3417 | 0.0599 | 0.2553 | 0.4223 | 0.2547 |
| HY077 | Rs8 | 4,362,064 | 2 | 1.9125 | 0.1256 | 0.4782 | 0.6701 | 0.4771 |
| HY088 | Rs9 | 13,982,718 | 2 | 1.6714 | 0.1792 | 0.4026 | 0.5913 | 0.4017 |
| HY092 | Rs9 | 43,709,005 | 2 | 1.6815 | 0.1204 | 0.4062 | 0.5952 | 0.4053 |
| Mean | 2 | 1.7120 | 0.1430 | 0.4018 | 0.5853 | 0.4011 |
| d.f. | SS | Est. Var. | Percentage of Variation | |
|---|---|---|---|---|
| Among Pops | 5 | 255.901 | 1.236 | 7.36% |
| Within Pops | 211 | 3283.076 | 15.560 | 93.64% |
| Total | 216 | 3538.977 | 16.795 | 100% |
| Screening Ratio | Na | Ne | I | Ho | He | uHe |
|---|---|---|---|---|---|---|
| 10% | 1.3958 * | 1.3112 | 0.2460 | 0.0569 ** | 0.1710 | 0.2024 |
| 20% | 1.5833 | 1.4511 | 0.3578 | 0.0682 ** | 0.2482 | 0.2781 |
| 30% | 1.7014 | 1.5176 | 0.4159 | 0.0899 * | 0.2866 | 0.3160 |
| 40% | 1.7361 | 1.5289 | 0.4291 | 0.0903 * | 0.2946 | 0.3160 |
| 50% | 1.8125 | 1.5548 | 0.4588 | 0.1056 * | 0.3127 | 0.3341 |
| ALL | 1.8750 | 1.5793 | 0.4834 | 0.1438 | 0.3283 | 0.3463 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Cui, L.; Zhang, L.; Huang, Y.; Zhang, S.; Chen, W.; Deng, X.; Jiao, Z.; Yang, W.; Qiu, Z.; et al. Genetic Diversity Analysis and Core Germplasm Collection Construction of Radish Cultivars Based on Structure Variation Markers. Int. J. Mol. Sci. 2023, 24, 2554. https://doi.org/10.3390/ijms24032554
Li X, Cui L, Zhang L, Huang Y, Zhang S, Chen W, Deng X, Jiao Z, Yang W, Qiu Z, et al. Genetic Diversity Analysis and Core Germplasm Collection Construction of Radish Cultivars Based on Structure Variation Markers. International Journal of Molecular Sciences. 2023; 24(3):2554. https://doi.org/10.3390/ijms24032554
Chicago/Turabian StyleLi, Xiaoyao, Lei Cui, Lei Zhang, Yan Huang, Shuting Zhang, Weifang Chen, Xiaohui Deng, Zhenbiao Jiao, Wenjie Yang, Zhengming Qiu, and et al. 2023. "Genetic Diversity Analysis and Core Germplasm Collection Construction of Radish Cultivars Based on Structure Variation Markers" International Journal of Molecular Sciences 24, no. 3: 2554. https://doi.org/10.3390/ijms24032554
APA StyleLi, X., Cui, L., Zhang, L., Huang, Y., Zhang, S., Chen, W., Deng, X., Jiao, Z., Yang, W., Qiu, Z., & Yan, C. (2023). Genetic Diversity Analysis and Core Germplasm Collection Construction of Radish Cultivars Based on Structure Variation Markers. International Journal of Molecular Sciences, 24(3), 2554. https://doi.org/10.3390/ijms24032554