

Reactivity Graph Yields Interpretable IgM Repertoire Signatures as Potential Tumor Biomarkers
 ,
,
Abstract
:1. Introduction
2. Results
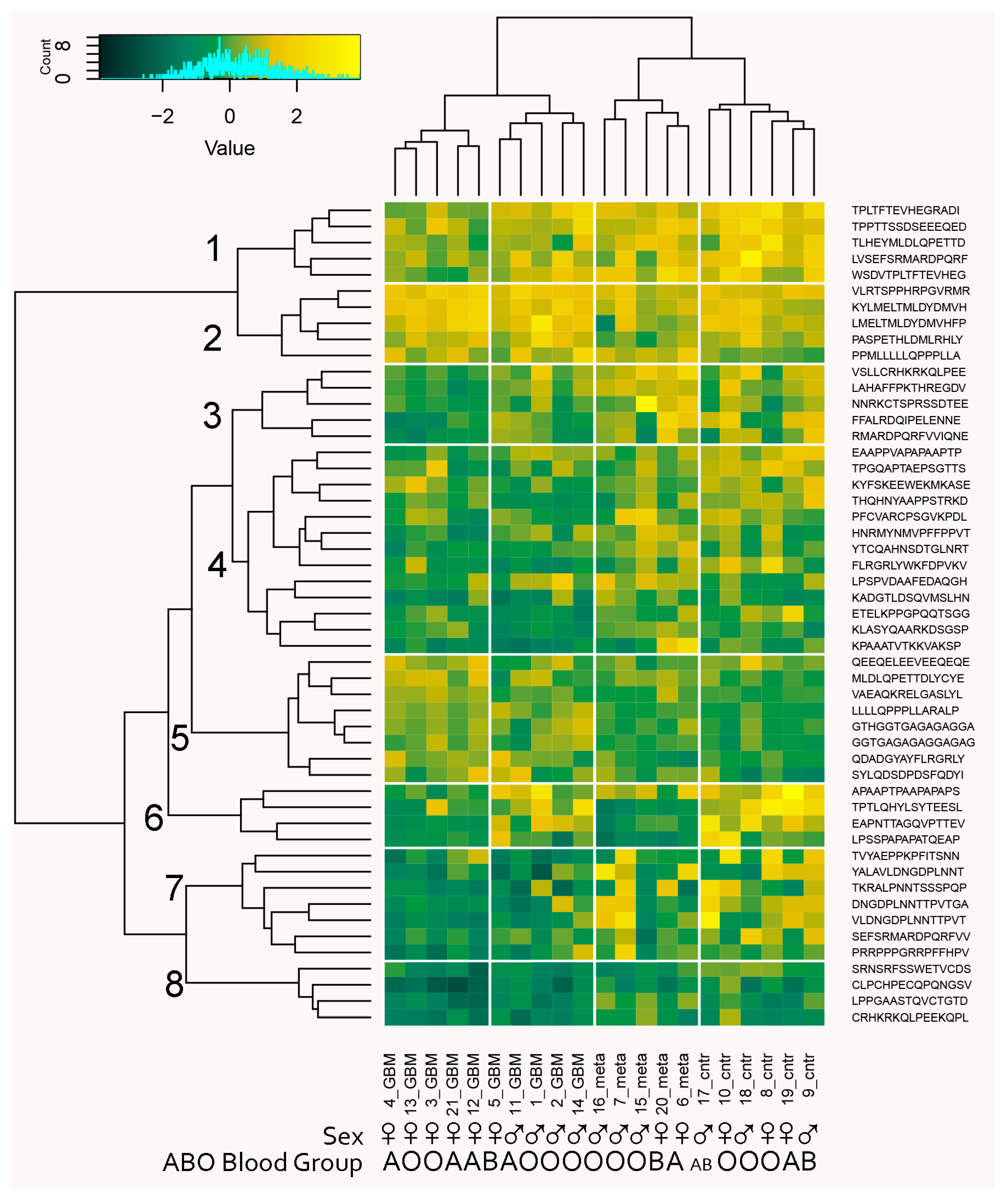
2.1. The Microarray Binding Data
2.2. The Reactivity Graph
2.2.1. Construction of the Graph
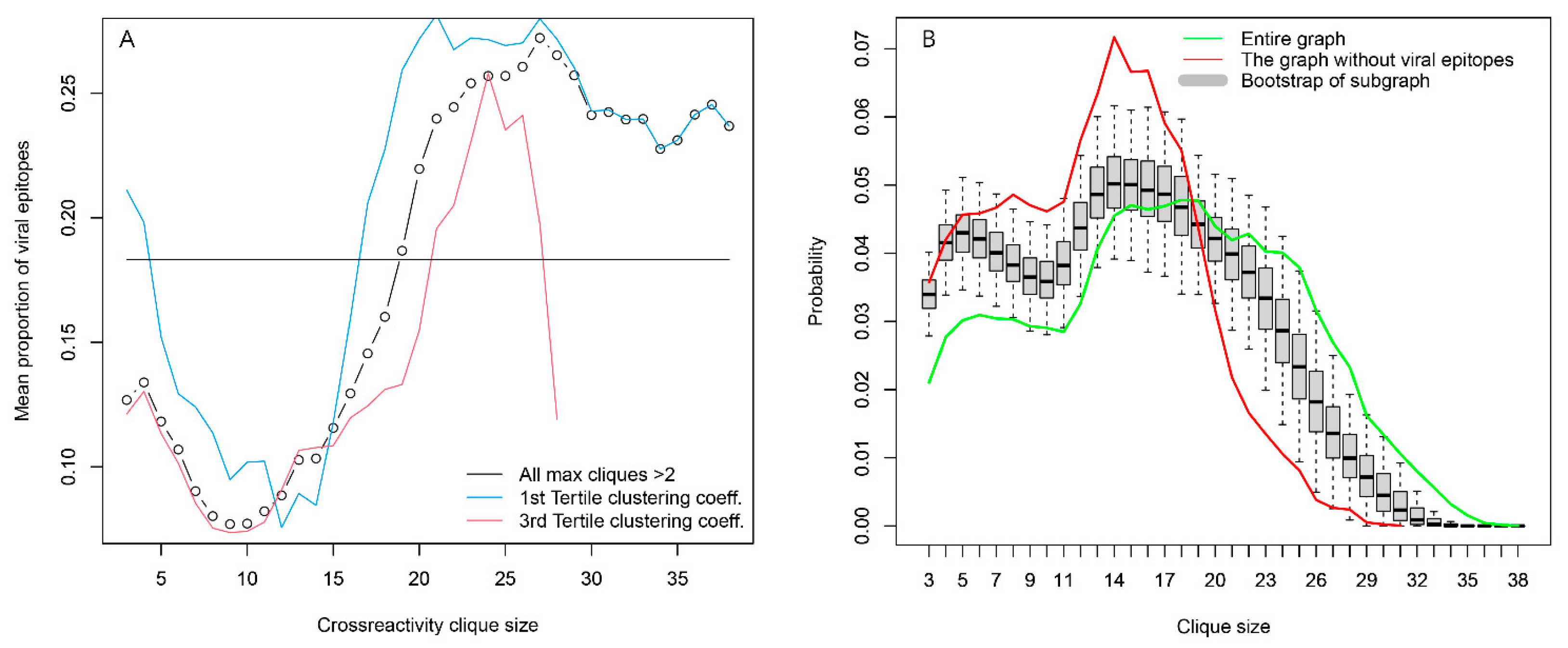
2.2.2. Overview of the Properties of the Reactivity Graph
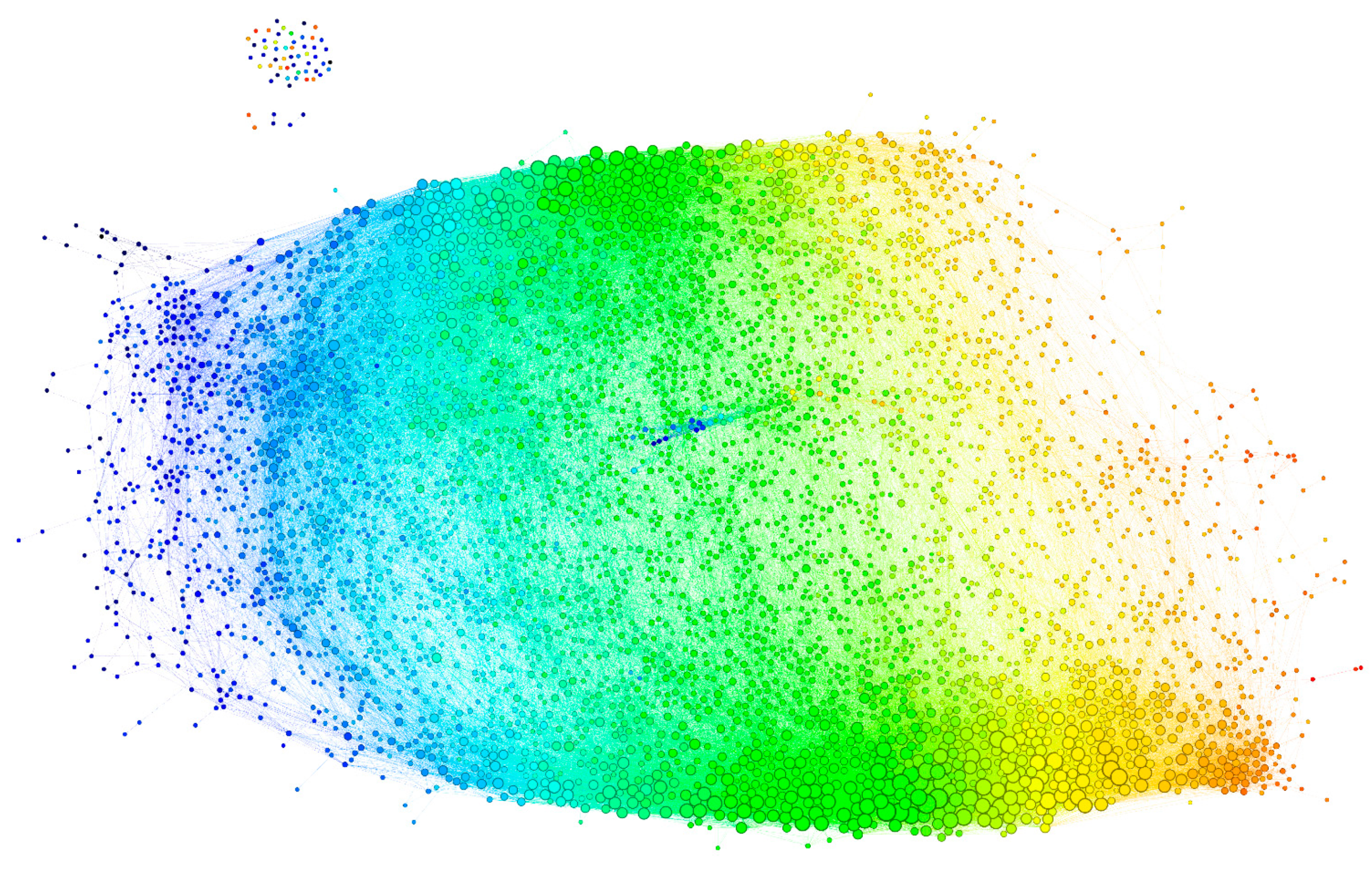
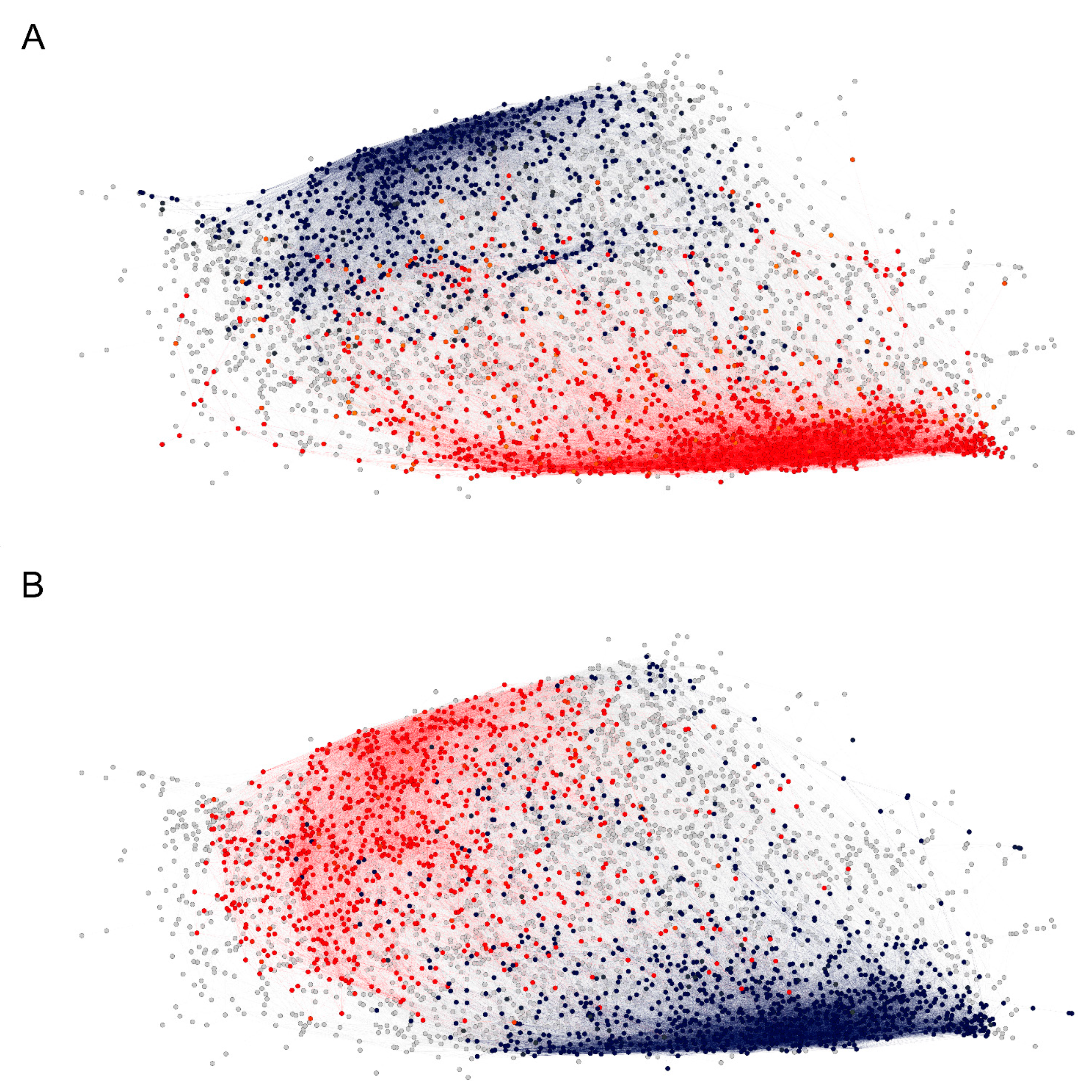
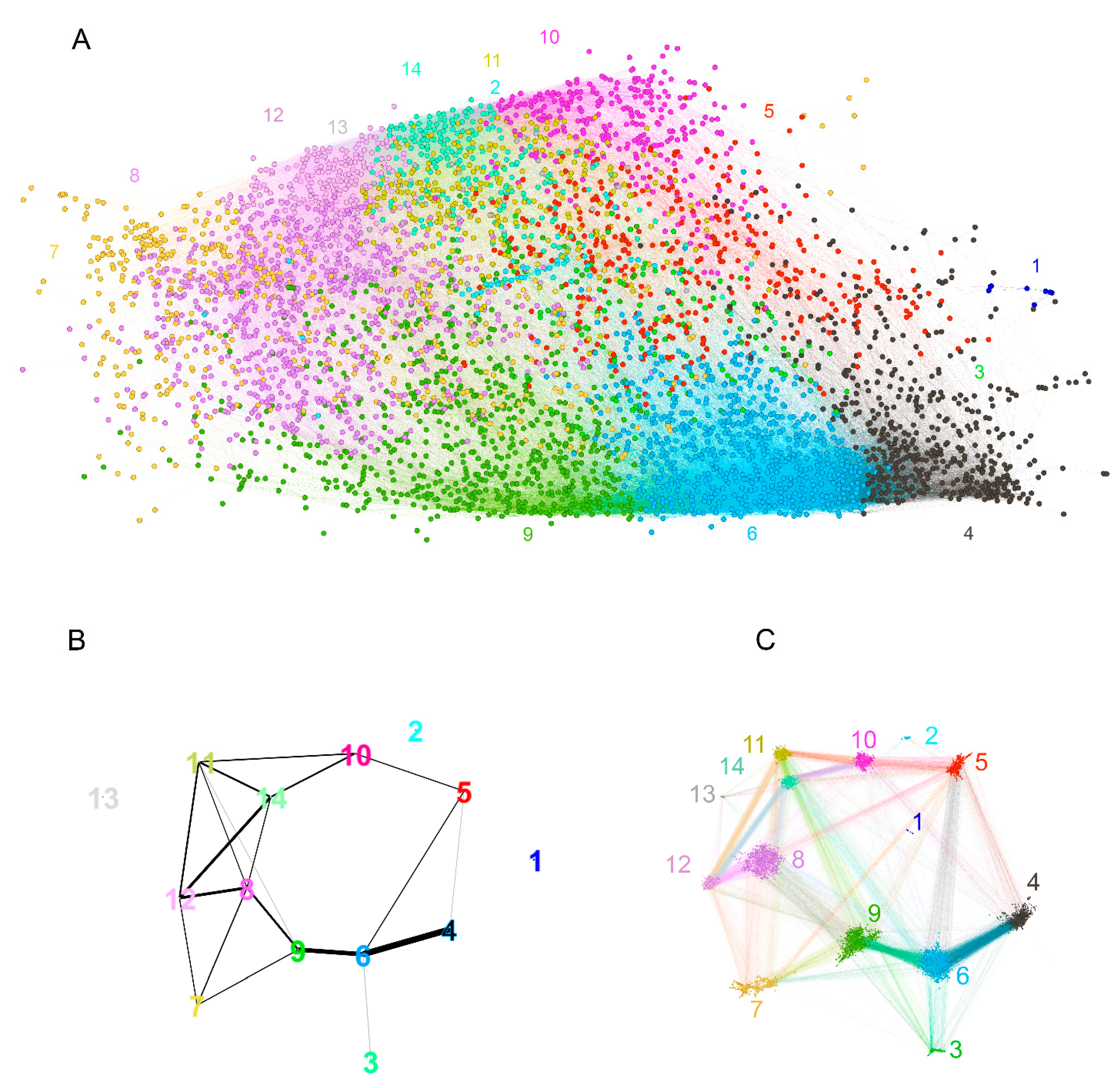
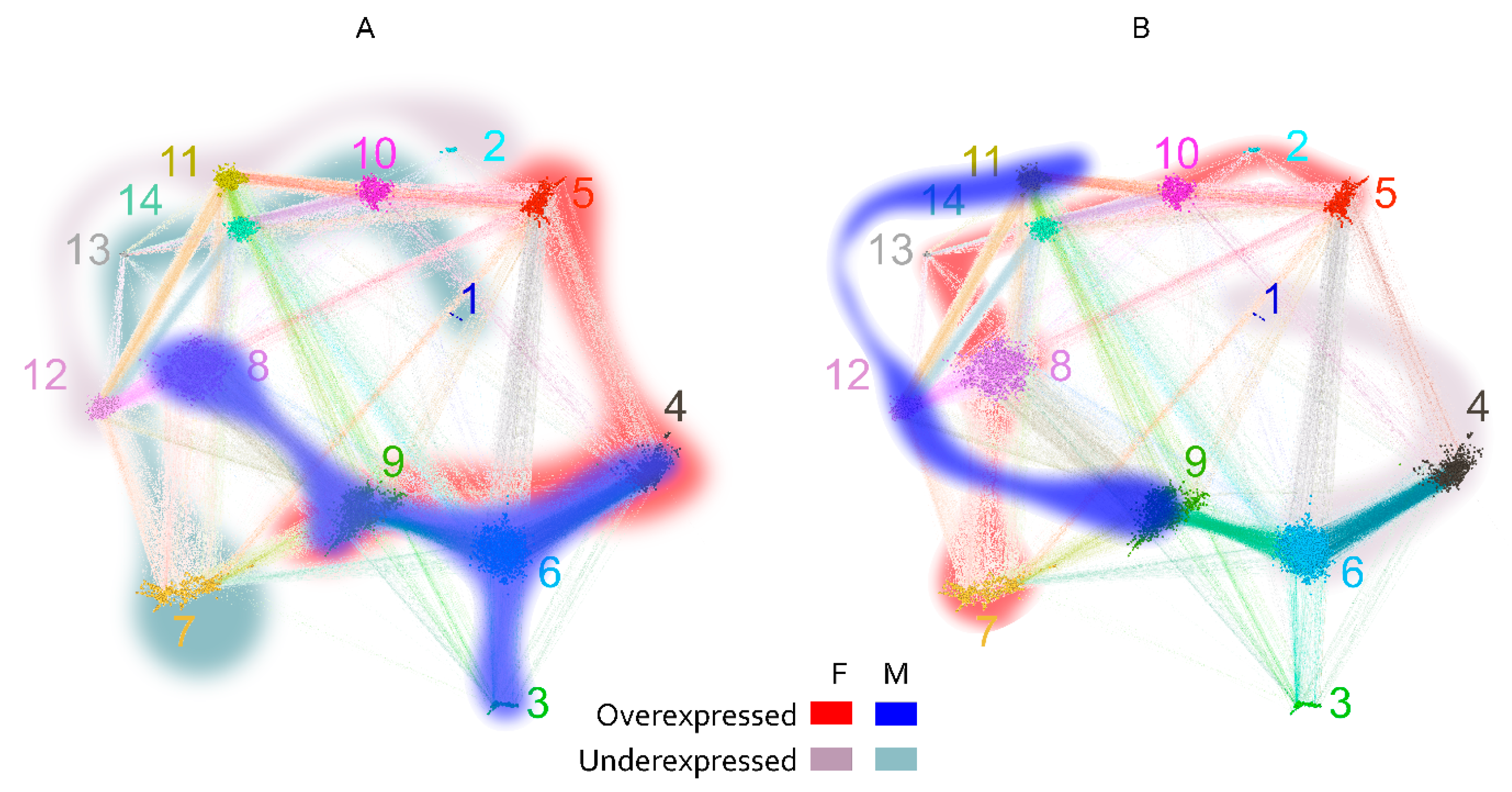
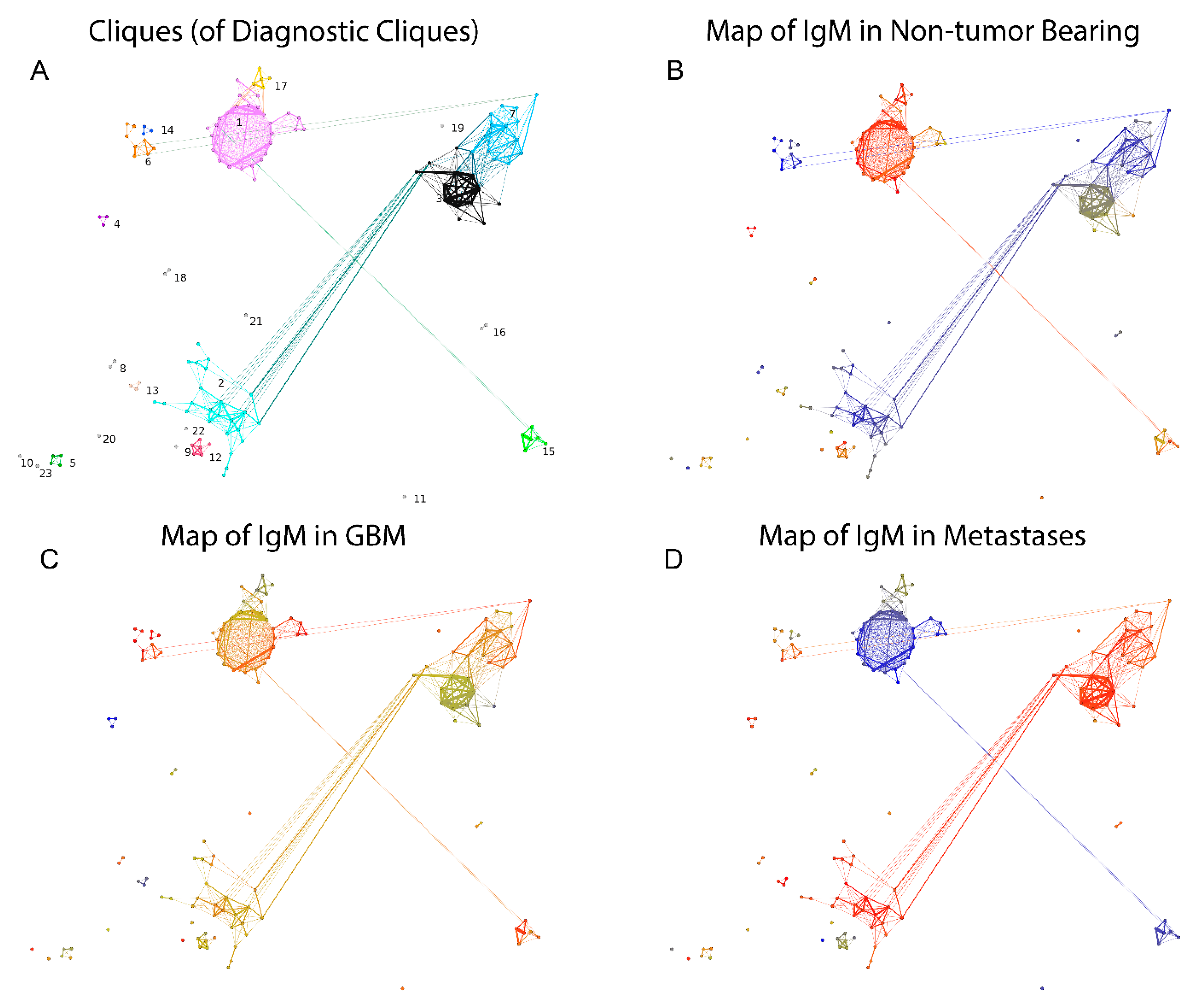
2.2.3. Visualization of the Reactivity Graph
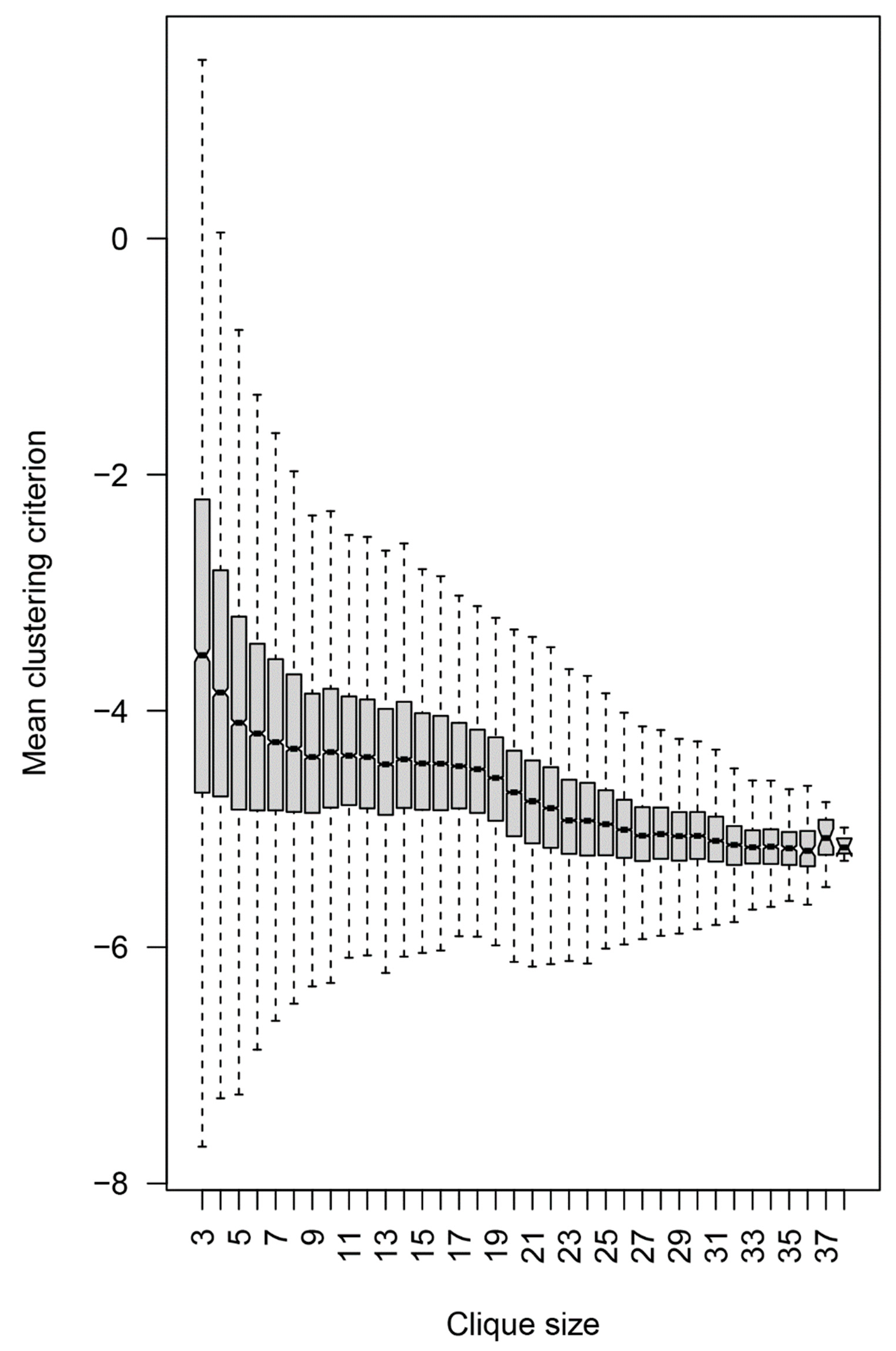
2.2.4. Topology of the Reactivity Graph


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Graph Clustering Based On | Modularity | Z-score | Mean Degree | Viral |
|---|---|---|---|---|
| Louvain Clustering | 5.72 × 10−1 | 514.683 | 34.536 | |
| LANA Human herpesvirus 8 | 1.75 × 10−3 | 43.096 | 34.462 | |
| Cancer/testis antigen 1 (NY-ESO-1) (UniProt P78358) | 5.42 × 10−3 | 38.299 | 35.247 | |
| Histone H1.2 (UniProt P16403) | 5.14 × 10−3 | 34.129 | 23.245 | |
| Histone H4 (UniProt P62805) | 1.84 × 10−3 | 28.544 | 43.268 | |
| All Viral | 2.20 × 10−2 | 27.886 | 34.172 | |
| Protein SSX2 (Cancer/testis antigen 5.2) (UniProt Q16385) | 3.31 × 10−3 | 24.357 | 56.138 | |
| L-dopachrome tautomerase (UniProt P40126) | 7.87 × 10−3 | 20.367 | 32.817 | |
| envelope glycoprotein Human T-lymphotropic virus 1 | 4.24 × 10−3 | 20.358 | 44.568 | |
| Carcinoembryonic antigen-related cell adhesion molecule 5 (UniProt P06731) | 9.30 × 10−3 | 18.928 | 31.683 | |
| G2/mitotic-specific cyclin-B1 (UniProt P14635) | 5.95 × 10−3 | 18.584 | 32.641 | |
| Melanocyte protein PMEL (UniProt P40967) | 7.40 × 10−3 | 15.590 | 36.761 | |
| Epstein-Barr nuclear antigen 1 Human herpesvirus 4 | 1.04 × 10−3 | 14.264 | 27.200 | |
| Transcription factor SOX-2 (UniProt P48431) | 3.23 × 10−3 | 13.371 | 42.364 | |
| Cellular tumor antigen p53 (UniProt Q2XN98) | 4.12 × 10−3 | 13.130 | 33.090 | |
| Mammaglobin-1 (UniProt Q13296) | 8.21 × 10−3 | 12.978 | 17.385 | |
| Stromelysin-3 (MMP11) (UniProt P24347) | 4.66 × 10−3 | 12.825 | 34.304 | |
| Myc proto-oncogene protein (UniProt P01106) | 3.92 × 10−3 | 12.220 | 36.788 | |
| HLA class I histocompatibility antigen A-36 alpha chain (UniProt P30455) | 3.23 × 10−3 | 12.127 | 32.730 | |
| Receptor tyrosine-protein kinase erbB-2 (UniProt P04626) | 1.05 × 10−2 | 12.043 | 32.769 | |
| E7 protein Human papillomavirus type 16 | 7.10 × 10−4 | 11.119 | 27.237 | |
| L2 protein Human papillomavirus type 16 | 2.00 × 10−4 | 11.004 | 19.667 | |
| Probable protein E4 Human papillomavirus type 16 | 3.40 × 10−4 | 10.493 | 54.455 | |
| BZLF1 Human herpesvirus 4 | 1.34 × 10−3 | 10.168 | 31.873 | |
| Putative HTLV-1-related endogenous sequence (p25) Homo sapiens | 2.37 × 10−5 | 9.643 | 4.000 | |
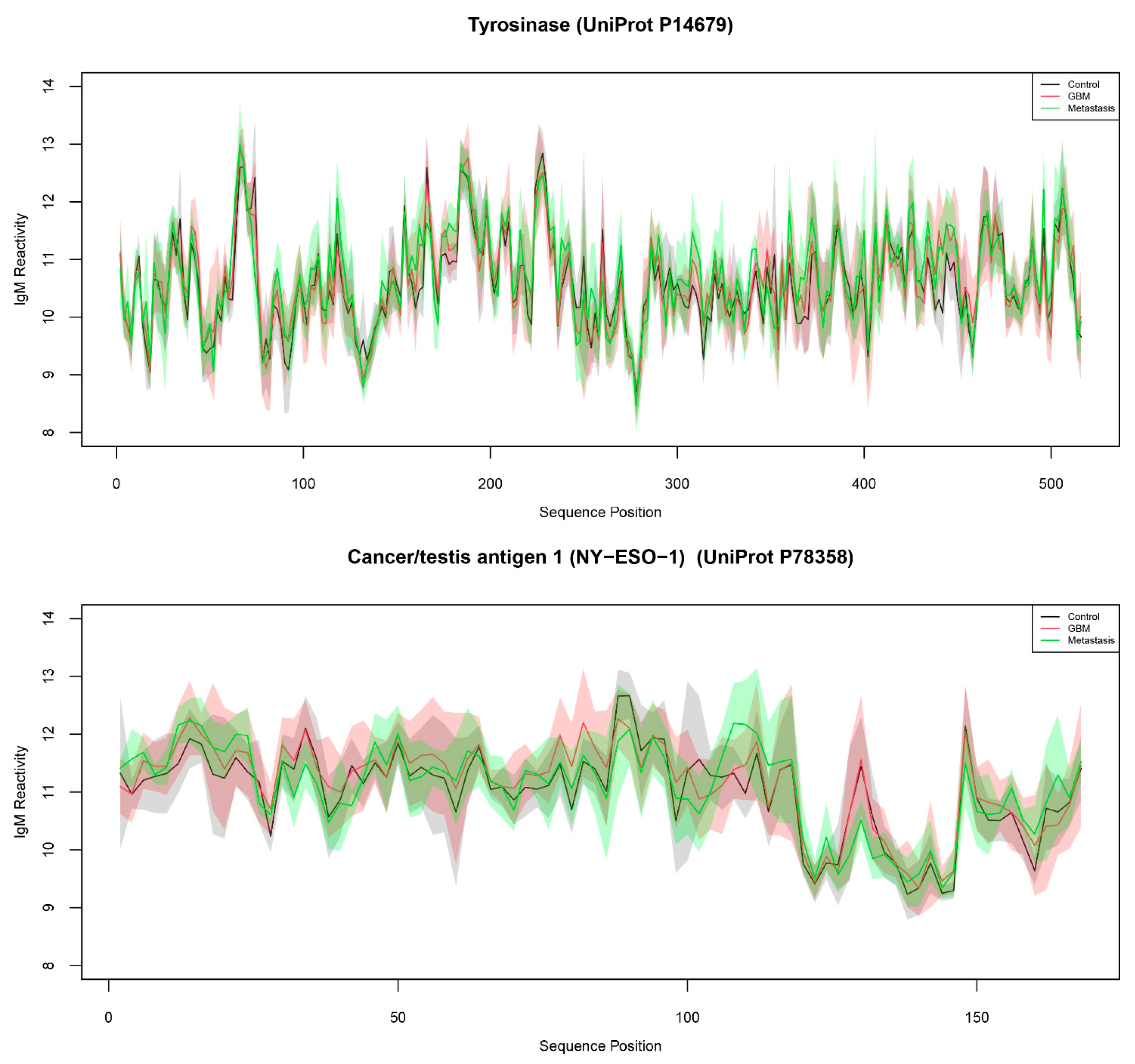
| Tyrosinase (UniProt P14679) | 3.31 × 10−3 | 8.536 | 34.015 | |
| Replication protein E1 Human papillomavirus type 16 | 6.30 × 10−5 | 8.226 | 101.667 | |
| Myelin-oligodendrocyte glycoprotein (UniProt Q16653) | 1.38 × 10−3 | 7.624 | 29.293 | |
| Claudin-6 (UniProt P56747) | 1.25 × 10−3 | 7.552 | 33.951 | |
| Capsid protein VP26 Human herpesvirus 4 (strain B95-8) | 1.21 × 10−4 | 7.409 | 20.091 | |
| Prostate-specific antigen (UniProt P07288) | 1.37 × 10−3 | 6.925 | 34.821 | |
| envelope glycoprotein Human T-lymphotropic virus 2 | 1.15 × 10−4 | 6.688 | 45.455 | |
| Ricin precursor Ricinus communis | 2.22 × 10−4 | 5.372 | 22.964 | |
| L1 protein Human papillomavirus type 16 | 4.81 × 10−4 | 5.340 | 30.340 | |
| Carbonic anhydrase 1 Homo sapiens | 1.83 × 10−4 | 5.096 | 41.682 | |
| tax protein Human T-lymphotropic virus 1 | 7.98 × 10−5 | 4.650 | 48.000 | |
| Pr gag-pro-pol Human T-lymphotropic virus 1 | 1.92 × 10−4 | 4.337 | 24.933 | |
| small viral capsid antigen Human herpesvirus 8 | 1.31 × 10−4 | 4.197 | 32.524 | |
| TCR gamma alternate reading frame protein (UniProt A2JGV3) | 1.23 × 10−4 | 4.114 | 34.727 | |
| L1 Human papillomavirus type 33 | 2.39 × 10−5 | 3.795 | 46.250 | |
| E7 protein Human papillomavirus type 18 | 2.27 × 10−5 | 2.807 | 45.667 | |
| Protein X Hepatitis B virus | 3.51 × 10−5 | 2.052 | 28.000 | |
| major capsid protein Human papillomavirus type 6 | 1.43 × 10−5 | 1.921 | 51.000 | |
| E2 protein Human papillomavirus type 16 | 2.37 × 10−5 | 1.801 | 19.222 | |
| ribonucleoside-diphosphate reductase large chain Human herpesvirus 4 | 1.60 × 10−5 | 1.650 | 30.500 | |
| Plasminogen-binding protein pgbA Helicobacter pylori | −1.39 × 10−8 | 0.353 | 13.000 | |
| hippocampal 38K autoantigen protein Homo sapiens | −7.91 × 10−8 | 0.163 | 15.500 | |
| Early antigen protein R Human herpesvirus 4 (strain B95-8) | −1.06 × 10−7 | 0.155 | 12.667 | |
| E2 Human papillomavirus type 18 | −1.23 × 10−7 | 0.100 | 21.500 | |
| Hbx protein Hepatitis B virus | −1.88 × 10−7 | 0.076 | 25.500 | |
| Latent membrane protein 2 Human herpesvirus 4 (strain B95-8) | −1.44 × 10−7 | 0.051 | 14.667 | |
| L2 Human papillomavirus type 6 | −5.81 × 10−7 | 0.040 | 23.000 | |
| E2 protein Human papillomavirus type 6 | −3.96 × 10−7 | 0.035 | 36.500 | |
| rex protein Human T-lymphotropic virus 1 | −3.03 × 10−6 | −0.008 | 21.100 | |
| T-cell receptor beta chain; TCR Homo sapiens | −5.53 × 10−7 | −0.020 | 43.000 | |
| E6 protein Human papillomavirus type 16 | −2.41 × 10−6 | −0.034 | 18.400 | |
| K8.1 Human herpesvirus 8 | −3.18 × 10−6 | −0.183 | 29.857 |
| Protein | N | |
|---|---|---|
| Mammaglobin-1 (UniProt Q13296) | SCGB2A2 | 39 |
| T-cell receptor beta chain; TCR Homo sapiens | TCRb | 2 |
| Carbonic anhydrase 1 Homo sapiens | CAH1 | 21 |
| TCR gamma alternate reading frame protein (UniProt A2JGV3) | TCRg_alt | 22 |
| Tyrosinase (UniProt P14679) | TYRO | 267 |
| Myc proto-oncogene protein (UniProt P01106) | Myc | 212 |
| Prostate-specific antigen (UniProt P07288) | PSA | 123 |
| Carcinoembryonic antigen-related cell adhesion molecule 5 (UniProt P06731) | CEA | 325 |
| Protein SSX2 (Cancer/testis antigen 5.2) (UniProt Q16385) | SSX2 | 87 |
| Myelin-oligodendrocyte glycoprotein (UniProt Q16653) | MOG | 116 |
| Transcription factor SOX-2 (UniProt P48431) | SOX2 | 151 |
| Claudin-6 (UniProt P56747) | CLDN6 | 103 |
| Receptor tyrosine-protein kinase erbB-2 (UniProt P04626) | erbB2 | 620 |
| L-dopachrome tautomerase (UniProt P40126) | DCT | 252 |
| HLA class I histocompatibility antigen A-36 alpha chain (UniProt P30455) | HLA_A36 | 178 |
| Stromelysin-3 (UniProt P24347) | MMP11 | 237 |
| Melanocyte protein PMEL (UniProt P40967) | PMEL | 323 |
| Cellular tumor antigen p53 (UniProt Q2XN98) | p53 | 200 |
| Histone H1.2 (UniProt P16403) | H1_2 | 98 |
| Hippocampal 38K autoantigen protein Homo sapiens | ELAVL2 | 2 |
| Cancer/testis antigen 1 (NY-ESO-1) (UniProt P78358) | NY-ESO-1 | 89 |
| G2/mitotic-specific cyclin-B1 (UniProt P14635) | CCNB1 | 209 |
| Histone H4 (UniProt P62805) | H4 | 41 |
| Putative HTLV-1-related endogenous sequence (p25) Homo sapiens | HRES1 | 2 |
2.2.5. Mapping Sequence and Specificity Data onto the Graph Topology
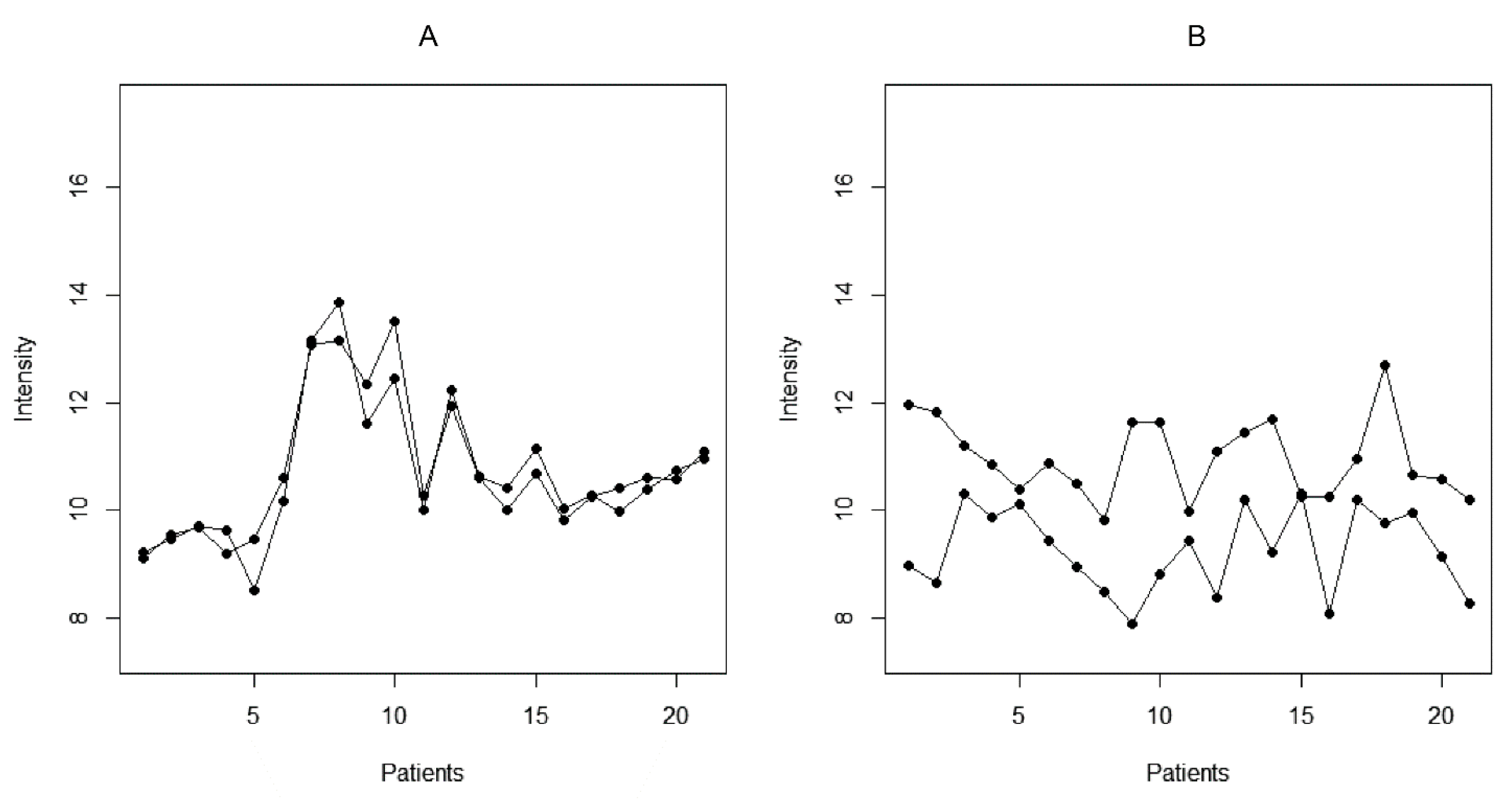
2.3. Reactivity Graph and the IgM Repertoire Changes in Brain Tumors
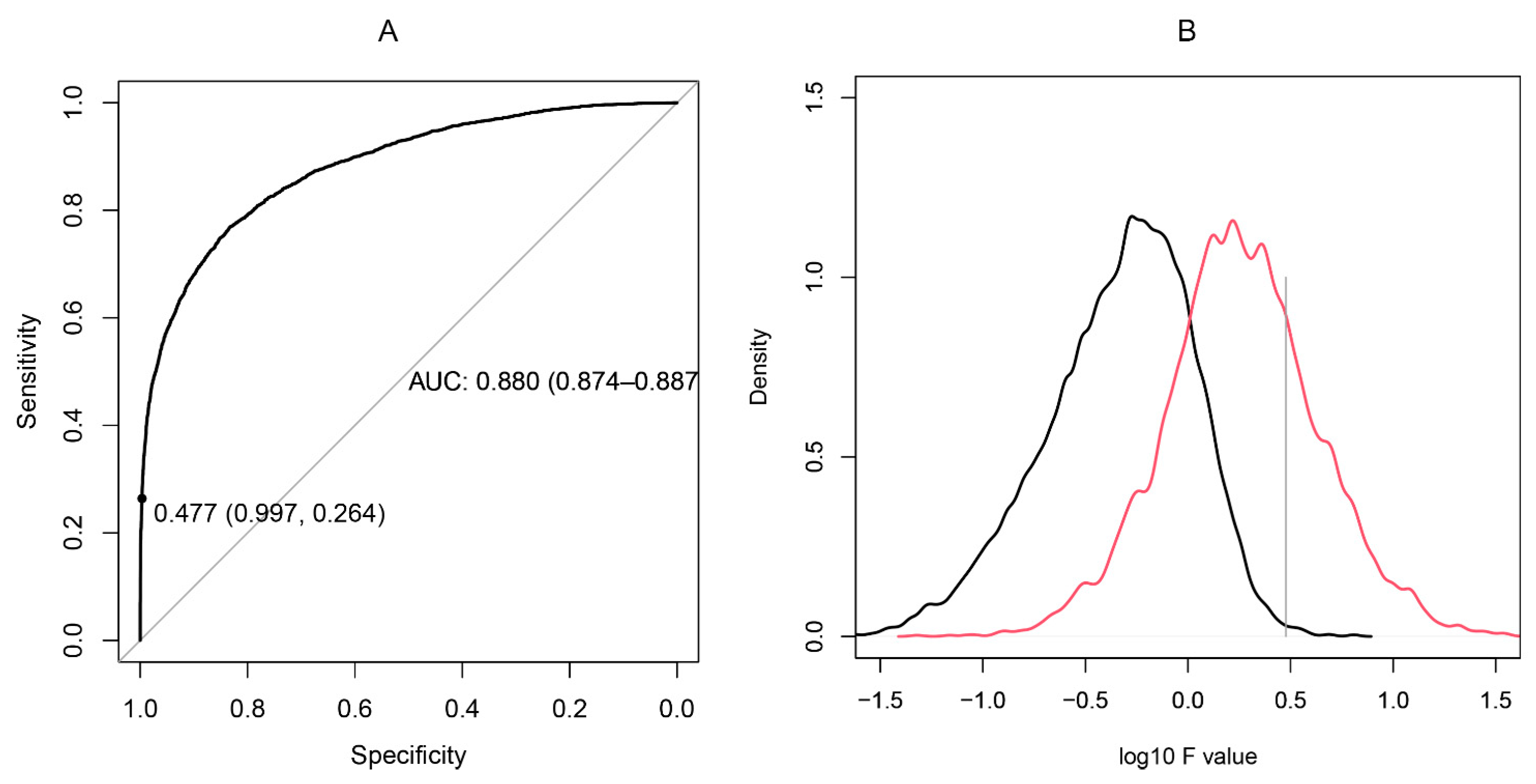
2.4. Proof-of-Principle Classifier
3. Discussion
4. Materials and Methods
4.1. Patients’ Sera
4.2. Peptide Microarray
4.3. Microarray Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Benichou, J.; Ben-Hamo, R.; Louzoun, Y.; Efroni, S. Rep-Seq: Uncovering the immunological repertoire through next-generation sequencing. Immunology 2012, 135, 183–191. [Google Scholar] [CrossRef]
- Greiff, V.; Bhat, P.; Cook, S.C.; Menzel, U.; Kang, W.; Reddy, S.T. A bioinformatic framework for immune repertoire diversity profiling enables detection of immunological status. Genome Med. 2015, 7, 49. [Google Scholar] [CrossRef]
- Miho, E.; Roškar, R.; Greiff, V.; Reddy, S.T. Large-scale network analysis reveals the sequence space architecture of antibody repertoires. Nat. Commun. 2019, 10, 1321. [Google Scholar] [CrossRef] [Green Version]
- Frietze, K.M.; Pascale, J.M.; Moreno, B.; Chackerian, B.; Peabody, D.S. Pathogen-specific deep sequence-coupled biopanning: A method for surveying human antibody responses. PLoS ONE 2017, 12, e0171511. [Google Scholar] [CrossRef] [Green Version]
- Frietze, K.M.; Core, S.B.; Linville, A.; Chackerian, B.; Peabody, D.S. Assessing Antibody Specificity in Human Serum Using Deep Sequence-Coupled Biopanning. In Genotype Phenotype Coupling: Methods and Protocols; Zielonka, S., Krah, S., Eds.; Springer: New York, NY, USA, 2020; pp. 157–171. [Google Scholar]
- Megremis, S.; Walker, T.D.J.; He, X.; O’Sullivan, J.; Ollier, W.E.R.; Chinoy, H.; Pendleton, N.; Payton, A.; Hampson, L.; Hampson, I.; et al. Analysis of human total antibody repertoires in TIF1γ autoantibody positive dermatomyositis. Commun. Biol. 2021, 4, 419. [Google Scholar] [CrossRef] [PubMed]
- Ryvkin, A.; Ashkenazy, H.; Smelyanski, L.; Kaplan, G.; Penn, O.; Weiss-Ottolenghi, Y.; Privman, E.; Ngam, P.B.; Woodward, J.E.; May, G.D.; et al. Deep Panning: Steps towards probing the IgOme. PLoS ONE 2012, 7, e41469. [Google Scholar] [CrossRef] [PubMed]
- Pashov, A.; Shivarov, V.; Hadzhieva, M.; Kostov, V.; Ferdinandov, D.; Heintz, K.-M.; Pashova, S.; Todorova, M.; Vassilev, T.; Kieber-Emmons, T.; et al. Diagnostic Profiling of the Human Public IgM Repertoire With Scalable Mimotope Libraries. Front. Immunol. 2019, 10, 2796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stafford, P.; Wrapp, D.; Johnston, S.A. General Assessment of Humoral Activity in Healthy Humans. Mol Cell Proteom. 2016, 15, 1610–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.; Smith, D.F.; Cummings, R.D.; Boligan, K.F.; Hamilton, R.G.; Bochner, B.S.; Miescher, S.; Simon, H.-U.; Pashov, A.; Vassilev, T.; et al. The human IgG anti-carbohydrate repertoire exhibits a universal architecture and contains specificity for microbial attachment sites. Sci. Transl. Med. 2015, 7, 269ra1. [Google Scholar] [CrossRef] [Green Version]
- Merbl, Y.; Itzchak, R.; Vider-Shalit, T.; Louzoun, Y.; Quintana, F.J.; Vadai, E.; Eisenbach, L.; Cohen, I.R. A Systems Immunology Approach to the Host-Tumor Interaction: Large-Scale Patterns of Natural Autoantibodies Distinguish Healthy and Tumor-Bearing Mice. PLoS ONE 2009, 4, e6053. [Google Scholar] [CrossRef]
- Janeway, J.C.A. Immunobiology: The Immune System in Health and Disease, 2nd ed.; Current Biology Ltd.: London, UK, 1996; p. 389. [Google Scholar]
- Baumgarth, N. A Hard(y) Look at B-1 Cell Development and Function. J. Immunol. 2017, 199, 3387–3394. [Google Scholar] [CrossRef] [Green Version]
- Reyneveld, G.I.; Savelkoul, H.F.J.; Parmentier, H.K. Current Understanding of Natural Antibodies and Exploring the Possibilities of Modulation Using Veterinary Models. A Review. Front. Immunol. 2020, 11, 2139. [Google Scholar] [CrossRef] [PubMed]
- Quách, T.D.; Rodríguez-Zhurbenko, N.; Hopkins, T.J.; Guo, X.; Hernández, A.M.; Li, W.; Rothstein, T.L. Distinctions among Circulating Antibody-Secreting Cell Populations, Including B-1 Cells, in Human Adult Peripheral Blood. J. Immunol. 2016, 196, 1060–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagnara, D.; Squillario, M.; Kipling, D.; Mora, T.; Walczak, A.M.; Da Silva, L.; Weller, S.; Dunn-Walters, D.K.; Weill, J.C.; Reynaud, C.A. A Reassessment of IgM Memory Subsets in Humans. J. Immunol. 2015, 195, 3716–3724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grönwall, C.; Silverman, G.J. Natural IgM: Beneficial autoantibodies for the control of inflammatory and autoimmune disease. J. Clin. Immunol. 2014, 34 (Suppl. S1), S12–S21. [Google Scholar] [CrossRef] [Green Version]
- Hughes, A.K.; Cichacz, Z.; Scheck, A.; Coons, S.W.; Johnston, S.A.; Stafford, P. Immunosignaturing Can Detect Products from Molecular Markers in Brain Cancer. PLoS ONE 2012, 7, e40201. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.; Ladd, J.; Gad, E.; Rastetter, L.; Johnson, M.M.; Marzbani, E.; Childs, J.S.; Lu, H.; Dang, Y.; Broussard, E.; et al. Mining the pre-diagnostic antibody repertoire of TgMMTV-neu mice to identify autoantibodies useful for the early detection of human breast cancer. J. Transl. Med. 2014, 12, 121. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Mukherjee, S.; Syed, P.; Pandala, N.G.; Choudhary, S.; Singh, V.A.; Singh, N.; Zhu, H.; Epari, S.; Noronha, S.B.; et al. Evaluation of autoantibody signatures in meningioma patients using human proteome arrays. Oncotarget 2017, 8, 58443–58456. [Google Scholar] [CrossRef] [Green Version]
- Mock, A.; Warta, R.; Geisenberger, C.; Bischoff, R.; Schulte, A.; Lamszus, K.; Stadler, V.; Felgenhauer, T.; Schichor, C.; Schwartz, C.; et al. Printed peptide arrays identify prognostic TNC serumantibodies in glioblastoma patients. Oncotarget 2015, 6, 13579–13590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, P.; Gupta, S.; Choudhary, S.; Pandala, N.G.; Atak, A.; Richharia, A.; K, P.M.; Zhu, H.; Epari, S.; Noronha, S.B.; et al. Autoantibody Profiling of Glioma Serum Samples to Identify Biomarkers Using Human Proteome Arrays. Sci. Rep. 2015, 5, 13895. [Google Scholar] [CrossRef]
- Rawat, K.; Soucy, S.M.; Kolling, F.W.; Diaz, K.M.; King, W.T.; Tewari, A.; Jakubzick, C.V. Natural Antibodies Alert the Adaptive Immune System of the Presence of Transformed Cells in Early Tumorigenesis. J. Immunol. 2022, 209, 1252–1259. [Google Scholar] [CrossRef] [PubMed]
- Hennings, L.; Artaud, C.; Jousheghany, F.; Monzavi-Karbassi, B.; Pashov, A.; Kieber-Emmons, T. Carbohydrate Mimetic Peptides Augment Carbohydrate-Reactive Immune Responses in the Absence of Immune Pathology. Cancers 2011, 3, 4151–4169. [Google Scholar] [CrossRef] [Green Version]
- Vollmers, H.P.; Brandlein, S. The “early birds”: Natural IgM antibodies and immune surveillance. Histol Histopathol 2005, 20, 927–937. [Google Scholar] [PubMed]
- Pashova, S.; Schneider, C.; von Gunten, S.; Pashov, A. Antibody repertoire profiling with mimotope arrays. Hum. Vaccines Immunother. 2017, 13, 314–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pashova, S.; Balabanski, L.; Elmadjian, G.; Savov, A.; Stoyanova, E.; Shivarov, V.; Petrov, P.; Pashov, A. Restriction of the Global IgM Repertoire in Antiphospholipid Syndrome. Front. Immunol. 2022, 13, 865232. [Google Scholar] [CrossRef]
- Barabási, A.L.; Posfai, M.Ã. Network Science; Cambridge University Press: Cambridge, UK, 2016. [Google Scholar]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. In Proceedings of the Third International AAAI Conference on Weblogs and Social Media, San Jose, CA, USA, 17–20 May 2009. [Google Scholar]
- Edelsbrunner, H.; Harer, J. Computational Topology: An Introduction; American Mathematical Society: Providence, RI, USA, 2010. [Google Scholar]
- Grant, C.E.; Bailey, T.L. XSTREME: Comprehensive motif analysis of biological sequence datasets. bioRxiv 2021. [Google Scholar] [CrossRef]
- Gessain, A.; Cassar, O. Epidemiological Aspects and World Distribution of HTLV-1 Infection. Front. Microbiol. 2012, 3, 388. [Google Scholar] [CrossRef] [Green Version]
- Legutki, J.B.; Magee, D.M.; Stafford, P.; Johnston, S.A. A general method for characterization of humoral immunity induced by a vaccine or infection. Vaccine 2010, 28, 4529–4537. [Google Scholar] [CrossRef]
- Roffo, G.; Melzi, S.; Castellani, U.; Vinciarelli, A.; Cristani, M. Infinite Feature Selection: A Graph-based Feature Filtering Approach. arXiv 2020, arXiv:2006.08184. [Google Scholar] [CrossRef]
- Cerruela-García, G.; Cuevas-Muñoz, J.M.; García-Pedrajas, N. Graph-Based Feature Selection Approach for Molecular Activity Prediction. J. Chem. Inf. Model. 2022, 62, 1618–1632. [Google Scholar] [CrossRef]
- Yam-Puc, J.C.; Zhang, L.; Maqueda-Alfaro, R.A.; Garcia-Ibanez, L.; Zhang, Y.; Davies, J.; Senis, Y.A.; Snaith, M.; Toellner, K.M. Enhanced BCR signaling inflicts early plasmablast and germinal center B cell death. iScience 2021, 24, 102038. [Google Scholar] [CrossRef] [PubMed]
- Vollmers, H.P.; Brandlein, S. Natural antibodies and cancer. New Biotechnol. 2009, 25, 294–298. [Google Scholar] [CrossRef]
- Díaz-Zaragoza, M.; Hernández-Ávila, R.; Viedma-Rodríguez, R.; Arenas-Aranda, D.; Ostoa-Saloma, P. Natural and adaptive IgM antibodies in the recognition of tumor-associated antigens of breast cancer (Review). Oncol. Rep. 2015, 34, 1106–1114. [Google Scholar] [CrossRef] [Green Version]
- Roney, M.S.I.; Lanagan, C.; Sheng, Y.H.; Lawler, K.; Schmidt, C.; Nguyen, N.T.; Begun, J.; Kijanka, G.S. IgM and IgA augmented autoantibody signatures improve early-stage detection of colorectal cancer prior to nodal and distant spread. Clin. Transl. Immunol. 2021, 10, e1330. [Google Scholar] [CrossRef]
- Vale, A.M.; Kearney, J.F.; Nobrega, A.; Schroeder, H.W. Chapter 7—Development and Function of B Cell Subsets. In Molecular Biology of B Cells, 2nd ed.; Alt, F.W., Honjo, T., Radbruch, A., Reth, M., Eds.; Academic Press: London, UK, 2015; pp. 99–119. [Google Scholar]
- Garaud, S.; Morva, A.; Lemoine, S.; Hillion, S.; Bordron, A.; Pers, J.O.; Berthou, C.; Mageed, R.A.; Renaudineau, Y.; Youinou, P. CD5 promotes IL-10 production in chronic lymphocytic leukemia B cells through STAT3 and NFAT2 activation. J. Immunol. 2011, 186, 4835–4844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, K.; Asano, M.; Shinton, S.A.; Gui, M.; Wen, L.J.; Dashoff, J.; Hardy, R.R. Positive selection of anti-thy-1 autoreactive B-1 cells and natural serum autoantibody production independent from bone marrow B cell development. J. Exp. Med. 2003, 197, 87–99. [Google Scholar] [CrossRef]
- Baumgarth, N. Breaking the Paradigm: Selection of Self-Reactive Natural Antibodies. J. Immunol. 2022, 209, 1621–1623. [Google Scholar] [CrossRef]
- Übelhart, R.; Jumaa, H. Autoreactivity and the positive selection of B cells. Eur. J. Immunol. 2015, 45, 2971–2977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, L.; Brill-Dashoff, J.; Shinton, S.A.; Asano, M.; Hardy, R.R.; Hayakawa, K. Evidence of marginal-zone B cell-positive selection in spleen. Immunity 2005, 23, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudin, E.; Hao, Y.; Rosado, M.M.; Chaby, R.; Girard, R.; Freitas, A.A. Positive Selection of B Cells Expressing Low Densities of Self-reactive BCRs. J. Exp. Med. 2004, 199, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Varela, F.; Andersson, A.; Dietrich, G.; Sundblad, A.; Holmberg, D.; Kazatchkine, M.; Coutinho, A. Population dynamics of natural antibodies in normal and autoimmune individuals. Proc. Natl. Acad. Sci. USA 1991, 88, 5917–5921. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wang, P.; Kim, Y.; Haste-Andersen, P.; Beaver, J.; Bourne, P.E.; Bui, H.H.; Buus, S.; Frankild, S.; Greenbaum, J.; et al. Immune epitope database analysis resource (IEDB-AR). Nucleic Acids Res. 2008, 36, W513–W518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, F.B.; Hubert, L.J. Measuring the Power of Hierarchical Cluster Analysis. J. Am. Stat. Assoc. 1975, 70, 31–38. [Google Scholar] [CrossRef]
- Dunn, J.C. Well-Separated Clusters and Optimal Fuzzy Partitions. J. Cybern. 1974, 4, 95–104. [Google Scholar] [CrossRef]
- Handl, J.; Knowles, J.; Kell, D.B. Computational cluster validation in post-genomic data analysis. Bioinformatics 2005, 21, 3201–3212. [Google Scholar] [CrossRef] [PubMed]











| Protein | N | ||
|---|---|---|---|
| HHV4 (EBV) | BZLF1 Human herpesvirus 4 | EBV-BZLF1 | 79 |
| Capsid protein VP26 Human herpesvirus 4 (strain B95-8) | EBV_VP26 | 11 | |
| Early antigen protein R Human herpesvirus 4 (strain B95-8) | EBV_EA | 3 | |
| Epstein–Barr nuclear antigen 1 Human herpesvirus 4 | EBV_EBNA1 | 45 | |
| Latent membrane protein 2 Human herpesvirus 4 (strain B95-8) | EBV_LMP2 | 3 | |
| ribonucleoside-diphosphate reductase large chain Human herpesvirus 4 | EBV_RIR1 | 8 | |
| HPV ( types 6, 16, 18, and 33) | E2 protein Human papillomavirus type 6 | HPV6_E2 | 2 |
| major capsid protein Human papillomavirus type 6 | HPV6_L1 | 6 | |
| L2 Human papillomavirus type 6 | HPV6_L2 | 4 | |
| Replication protein E1 Human papillomavirus type 16 | HPV16_E1 | 6 | |
| E2 protein Human papillomavirus type 16 | HPV16_E2 | 9 | |
| Probable protein E4 Human papillomavirus type 16 | HPV16_E4 | 22 | |
| E6 protein Human papillomavirus type 16 | HPV16_E6 | 10 | |
| E7 protein Human papillomavirus type 16 | HPV16_E7 | 38 | |
| L1 protein Human papillomavirus type 16 | HPV16_L1 | 53 | |
| L2 Human papillomavirus type 16 | HPV16_L2 | 12 | |
| E2 Human papillomavirus type 18 | HPV18_E2 | 2 | |
| E7 protein Human papillomavirus type 18 | HPV18_E7 | 6 | |
| L1 Human papillomavirus type 33 | HPV33_L1 | 4 | |
| Envelope glycoprotein gp62 precursor Human T-lymphotropic virus 1 | HTLV1_env | 125 | |
| HTLV1 and 2 | Pr gag-pro-pol Human T-lymphotropic virus 1 | HTLV1_gag | 30 |
| rex protein Human T-lymphotropic virus 1 | HTLV1_rex | 10 | |
| tax protein Human T-lymphotropic virus 1 | HTLV1_tax | 12 | |
| envelope glycoprotein Human T-lymphotropic virus 2 | HTLV2_env | 11 | |
| HBV | Hbx protein Hepatitis B virus | HBV_X | 13 |
| HHV8 | K8.1 Human herpesvirus 8 | HHV8_K8_1 | 7 |
| LANA Human herpesvirus 8 | HHV8_LANA | 26 | |
| small viral capsid antigen Human herpesvirus 8 | HHV8_ORF65 | 21 | |
| H. pylori | Plasminogen-binding protein pgbA Helicobacter pylori | Hp_pgbA | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferdinandov, D.; Kostov, V.; Hadzhieva, M.; Shivarov, V.; Petrov, P.; Bussarsky, A.; Pashov, A.D. Reactivity Graph Yields Interpretable IgM Repertoire Signatures as Potential Tumor Biomarkers. Int. J. Mol. Sci. 2023, 24, 2597. https://doi.org/10.3390/ijms24032597
Ferdinandov D, Kostov V, Hadzhieva M, Shivarov V, Petrov P, Bussarsky A, Pashov AD. Reactivity Graph Yields Interpretable IgM Repertoire Signatures as Potential Tumor Biomarkers. International Journal of Molecular Sciences. 2023; 24(3):2597. https://doi.org/10.3390/ijms24032597
Chicago/Turabian StyleFerdinandov, Dilyan, Viktor Kostov, Maya Hadzhieva, Velizar Shivarov, Peter Petrov, Assen Bussarsky, and Anastas Dimitrov Pashov. 2023. "Reactivity Graph Yields Interpretable IgM Repertoire Signatures as Potential Tumor Biomarkers" International Journal of Molecular Sciences 24, no. 3: 2597. https://doi.org/10.3390/ijms24032597
APA StyleFerdinandov, D., Kostov, V., Hadzhieva, M., Shivarov, V., Petrov, P., Bussarsky, A., & Pashov, A. D. (2023). Reactivity Graph Yields Interpretable IgM Repertoire Signatures as Potential Tumor Biomarkers. International Journal of Molecular Sciences, 24(3), 2597. https://doi.org/10.3390/ijms24032597