

Attenuation of Tumor Burden in Response to Rucaparib in Lung Adenocarcinoma: The Contribution of Oxidative Stress, Apoptosis, and DNA Damage

 and
and 
Abstract
:1. Introduction
2. Results
2.1. Physiological and Tumor Characteristics in LC Mice
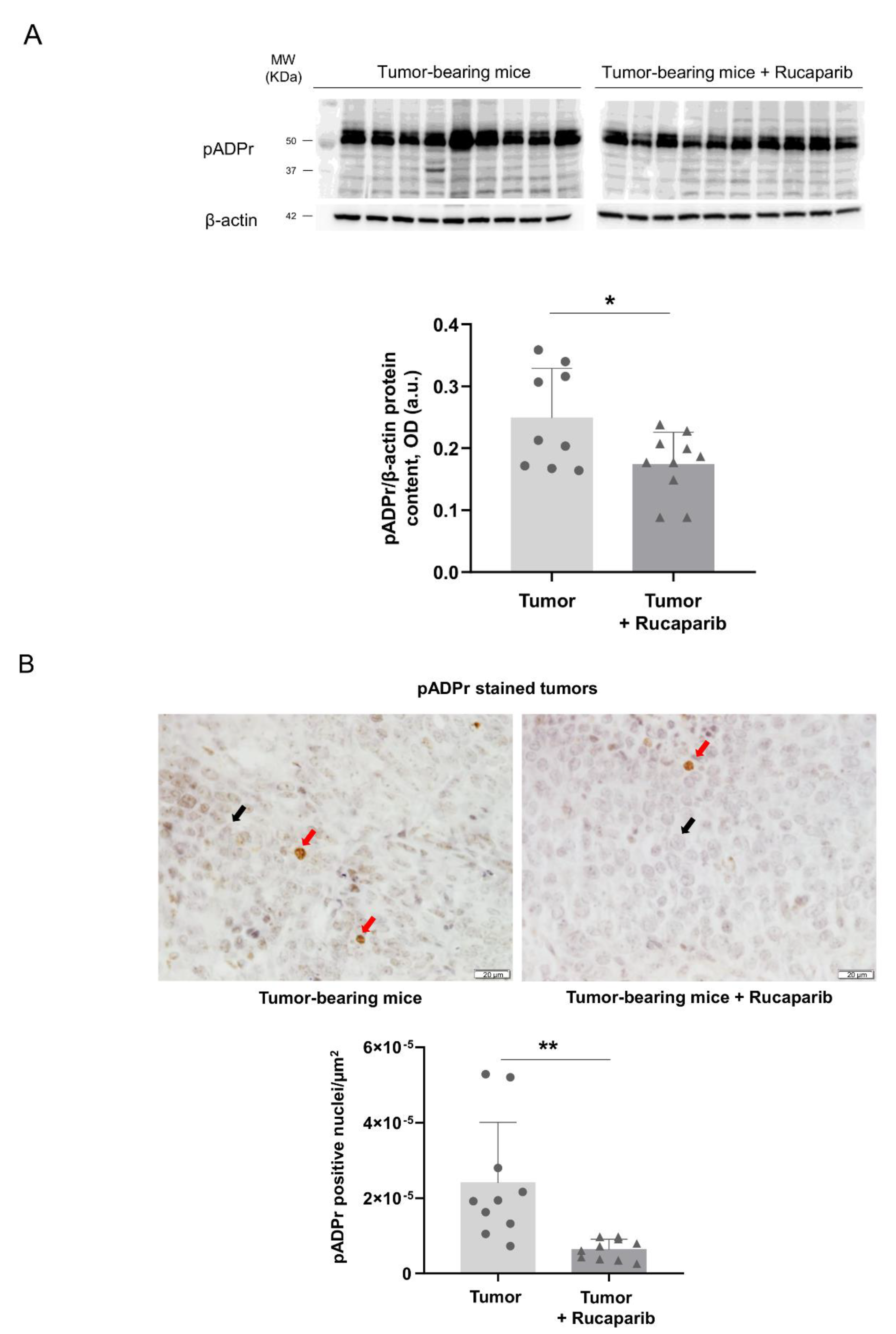
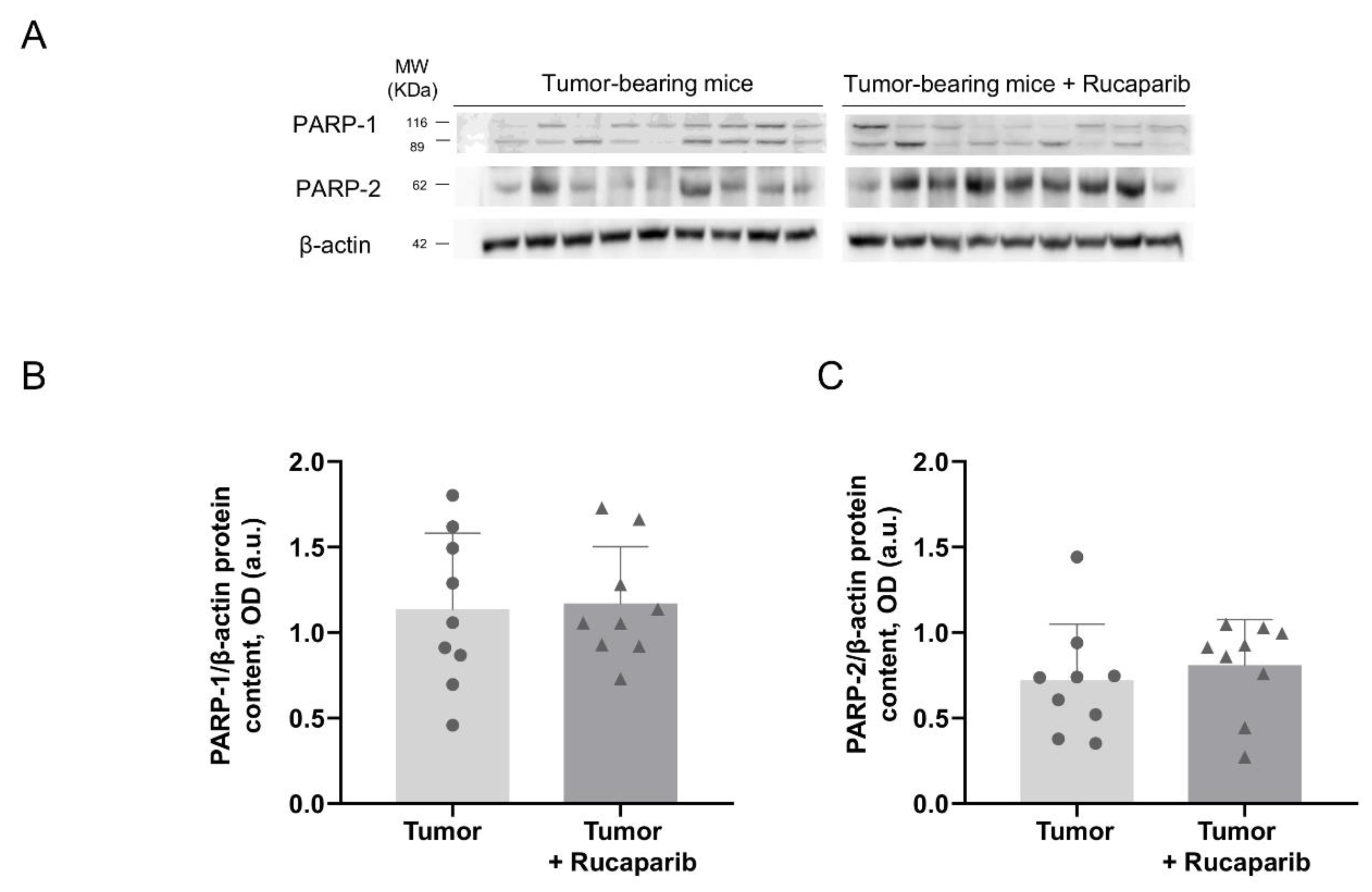
2.2. PARP Activity Decreased in Tumors Treated with Rucaparib
2.3. DNA Damage Increased in Tumors Treated with Rucaparib
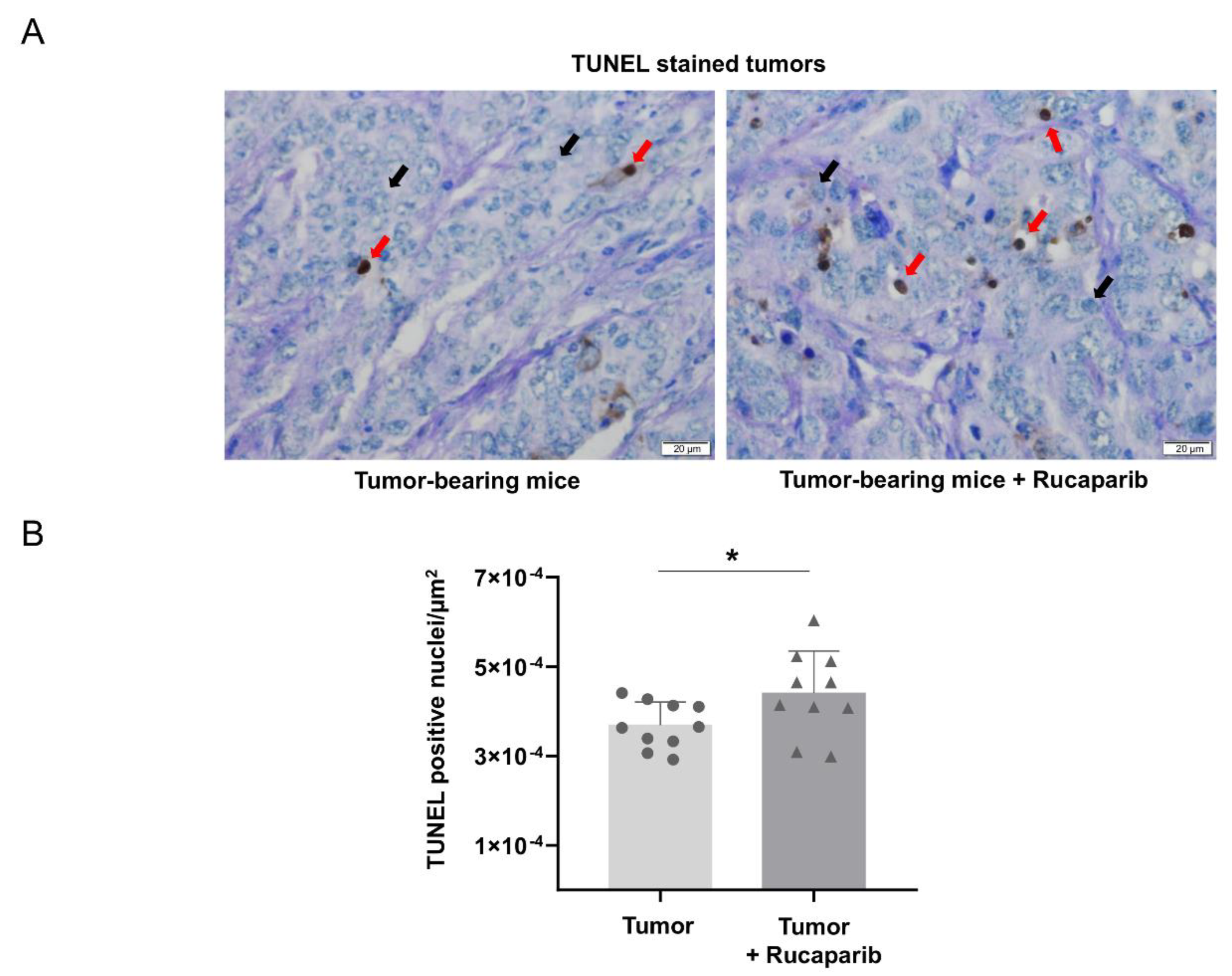
2.4. TUNEL-Positive Nuclei in Tumors Treated with Rucaparib
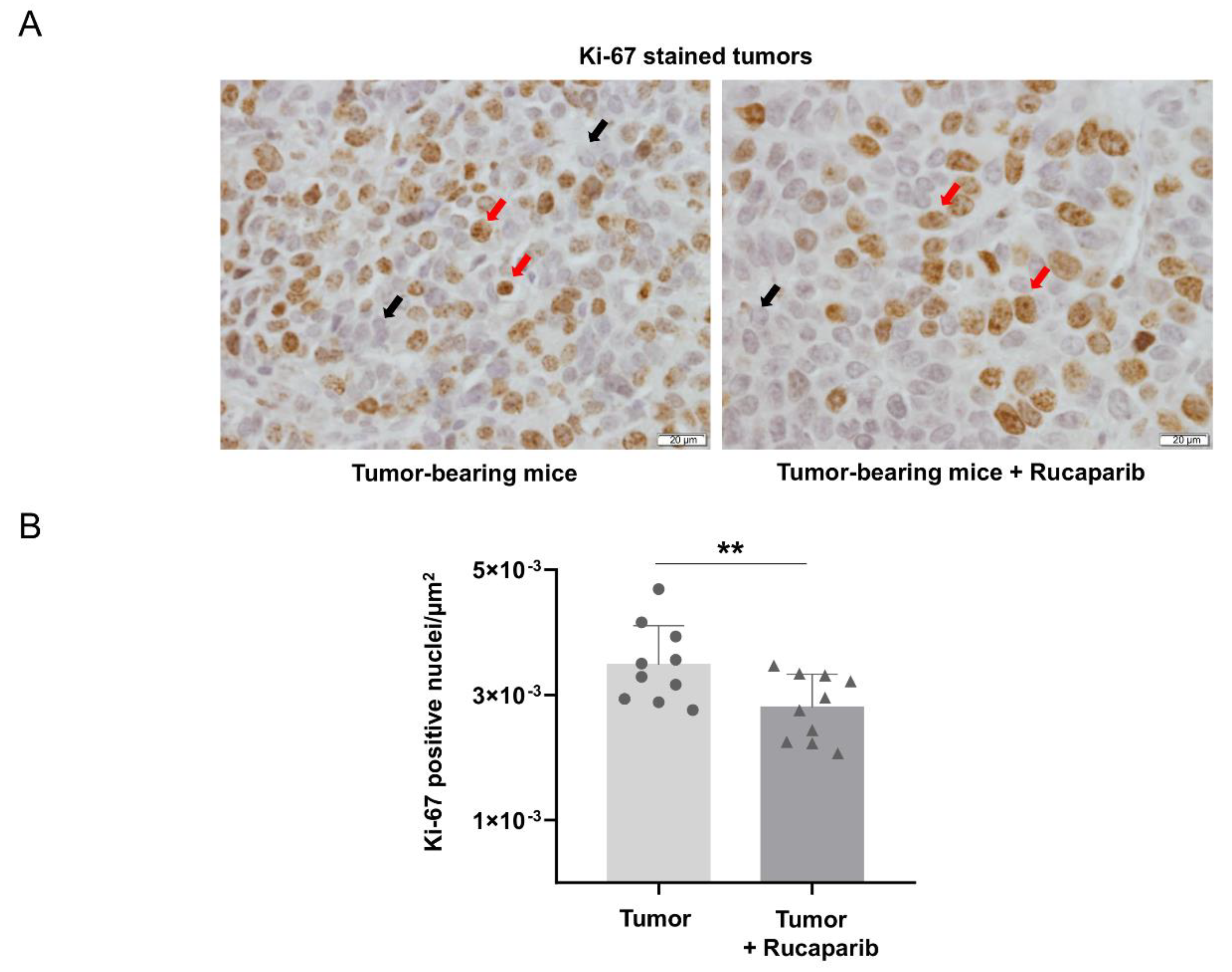
2.5. Cellular Proliferation Decreased in Tumors Treated with Rucaparib
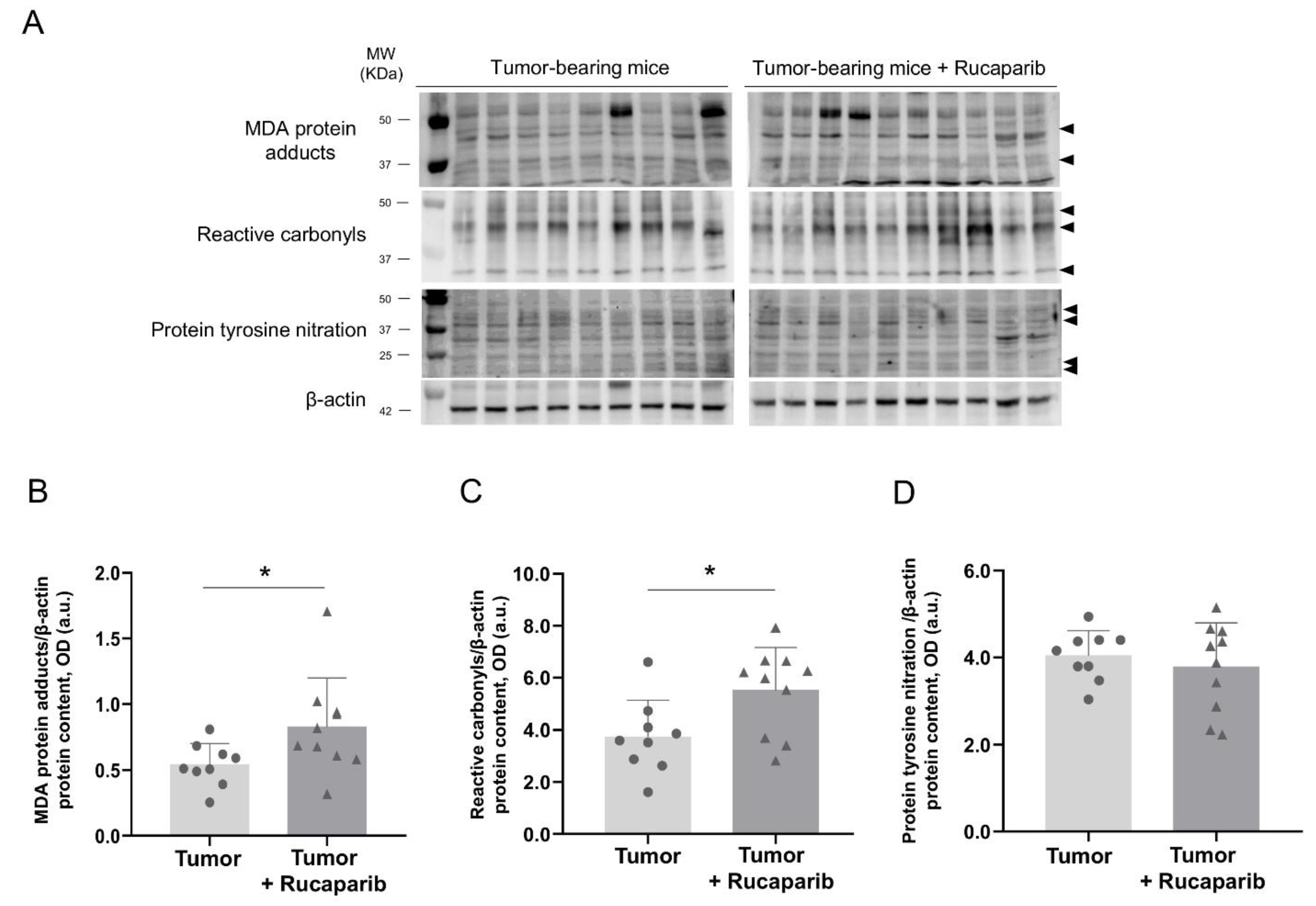
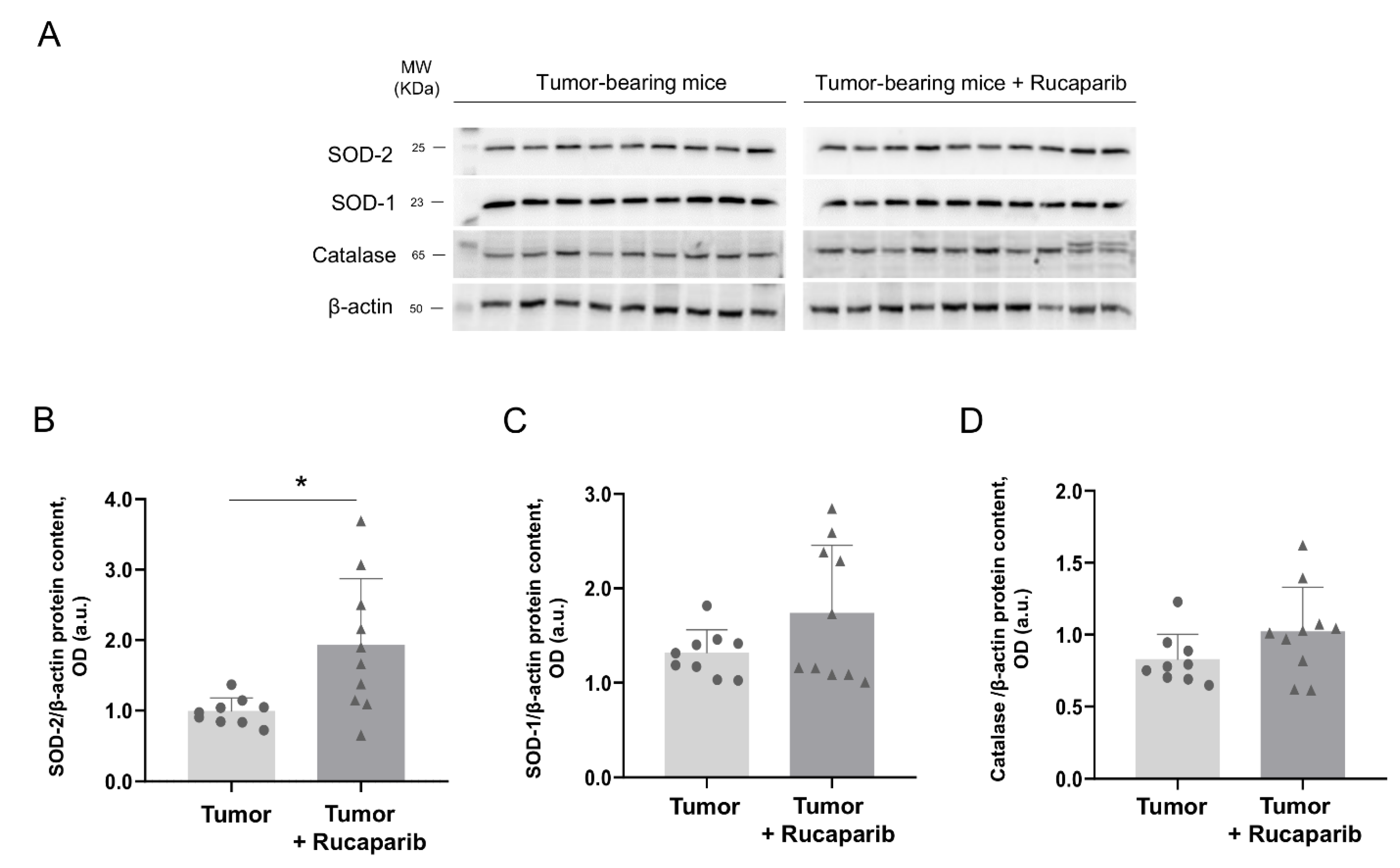
2.6. Redox Balance in Tumors in Response to Rucaparib Treatment
3. Discussion
Study Limitations
4. Materials and Methods
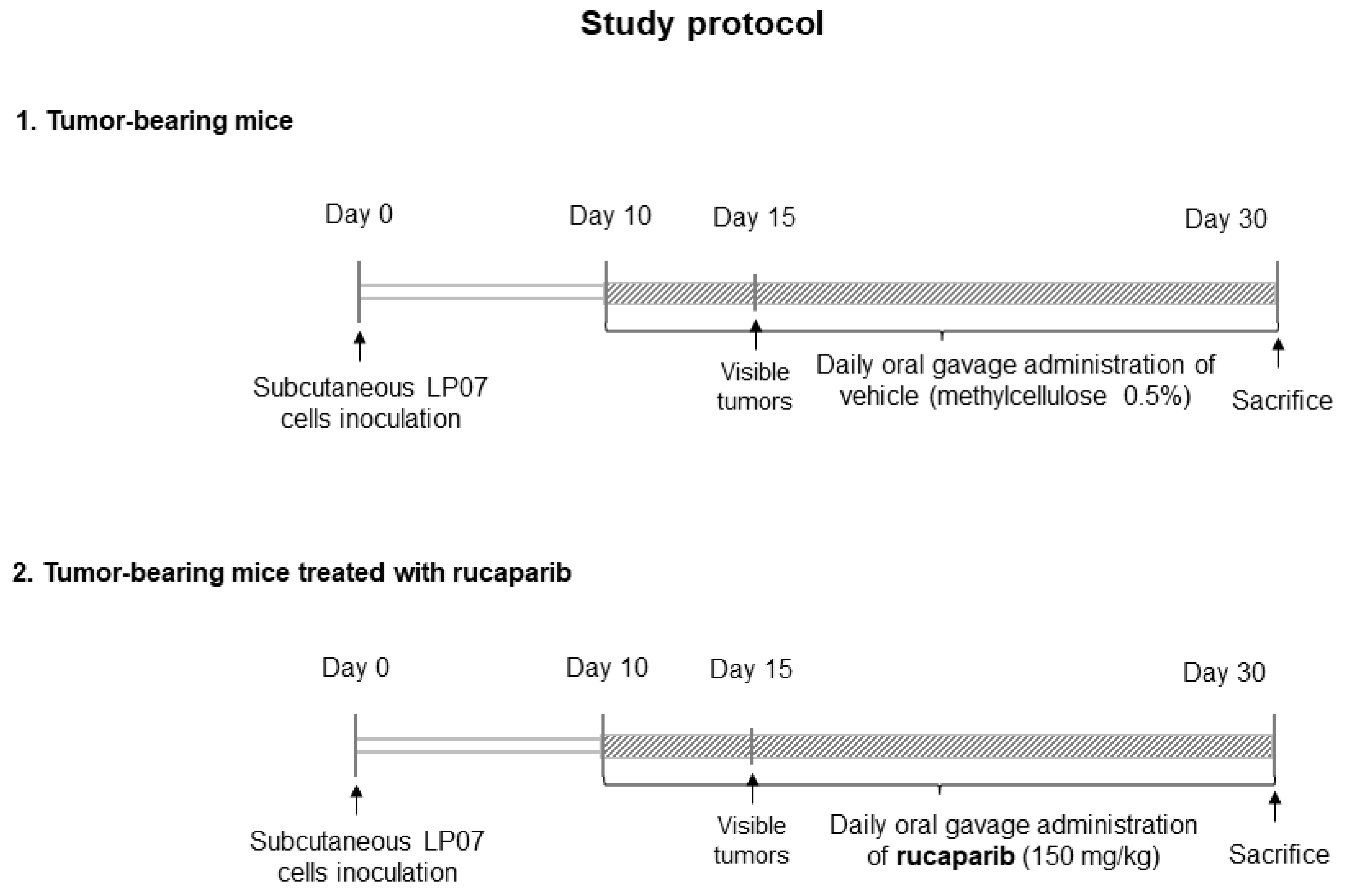
4.1. Experimental Model and Design
4.2. Sacrifice and Sample Collection
4.3. In Vivo Measurements in the Mice
4.4. Biological Analysis
4.4.1. Histological Analyses of Tumor Samples
4.4.2. Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick-End Labeling (TUNEL) Assay
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De-Torres, J.P.; Wisnivesky, J.P.; Bastarrika, G.; Wilson, D.O.; Celli, B.R.; Zulueta, J.J. Exploring the Impact of Lung Cancer Screening on Lung Cancer Mortality of Smokers with Obstructive Lung Disease: Analysis of the NLST-ACRIN Cohort. Arch. Bronconeumol. 2021, 57, 36–41. [Google Scholar] [CrossRef]
- Clofent, D.; Culebras, M.; Loor, K.; Cruz, M.J. Environmental Pollution and Lung Cancer: The Carcinogenic Power of the Air We Breathe. Arch. Bronconeumol. 2021, 57, 317–318. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, F.R.; Scagliotti, G.V.; Mulshine, J.L.; Kwon, R.; Curran, W.J.; Wu, Y.L.; Paz-Ares, L. Lung cancer: Current therapies and new targeted treatments. Lancet 2017, 389, 299–311. [Google Scholar] [CrossRef]
- Cayuela, L.; López-Campos, J.L.; Otero, R.; Rodriguez Portal, J.A.; Rodríguez-Domínguez, S.; Cayuela, A. The Beginning of the Trend Change in Lung Cancer Mortality Trends in Spain, 1980–2018. Arch. Bronconeumol. 2021, 57, 115–121. [Google Scholar] [CrossRef]
- Fraile Olivero, C.A.; Pardina Solano, M.A.; Milla Collado, L. Intraoperatory Diagnosis of Partial Anomalous Pulmonary Venous Return During Pulmonary Resection Surgery in a Non-Small Cell Lung Cancer Patient. Arch. Bronconeumol. 2021, 57, 703. [Google Scholar] [CrossRef]
- Malhotra, J.; Malvezzi, M.; Negri, E.; La Vecchia, C.; Boffetta, P. Risk factors for lung cancer worldwide. Eur. Respir. J. 2016, 48, 889–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesa-Guzmán, M.; González, J.; Alcaide, A.B.; Bertó, J.; de-Torres, J.P.; Campo, A.; Seijo, L.M.; Ocón, M.M.; Pueyo, J.C.; Bastarrika, G.; et al. Surgical Outcomes in a Lung Cancer-Screening Program Using Low Dose Computed Tomography. Arch. Bronconeumol. 2021, 57, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, G.; Andrini, E.; Sisi, M.; Rizzo, A.; Parisi, C.; Di Federico, A.; Gelsomino, F.; Ardizzoni, A. Beyond EGFR, ALK and ROS1: Current evidence and future perspectives on newly targetable oncogenic drivers in lung adenocarcinoma. Crit. Rev. Oncol. Hematol. 2020, 156, 103119. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Cusmai, A.; Giovannelli, F.; Acquafredda, S.; Rinaldi, L.; Misino, A.; Montagna, E.S.; Ungaro, V.; Lorusso, M.; Palmiotti, G. Impact of Proton Pump Inhibitors and Histamine-2-Receptor Antagonists on Non-Small Cell Lung Cancer Immunotherapy: A Systematic Review and Meta-Analysis. Cancers 2022, 14, 1404. [Google Scholar] [CrossRef] [PubMed]
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-ribosyl)ation by PARP1: Reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019, 47, 3811–3827. [Google Scholar] [CrossRef] [PubMed]
- Bai, P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef] [Green Version]
- Yélamos, J.; Schreiber, V.; Dantzer, F. Toward specific functions of poly(ADP-ribose) polymerase-2. Trends Mol. Med. 2008, 14, 169–178. [Google Scholar] [CrossRef] [PubMed]
- De, P.; Sun, Y.; Carlson, J.H.; Friedman, L.S.; Leyland-Jones, B.R.; Dey, N. Doubling down on the PI3K-AKT-mTOR pathway enhances the antitumor efficacy of PARP inhibitor in triple negative breast cancer model beyond BRCA-ness. Neoplasia 2014, 16, 43–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, H.; Shi, C.; Yan, Z.; Li, H. Combination of erlotinib and a PARP inhibitor inhibits growth of A2780 tumor xenografts due to increased autophagy. Drug Des. Devel. Ther. 2015, 9, 3183–3190. [Google Scholar] [CrossRef] [Green Version]
- Tewari, K.S.; Eskander, R.N.; Monk, B.J. Development of olaparib for BRCA-deficient recurrent epithelial ovarian cancer. Clin. Cancer Res. 2015, 21, 3829–3835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Noll, R.; Marchetti, S.; Steeghs, N.; Beijnen, J.H.; Mergui-Roelvink, M.W.J.; Harms, E.; Rehorst, H.; Sonke, G.S.; Schellens, J.H.M. Long-term safety and anti-tumour activity of olaparib monotherapy after combination with carboplatin and paclitaxel in patients with advanced breast, ovarian or fallopian tube cancer. Br. J. Cancer 2015, 113, 396–402. [Google Scholar] [CrossRef]
- Schiewer, M.J.; Goodwin, J.F.; Han, S.; Chad Brenner, J.; Augello, M.A.; Dean, J.L.; Liu, F.; Planck, J.L.; Ravindranathan, P.; Chinnaiyan, A.M.; et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012, 2, 1134. [Google Scholar] [CrossRef] [Green Version]
- Rojo, F.; García-Parra, J.; Zazo, S.; Tusquets, I.; Ferrer-Lozano, J.; Menendez, S.; Eroles, P.; Chamizo, C.; Servitja, S.; Ramírez-Merino, N.; et al. Nuclear PARP-1 protein overexpression is associated with poor overall survival in early breast cancer. Ann. Oncol. 2011, 23, 1156–1164. [Google Scholar] [CrossRef]
- Césaire, M.; Thariat, J.; Candéias, S.M.; Stefan, D.; Saintigny, Y.; Chevalier, F. Combining PARP inhibition, radiation, and immunotherapy: A possible strategy to improve the treatment of cancer? Int. J. Mol. Sci. 2018, 19, 3793. [Google Scholar] [CrossRef] [Green Version]
- Weil, M.K.; Chen, A.P. PARP Inhibitor Treatment in Ovarian and Breast Cancer. Curr. Probl. Cancer 2011, 35, 7. [Google Scholar] [CrossRef]
- Lallo, A.; Frese, K.K.; Morrow, C.J.; Sloane, R.; Gulati, S.; Schenk, M.W.; Trapani, F.; Simms, N.; Galvin, M.; Brown, S.; et al. The Combination of the PARP Inhibitor Olaparib and the WEE1 Inhibitor AZD1775 as a New Therapeutic Option for Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 5153–5164. [Google Scholar] [CrossRef] [Green Version]
- Mateu-Jiménez, M.; Cucarull-Martínez, B.; Yelamos, J.; Barreiro, E. Reduced tumor burden through increased oxidative stress in lung adenocarcinoma cells of PARP-1 and PARP-2 knockout mice. Biochimie 2016, 121, 278–286. [Google Scholar] [CrossRef] [Green Version]
- Davar, D.; Beumer, L.J.H.; Hamieh, L.; Tawbi, H. Role of PARP inhibitors in cancer biology and therapy. Curr. Med. Chem. 2012, 19, 3907–3921. [Google Scholar] [CrossRef] [PubMed]
- Domagala, P.; Huzarski, T.; Lubinski, J.; Gugala, K.; Domagala, W. PARP-1 expression in breast cancer including BRCA1-associated, triple negative and basal-like tumors: Possible implications for PARP-1 inhibitor therapy. Breast Cancer Res. Treat. 2011, 127, 861–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.M.; Ledermann, J.A.; Kohn, E.C. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann. Oncol. 2014, 25, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Yu, D.S.; Liang, Y.C.; Huang, K.F.; Chou, S.J.; Chen, T.C.; Lee, C.C.; Chen, C.L.; Chiou, S.H.; Huang, H.S. New approaches of PARP-1 inhibitors in human lung cancer cells and cancer stem-like cells by some selected anthraquinone-derived small molecules. PLoS ONE 2013, 8, e56284. [Google Scholar] [CrossRef]
- Postel-Vinay, S.; Bajrami, I.; Friboulet, L.; Elliott, R.; Fontebasso, Y.; Dorvault, N.; Olaussen, K.A.; André, F.; Soria, J.C.; Lord, C.J.; et al. A high-throughput screen identifies PARP1/2 inhibitors as a potential therapy for ERCC1-deficient non-small cell lung cancer. Oncogene 2013, 32, 5377–5387. [Google Scholar] [CrossRef] [Green Version]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Liu, C.; Chang, C.; Shen, C.; Yin, Y.; Yin, X.; Jiang, Z.; Zhao, Z.; Mu, M.; Cao, D.; et al. Comparative safety and tolerability of approved PARP inhibitors in cancer: A systematic review and network meta-analysis. Pharmacol. Res. 2021, 172, 105808. [Google Scholar] [CrossRef]
- Pérez-Peiró, M.; Duran, X.; Yélamos, J.; Barreiro, E. Attenuation of Muscle Damage, Structural Abnormalities, and Physical Activity in Respiratory and Limb Muscles following Treatment with Rucaparib in Lung Cancer Cachexia Mice. Cancers 2022, 14, 2894. [Google Scholar] [CrossRef]
- Dockery, L.E.; Gunderson, C.C.; Moore, K.N. Rucaparib: The past, present, and future of a newly approved PARP inhibitor for ovarian cancer. Onco Targets Ther. 2017, 10, 3029–3037. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Zhang, B.; Chen, Y.; Liu, H.; Liu, Y.; Li, X.; Bao, Z.; Song, Z.; Wang, Z. Poly(adp-ribose) polymerase inhibitor pj34 attenuated hepatic triglyceride accumulation in alcoholic fatty liver disease in mice. J. Pharmacol. Exp. Ther. 2018, 364, 364–452. [Google Scholar] [CrossRef] [PubMed]
- Albert, J.M.; Cao, C.; Kwang, W.K.; Willey, C.D.; Geng, L.; Xiao, D.; Wang, H.; Sandler, A.; Johnson, D.H.; Colevas, A.D.; et al. Inhibition of poly(ADP-ribose) polymerase enhances cell death and improves tumor growth delay in irradiated lung cancer models. Clin. Cancer Res. 2007, 13, 3033–3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Xiao, S.; Yuan, M.; Li, Q.; Xiao, G.; Wu, W.; Ouyang, Y.; Huang, L.; Yao, C. PARP inhibitor re-sensitizes Adriamycin resistant leukemia cells through DNA damage and apoptosis. Mol. Med. Rep. 2019, 19, 75–84. [Google Scholar] [CrossRef] [Green Version]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. gammaH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Hou, D.; Liu, Z.; Xu, X.; Liu, Q.; Zhang, X.; Kong, B.; Wei, J.J.; Gong, Y.; Shao, C. Increased oxidative stress mediates the antitumor effect of PARP inhibition in ovarian cancer. Redox Biol. 2018, 17, 99–111. [Google Scholar] [CrossRef]
- Gangopadhyay, N.N.; Luketich, J.D.; Opest, A.; Visus, C.; Meyer, E.M.; Landreneau, R.; Schuchert, M.J. Inhibition of poly(ADP-ribose) polymerase (PARP) induces apoptosis in lung cancer cell lines. Cancer Investig. 2011, 29, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.L.; Young, C.D.; Bian, L.; Weigel, K.; Nolan, K.; Frederick, B.; Han, G.; He, G.; Devon Trahan, G.; Rudolph, M.C.; et al. PARP inhibition enhances radiotherapy of SMAD4 deficient human head and neck squamous cell carcinomas in experimental models. Clin. Cancer Res. 2020, 26, 3058. [Google Scholar] [CrossRef] [Green Version]
- Ordóñez, J.L.; Amaral, A.T.; Carcaboso, A.M.; Herrero-Martín, D.; Del Carmen García-Macías, M.; Sevillano, V.; Alonso, D.; Pascual-Pasto, G.; San-Segundo, L.; Vila-Ubach, M.; et al. The PARP inhibitor olaparib enhances the sensitivity of Ewing sarcoma to trabectedin. Oncotarget 2015, 6, 18875. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, L.; Chen, S.; Huang, P.; Ma, L.; Ding, H.; Basappa, B.; Zhu, T.; Lobie, P.E.; Pandey, V. Combined inhibition of BADSer99 phosphorylation and PARP ablates models of recurrent ovarian carcinoma. Commun. Med. 2022, 2, 82. [Google Scholar] [CrossRef]
- Urtreger, A.J.; Diament, M.J.; Ranuncolo, S.M.; Del, C.; Vidal, M.; Puricelli, L.I.; Klein, S.M.; De Kier Joffe, E.D. New murine cell line derived from a spontaneous lung tumor induces paraneoplastic syndromes. Int. J. Oncol. 2001, 18, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Degracia, A.; Granado-Martínez, P.; Millán-Sánchez, A.; Tang, J.; Pons-Carreto, A.; Barreiro, E. Reduced lung cancer burden by selective immunomodulators elicits improvements in muscle proteolysis and strength in cachectic mice. J. Cell. Physiol. 2019, 234, 18041–18052. [Google Scholar] [CrossRef] [PubMed]
- Mañas-García, L.; Penedo-Vázquez, A.; López-Postigo, A.; Deschrevel, J.; Durán, X.; Barreiro, E. Prolonged Immobilization Exacerbates the Loss of Muscle Mass and Function Induced by Cancer-Associated Cachexia through Enhanced Proteolysis in Mice. Int. J. Mol. Sci. 2020, 21, 8167. [Google Scholar] [CrossRef] [PubMed]
- Penedo-Vázquez, A.; Duran, X.; Mateu, J.; López-Postigo, A.; Barreiro, E. Curcumin and Resveratrol Improve Muscle Function and Structure through Attenuation of Proteolytic Markers in Experimental Cancer-Induced Cachexia. Molecules 2021, 26, 4904. [Google Scholar] [CrossRef] [PubMed]
- Chacon-Cabrera, A.; Fermoselle, C.; Salmela, I.; Yelamos, J.; Barreiro, E. MicroRNA expression and protein acetylation pattern in respiratory and limb muscles of Parp-1(−/−) and Parp-2(−/−) mice with lung cancer cachexia. Biochim. Biophys. Acta 2015, 1850, 2530–2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, D.; Xu, G.; Zhang, C.; Li, B.; Qin, J.; Hao, X.; Liu, Q.; Zhang, X.; Liu, J.; Wei, J.; et al. Berberine induces oxidative DNA damage and impairs homologous recombination repair in ovarian cancer cells to confer increased sensitivity to PARP inhibition. Cell Death Dis. 2017, 8, e3070. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Peng, X.; Li, X.; Liu, H.; Zhao, B.; Elkabets, M.; Liu, Y.; Wang, W.; Wang, R.; Zhong, Y.; et al. BKM120 sensitizes glioblastoma to the PARP inhibitor rucaparib by suppressing homologous recombination repair. Cell Death Dis. 2021, 12, 546. [Google Scholar] [CrossRef]
- Deschênes, F.; Garand, C.; Lebel, M. In vivo misregulation of genes involved in apoptosis, development and oxidative stress in mice lacking both functional Werner syndrome protein and poly(ADP-ribose) polymerase-1. Hum. Mol. Genet. 2005, 14, 3293–3308. [Google Scholar] [CrossRef] [PubMed]
- Mateu-Jimenez, M.; Fermoselle, C.; Rojo, F.; Mateu, J.; Peña, R.; Urtreger, A.; Diament, M.; Joffé, E.; Pijuan, L.; Herreros, A.; et al. Pharmacological Approaches in an Experimental Model of Non-Small Cell Lung Cancer: Effects on Tumor Biology. Curr. Pharm. Des. 2016, 22, 5300–5310. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Gheorghiu, L.; Drumm, M.; Clayman, R.; Eidelman, A.; Wszolek, M.F.; Olumi, A.; Feldman, A.; Wang, M.; Marcar, L.; et al. PARP-1 inhibition with or without ionizing radiation confers reactive oxygen species-mediated cytotoxicity preferentially to cancer cells with mutant TP53. Oncogene 2018, 37, 2793–2805. [Google Scholar] [CrossRef]
- Peluffo, G.D.; Stillitani, I.; Rodríguez, V.A.; Diament, M.J.; Klein, S.M. Reduction of tumor progression and paraneoplastic syndrome development in murine lung adenocarcinoma by nonsteroidal antiinflammatory drugs. Int. J. Cancer 2004, 110, 825–830. [Google Scholar] [CrossRef]
- Chacon-Cabrera, A.; Mateu-Jimenez, M.; Langohr, K.; Fermoselle, C.; García-Arumí, E.; Andreu, A.L.; Yelamos, J.; Barreiro, E. Role of PARP activity in lung cancer-induced cachexia: Effects on muscle oxidative stress, proteolysis, anabolic markers, and phenotype. J. Cell. Physiol. 2017, 232, 3744–3761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chacon-Cabrera, A.; Fermoselle, C.; Urtreger, A.J.; Mateu-Jimenez, M.; Diament, M.J.; de Kier Joffé, E.D.B.; Sandri, M.; Barreiro, E. Pharmacological strategies in lung cancer-induced cachexia: Effects on muscle proteolysis, autophagy, structure, and weakness. J. Cell. Physiol. 2014, 229, 1660–1672. [Google Scholar] [CrossRef] [Green Version]
- Aoufouchi, S. MAb A4.3.4, A6.4.12, B5.3.9, B15.4.13 anti-poly (ADP-ribose) polymerase. Hybridoma 1997, 16, 583. [Google Scholar] [CrossRef]
- Monreal, J.; Menissier, J.; Yélamos, J. Anti-Poly-ADPribose polymerase-2 (PARP-2) mouse mAb 4G8. Hybridoma 2006, 25, 102. [Google Scholar] [CrossRef]
- Busquets, S.; Pérez-Peiró, M.; Salazar-Degracia, A.; Argilés, J.M.; Serpe, R.; Rojano-Toimil, A.; López-Soriano, F.J.; Barreiro, E. Differential structural features in soleus and gastrocnemius of carnitine-treated cancer cachectic rats. J. Cell. Physiol. 2020, 235, 526–537. [Google Scholar] [CrossRef]
- Fermoselle, C.; García-Arumí, E.; Puig-Vilanova, E.; Andreu, A.L.; Urtreger, A.J.; de Kier Joffé, E.D.B.; Tejedor, A.; Puente-Maestu, L.; Barreiro, E. Mitochondrial dysfunction and therapeutic approaches in respiratory and limb muscles of cancer cachectic mice. Exp. Physiol. 2013, 98, 1349–1365. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef]
- Lai, Y.C.; Aneja, R.K.; Satchell, M.A.; Clark, R.S.B. Detecting and quantifying pADPr in vivo. Methods Mol. Biol. 2011, 780, 117–134. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Tumor- Bearing Mice | Tumor-Bearing Mice + Rucaparib |
|---|---|---|
| Tumor weight (g) | 1.63 (0.33) | 1.09 (0.40) ** |
| Tumor area (mm2) | 1755.06 (556.61) | 1216.06 (444.03) * |
| Body weight gain (%) | −8.65 (4.66) | +2.10 (9.01) ** |
| Body weight gain without tumor (%) | −12.96 (7.68) | −4.02 (11.21) * |
| Initial body weight (g) | 20.86 (0.80) | 20.43 (0.80) |
| Final body weight (g) | 19.78 (1.68) | 20.91 (2.11) |
| Final body weight without tumor (g) | 18.27 (1.81) | 19.57 (2.50) |
| Body Weight Gain (%) | Body Weight Gain without Tumor (%) | MDA-Protein Adducts (a.u) | Protein Tyrosine Nitration (a.u) | Ki-67 (Positive Nuclei/µm2) | |
|---|---|---|---|---|---|
| Tumor-bearing mice | |||||
| Tumor weight (g) | |||||
| r | −0.689 | −0.764 | 0.895 | −0.501 | - |
| p | 0.059 | 0.046 | 0.016 | 0.311 | |
| pADPr (positive nuclei/µm2) | |||||
| r | - | - | - | - | −0.573 |
| p | 0.084 | ||||
| Tumor-bearing mice + Rucaparib | |||||
| Tumor weight (g) | |||||
| r | −0.671 | −0.759 | 0.919 | 0.898 | - |
| p | 0.034 | 0.011 | 0.001 | 0.002 | |
| pADPr (positive nuclei/µm2) | |||||
| r | - | - | - | - | 0.580 |
| p | 0.048 |
| Antibody | Dilution | Catalog Number | Supplier |
|---|---|---|---|
| Anti-pADPr antibody | 1:500 | sc-56198 | Santa Cruz Biotechnology, Dallas, TX, USA |
| Anti-PARP-1 antibody | 1:20 | - | In-house generated affinity purified monoclonal antibody (clone A6.4.12) [54] |
| Anti-PARP-2 antibody | 1:1000 | - | In-house generated affinity purified monoclonal antibody (clone 4G8) [55] |
| Anti- MDA-protein adducts antibody | 1:2000 | MD20A-G1a | Academy Bio-Chemical Company, Houston, TX, USA) |
| Anti-3-nitrotyrosine antibody | 1:1000 | A-21285 | Thermo Fisher Scientific, Waltham, MA, USA |
| Protein carbonyl assay kit | 1:5000 | Ab178020 | Abcam, Cambridge, UK |
| Anti-SOD-1 antibody | 1:2000 | sc-17767 | Santa Cruz Biotechnology |
| Anti-SOD-2 antibody | 1:2000 | sc-30080 | Santa Cruz Biotechnology |
| Anti-catalase antibody | 1:4000 | 219010 | Merck KGaA, Darmstadt, Germany |
| Anti-β-actin, antibody | 1:5000 | sc-47778 | Santa Cruz Biotechnology |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Peiró, M.; Valentí-Serra, P.; León-González, B.; Ampurdanés, C.; Duran, X.; Yélamos, J.; Barreiro, E. Attenuation of Tumor Burden in Response to Rucaparib in Lung Adenocarcinoma: The Contribution of Oxidative Stress, Apoptosis, and DNA Damage. Int. J. Mol. Sci. 2023, 24, 2580. https://doi.org/10.3390/ijms24032580
Pérez-Peiró M, Valentí-Serra P, León-González B, Ampurdanés C, Duran X, Yélamos J, Barreiro E. Attenuation of Tumor Burden in Response to Rucaparib in Lung Adenocarcinoma: The Contribution of Oxidative Stress, Apoptosis, and DNA Damage. International Journal of Molecular Sciences. 2023; 24(3):2580. https://doi.org/10.3390/ijms24032580
Chicago/Turabian StylePérez-Peiró, Maria, Paula Valentí-Serra, Blanca León-González, Coral Ampurdanés, Xavier Duran, José Yélamos, and Esther Barreiro. 2023. "Attenuation of Tumor Burden in Response to Rucaparib in Lung Adenocarcinoma: The Contribution of Oxidative Stress, Apoptosis, and DNA Damage" International Journal of Molecular Sciences 24, no. 3: 2580. https://doi.org/10.3390/ijms24032580
APA StylePérez-Peiró, M., Valentí-Serra, P., León-González, B., Ampurdanés, C., Duran, X., Yélamos, J., & Barreiro, E. (2023). Attenuation of Tumor Burden in Response to Rucaparib in Lung Adenocarcinoma: The Contribution of Oxidative Stress, Apoptosis, and DNA Damage. International Journal of Molecular Sciences, 24(3), 2580. https://doi.org/10.3390/ijms24032580