


Metabolism-Disrupting Chemicals Affecting the Liver: Screening, Testing, and Molecular Pathway Identification

Abstract
:1. Background
2. Signaling Molecules and Receptors Regulating Hepatic Energy Metabolism

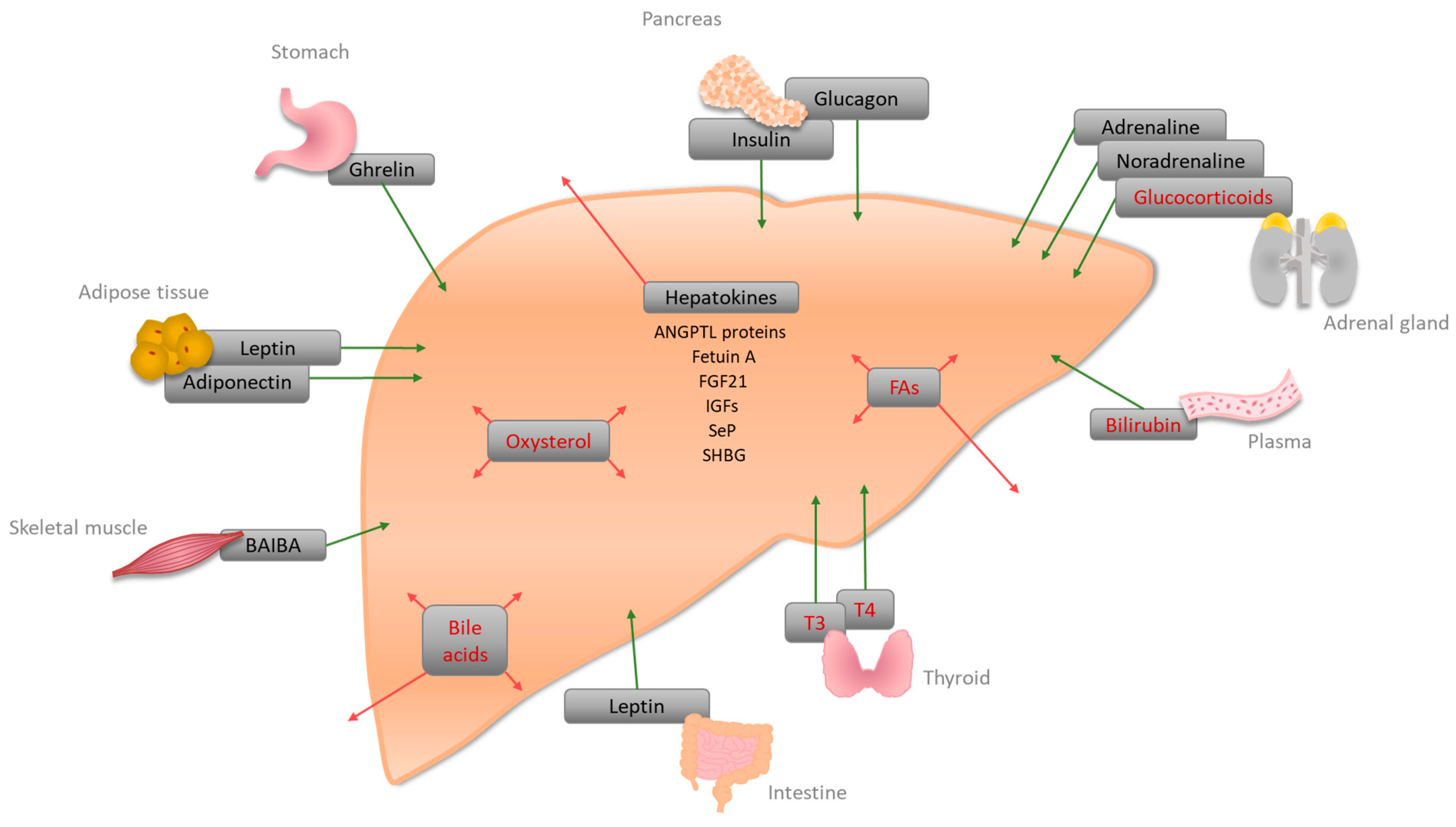{kind=link}
| Hormone | Site of Synthesis | Receptor(s) | Site of Action | Function |
|---|---|---|---|---|
| A | ||||
| Bile acid | Liver | FXR, PXR, VDR, GPCRs (TGR5, Sphingosine 1-phosphate receptor (S1P2)) | Liver, intestine | Bile acid homeostasis, lipid, glucose, and energy homeostasis [51] |
| Bilirubin | Plasma | CAR, PXR, PPARα | Liver | Conjugation and secretion of bilirubin [94], an increase of FA oxidation, and decrease of lipid accumulation [15] |
| Fatty acids (FAs) | Liver, Adipose tissue | PPARα, PPARβ/δ, PPARγ | PPARα: liver, muscle, BAT, heart; PPARβ/δ: ubiquitous; PPARγ: adipose tissue, weak in Liver | PPARα: increase of fatty acid oxidation (FAO), a decrease of glucose uptake; PPARβ/δ: increase of FAO and glucose metabolism, decrease of inflammation [95]; PPARγ: might be involved in FA uptake and DAG synthesis [33] |
| Glucocorticoids (corticosterone, cortisol) | Adrenal cortex | GRs | Liver | Gluconeogenesis by lipolysis and ketogenesis [38,41] |
| Oxysterol | Liver | LXRα/LXRβ | Liver | Activation of LXRα, regulation of cholesterol metabolism [96]; LXR-induced Srebp-1c increases de novo lipogenesis [50] |
| Thyroid hormones (T3, T4) | Thyroid | TRα, TRβ | Liver, kidney, bone, heart | Cholesterol metabolism, stimulation of FAO, activation of de novo lipogenesis, and glucose homeostasis [97] |
| B | ||||
| Adiponectin (adipokine) | White adipose tissue (WAT) | Adiponectin receptor 1 and 2 (AdipoR1/2) | Liver, skeletal muscle, WAT | Suppression of glucose production in the liver via activation of AMPK [67] |
| Adrenaline, noradrenaline | Adrenal medulla | Adrenoreceptors alpha1, alpha2, and beta | Liver | Glycogenolysis, increase of blood glucose [65,66] |
| Angiopoietin-like proteins (ANGPTL3, ANGPTL 6) * | Liver | - | Plasma | Increase of plasma TG level in mice via lipoprotein lipase inhibition [84]; activation of Angptl6 has been associated with protection from HFD-induced obesity, insulin resistance, and hepatic steatosis [87] |
| β-aminoisobutyric acid (BAIBA) | Skeletal muscle | AMPK, transcription factors | Liver, WAT, skeletal muscle | Improvement of hepatic lipid metabolism via PPAR-mediated β-oxidation [69,70] |
| Fetuin A (α2-HS-Glycoprotein) * | Liver | - | Plasma | Inhibition of insulin receptor tyrosine kinase [71] |
| Fibroblast growth factor 21 (FGF21) * | Liver | - | Plasma | Fasting-induced hormone enhancing insulin sensitivity, lowering body weight, and increasing gluconeogenesis [98] |
| Ghrelin | Stomach | Ghrelin receptor (GHSR1a) | Liver, Agouti-related protein (ARGP)/neuropeptide Y (NPY) neurons, adipocytes | Increase of triglycerides by induction of lipogenesis-related gene expression [64] |
| Glucagon | Pancreas | Glucagon receptor | Mainly liver, kidney | Gluconeogenesis [99] |
| Insulin | Pancreas | Insulin receptor | liver | Lipogenesis, cholesterol uptake, and synthesis [100] |
| Insulin-like growth factors-1 and -2 (IGFs) * | Liver | IGF receptors -1 and -2 | Plasma | IGF-1 decreases blood glucose levels, and improves insulin sensitivity [88,89,90]. IGF-2 can be a key factor in steatosis initiation [91] |
| Leptin | Adipose tissue, small intestine | Leptin receptor | Liver, hypothalamus, and several other tissues | Lack of hepatic leptin leads to increased lipid accumulation in the liver [62] |
| Selenoprotein P (SeP) * | Liver | - | Plasma | Glycoprotein; hepatic expression has been linked to insulin resistance [80] |
| Sex-hormone-binding globulin (SHBG) * | Liver | SHBG-receptor | Plasma | Circulating levels of SHBG are a biomarker for insulin resistance and type II diabetes [93] |
3. Examples of Compounds Affecting the Liver and Inducing Metabolic Changes
3.1. Bisphenols
3.2. (Tri-)azoles
3.3. Polyfluoroalkyl Substances (PFAS)
3.4. Polychlorinated Biphenyls (PCBs)
3.5. Phthalates
3.6. Dioxins
3.7. Alkylphenols
3.8. Organotins
3.9. Polycyclic Aromatic Hydrocarbons (PAHs)
3.10. Non-Steroidal Estrogens
3.11. Organochlorines
3.12. Organophosphates
3.13. Heavy Metals
| Substance | Putative Mechanism | Effect | Test System | Reference |
|---|---|---|---|---|
| Bisphenols (BPA) | ROS production | Lipid accumulation | In vitro (HepG2, 72 h) | [107] |
| Upregulation of genes involved in lipogenesis | Accumulation of liver TGs | In vivo (mice, 28 days) | [108] | |
| Inhibition of autophagy possibly via mTOR | Hepatic lipid accumulation | In vivo (male mice, 8 weeks) and in vitro (HepG2, primary hepatocytes) | [109] | |
| Upregulation of Pparγ | Increase of hepatic triglycerides | In vivo (in utero exposure of male mice, days 9 to 16 of pregnancy) | [110] | |
| Promoter methylation of hepatic glucokinase | Increase in hepatic glycogen content | In vivo (rats, throughout gestation and lactation) | [111] | |
| (Tri-)azoles (propiconazole, tebuconazole) | Activation of PXR, CAR, regulation of steatosis-related genes | Triglyceride accumulation | In vitro (HepG2/HepaRG, 24 h) | [117] |
| PFAS (PFOS, PFOA) | PPARα | Increase in liver weight and cell size, increased lipid accumulation, liver steatosis | In vivo (mice, 7 days) | [126] |
| Inhibition of mitochondrial FA β-oxidation | Hepatic steatosis | In vivo (mice, up to 21 days) | [125] | |
| Decrease of CYP7A1 | Decreased levels of bile acids | In vitro (HepaRG, 24 and 48 h) | [130] | |
| Modulation of PI3K-AKT pathway | Altered glucose homeostasis and induction of insulin sensitivity | In vivo (mice, 28 days) | [109] | |
| PCBs (PCB 126, Aroclor 1260) | PCB126: Increased expression of Nr1i3 (Car), induction of Cyp1a2, Cyp2b10, and genes involved in lipid metabolism | Increased TGs and free FAs leading to steatosis | In vivo (male mice, 2 weeks) | [137] |
| Aroclor 1260: PXR, CAR, AhR (agonistically) PPARα (antagonistically) | Induction of CYP1A1, CD36 (AhR), induction of CYP3A4 (PXR) | In vitro (HepG2 and primary human hepatocytes, 24 h) | [137] | |
| Phthalates (DEHP, DBP, MEHP) | Activation of SREBP-1c and PPARα | Lipid accumulation | In vitro (HepG2, 48 h) | [142] |
| Activation of CAR2, induction of CYP2B6 and CYP3A4 | - | In vitro (HepG2, 48 h) | [143] | |
| Dioxin (TCDD) | AhR | Insulin resistance-like phenotype | In vivo (mice, 18 days) | [146] |
| Inhibition of VLDL-TG secretion | In vivo (mice, 7 days) | [147] | ||
| Alkylphenols (4-NP) | Contributing factors: Fas Cell Surface Death Receptor (FAS)/FAS ligand (FASL), Tumor Necrosis Factor alpha (TNFα), Caspase-9 mRNA activation | Hepatic steatosis and apoptosis | In vivo (male rats, 30 days) | [150] |
| - | Steatosis and NAFLD | In vivo (male rats, 90 days chronic exposure) | [151] | |
| Increased activity of hexokinase and phosphofructokinase, a decrease of glycogen phosphorylase, increased H2O2 generation and lipid peroxidation, decreased protein level of insulin receptor (IR), IR substrate (IRS)-1 and IRS2 and PI3K | Short-term: impaired liver glucose homeostasis | In vivo (rats, 7 days) | [174] | |
| Long-term: downregulation of insulin signaling | In vivo (rats, 45 days) | [173] | ||
| Organotins (TBT) | Activation of PPARγ and RXR, increased gene expression of genes involved in lipogenesis, FA synthesis, glycerol uptake, lipolysis | Hepatic lipid accumulation | In vivo (adult mice upon in-utero exposure throughout pregnancy) | [153] |
| PPARγ/RXR-induced induction of lipogenesis | Increased hepatic TGs, steatosis | In vivo (mice upon in utero exposure from E12–18) and in vitro (HepaRG, 14 days) | [155,158] | |
| Reduction of hepatic resistin and adiponectin, an increase of plasma resistin and leptin | Hepatic steatosis, hyperinsulinemia, and hyperleptinemia | In vivo (male mice, 45 days) | [154,185] | |
| Dose- and sex-specific alterations of genes involved in lipogenesis | Accumulation of hepatic triglycerides in males, hepatomegaly in females | In vivo (zebrafish, pre-hatch-9 months) | [157] | |
| PAHs (BaP, fluoranthene) | AhR (some BaP metabolites [186]), gene expression related to FA β-oxidation | Hepatic steatosis | In vivo (mice upon in utero exposure from gestational days 7–16) | [163] |
| CAR (pyrene and fluoranthene) and CYP2B6 induction | In vitro (HepG2 and HepaRG, 24 h) | [164] | ||
| Non-steroidal estrogens (DES) | ERα, SHP | Increases liver weight, alteration in bile acid and triglyceride homeostasis | In vivo (mice: neonatal exposure, 5 days) | [167,168] |
| Suppression of ApoE secretion → reduction of serum High-Density-Lipoprotein (HDL)/cholesterol levels | Steroidogenesis disruption in adrenal glands | In vivo (male rats, 24 h) | [169] | |
| Organochlorines (Vinyl chloride) | Decreased mitochondrial respiration, endoplasmatic reticulum stress, impaired ALDH2 function | Enhanced TG accumulation in HFD-induced hepatic steatosis | In vivo (mice, 12 weeks) | [173,174] |
| Increase of FA synthesis, possibly via endoplasmatic reticulum- and oxidative stress | Hepatic steatosis | In vivo (mice: sub-chronic exposure, 16 weeks) | [175] | |
| Organophosphates (OPFRs, Malathion) | ERα/β, PXR, AR, GR | - | - | [176] |
| De novo FA synthesis, inhibition of β-oxidation, induction of total cholesterol deposition, mitochondrial dysfunction | Lipid accumulation | In vitro (HepG2, 24 h) | [177,178] | |
| Oxidative stress | Promotion of insulin resistance, hepatic steatosis | In vivo (rats, 28 days) | [179] | |
| Increased hepatic PEPCK and glycogen phosphorylase activity | Increased glucose release into the blood | In vivo (rats, sub-chronic exposure, 4 weeks) | [180] | |
| Heavy metals (Cadmium, cadmium chloride) | Increased activity of key enzymes involved in glucose production | Increased gluconeogenesis | In vivo (rats, 45 days) | [182] |
| HFD-related altered levels of metallothionein | Exacerbated (higher-dose exposure) and attenuated (low-dose exposure) HFD-induced steatosis | In vivo (mice, whole life exposure, starting in utero) | [184] | |
| Upregulation of SREBP1/2 and downregulation of PPARα, suppression of SIRT1/FXR axis | Induction of NAFLD | In vivo (rats, 10 weeks) | [183] | |
| Differential expression of NAFLD-associated genes | Increased liver lipids | In vivo (male mice, low-dose exposure) | [187] | |
| Accumulation of TG, upregulation of steatotic marker genes | In vitro (HepaRG and HepG2 cells) | [188] |
4. Testing Methods for MDC Identification
| Method | Principle | Effects Analyzed | Status | Reference |
|---|---|---|---|---|
| OECD standardized test guidelines for evaluating EDs | Repeated-dose 28-day/90-day study | Body and organ weight, (histo)pathology, clinical chemistry | Harmonized test guidelines approved for regulatory use | [190,191] |
| In vivo endpoints (to characterize metabolic phenotype) | Glucose and insulin tolerance test (GTT, ITT) | Blood glucose levels are measured upon administration of glucose/insulin | Additional techniques might be added as new endpoints | [199,200] |
| Non-targeted metabolomics | Non-targeted liquid chromatography/mass spectrometry (LC/MS) | [196,197,205,206] | ||
| Targeted metabolomics | Triglyceride measurement by gas chromatography | |||
| In silico approach | Computerized models (e.g., (Q)SAR) predicting physicochemical, biological, and environmental fate properties based on chemical structure | Interaction of a chemical with a defined biological target (modeling of molecular docking simulations to receptors) | Use for identification of MIEs of AOPs | [178] |
| Grouping of substances and read-across | Use of relevant information from tested substances to predict the properties of target substances | Alternative approach for filling data gaps | In registrations submitted under the REACH regulation | [204] |
| In vitro toolbox | AOP-based in vitro assays measuring MIEs or KEs | Combinations of NR activation, gene and protein expression, lipid accumulation, mitochondrial respiration/dysfunction, formation of fatty liver cells | Use for AOPs | [118,178,202] |
| Transcriptomic signatures | In vitro model | Gene expression markers for accumulation of triglycerides | [203] |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grun, F.; Blumberg, B. Environmental obesogens: Organotins and endocrine disruption via nuclear receptor signaling. Endocrinology 2006, 147, S50–S55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grun, F.; Blumberg, B. Endocrine disrupters as obesogens. Mol. Cell. Endocrinol. 2009, 304, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heindel, J.J.; Blumberg, B.; Cave, M.; Machtinger, R.; Mantovani, A.; Mendez, M.A.; Nadal, A.; Palanza, P.; Panzica, G.; Sargis, R.; et al. Metabolism disrupting chemicals and metabolic disorders. Reprod. Toxicol. 2017, 68, 3–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heindel, J.J.; Vom Saal, F.S.; Blumberg, B.; Bovolin, P.; Calamandrei, G.; Ceresini, G.; Cohn, B.A.; Fabbri, E.; Gioiosa, L.; Kassotis, C.; et al. Parma consensus statement on metabolic disruptors. Environ. Health 2015, 14, 54. [Google Scholar] [CrossRef] [Green Version]
- Nadal, A.; Quesada, I.; Tuduri, E.; Nogueiras, R.; Alonso-Magdalena, P. Endocrine-disrupting chemicals and the regulation of energy balance. Nat. Rev. Endocrinol. 2017, 13, 536–546. [Google Scholar] [CrossRef]
- Miyajima, A.; Tanaka, M.; Itoh, T. Stem/progenitor cells in liver development, homeostasis, regeneration, and reprogramming. Cell Stem Cell 2014, 14, 561–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lempradl, A.; Pospisilik, J.A.; Penninger, J.M. Exploring the emerging complexity in transcriptional regulation of energy homeostasis. Nat. Rev. Genet. 2015, 16, 665–681. [Google Scholar] [CrossRef]
- Stefan, N.; Haring, H.U. The role of hepatokines in metabolism. Nat. Rev. Endocrinol. 2013, 9, 144–152. [Google Scholar] [CrossRef]
- Han, H.S.; Kang, G.; Kim, J.S.; Choi, B.H.; Koo, S.H. Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med. 2016, 48, e218. [Google Scholar] [CrossRef] [Green Version]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride Metabolism in the Liver. Compr. Physiol. 2017, 8, 1–22. [Google Scholar] [CrossRef]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannon, W.B. Bodily Changes in Pain, Hunger, Fear, and Rage: An Account of Recent Researches into the Function of Emotional Excitement; D. Appleton and Company: New York, NY, USA, 1915. [Google Scholar]
- Selye, H. A syndrome produced by diverse nocuous agents. J. Neuropsychiatry Clin. Neurosci. 1998, 10, 230–231. [Google Scholar] [CrossRef] [Green Version]
- You, L. Steroid hormone biotransformation and xenobiotic induction of hepatic steroid metabolizing enzymes. Chem. Biol. Interact. 2004, 147, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Hamoud, A.R.; Weaver, L.; Stec, D.E.; Hinds, T.D., Jr. Bilirubin in the Liver-Gut Signaling Axis. Trends Endocrinol. Metab. 2018, 29, 140–150. [Google Scholar] [CrossRef]
- Garcia, M.; Thirouard, L.; Sedes, L.; Monrose, M.; Holota, H.; Caira, F.; Volle, D.H.; Beaudoin, C. Nuclear Receptor Metabolism of Bile Acids and Xenobiotics: A Coordinated Detoxification System with Impact on Health and Diseases. Int. J. Mol. Sci. 2018, 19, 3630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Y.; Liang, T.; Wang, G.; Li, Z. Ghrelin, a gastrointestinal hormone, regulates energy balance and lipid metabolism. Biosci. Rep. 2018, 38, BSR20181061. [Google Scholar] [CrossRef] [PubMed]
- Janah, L.; Kjeldsen, S.; Galsgaard, K.D.; Winther-Sorensen, M.; Stojanovska, E.; Pedersen, J.; Knop, F.K.; Holst, J.J.; Wewer Albrechtsen, N.J. Glucagon Receptor Signaling and Glucagon Resistance. Int. J. Mol. Sci. 2019, 20, 3314. [Google Scholar] [CrossRef] [Green Version]
- D’Souza A, M.; Neumann, U.H.; Glavas, M.M.; Kieffer, T.J. The glucoregulatory actions of leptin. Mol. Metab. 2017, 6, 1052–1065. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [Green Version]
- Babata, L.K.; Pedrosa, M.M.; Garcia, R.F.; Peicher, M.V.; de Godoi, V.A. Sustained Liver Glucose Release in Response to Adrenaline Can Improve Hypoglycaemic Episodes in Rats under Food Restriction Subjected to Acute Exercise. Int. J. Endocrinol. 2014, 2014, 969137. [Google Scholar] [CrossRef]
- Javed, K.; Fairweather, S.J. Amino acid transporters in the regulation of insulin secretion and signalling. Biochem. Soc. Trans. 2019, 47, 571–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karim, S.; Adams, D.H.; Lalor, P.F. Hepatic expression and cellular distribution of the glucose transporter family. World J. Gastroenterol. 2012, 18, 6771–6781. [Google Scholar] [CrossRef]
- Bradbury, M.W. Lipid metabolism and liver inflammation. I. Hepatic fatty acid uptake: Possible role in steatosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G194–G198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, M.S.; Goldstein, J.L. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, 34–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangelsdorf, D.J.; Evans, R.M. The RXR heterodimers and orphan receptors. Cell 1995, 83, 841–850. [Google Scholar] [CrossRef] [Green Version]
- Kliewer, S.A.; Goodwin, B.; Willson, T.M. The nuclear pregnane X receptor: A key regulator of xenobiotic metabolism. Endocr. Rev. 2002, 23, 687–702. [Google Scholar] [CrossRef] [Green Version]
- Honkakoski, P.; Zelko, I.; Sueyoshi, T.; Negishi, M. The nuclear orphan receptor CAR-retinoid X receptor heterodimer activates the phenobarbital-responsive enhancer module of the CYP2B gene. Mol. Cell. Biol. 1998, 18, 5652–5658. [Google Scholar] [CrossRef] [Green Version]
- Kersten, S.; Stienstra, R. The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie 2017, 136, 75–84. [Google Scholar] [CrossRef]
- Liu, S.; Hatano, B.; Zhao, M.; Yen, C.C.; Kang, K.; Reilly, S.M.; Gangl, M.R.; Gorgun, C.; Balschi, J.A.; Ntambi, J.M.; et al. Role of peroxisome proliferator-activated receptor δ/β in hepatic metabolic regulation. J. Biol. Chem. 2011, 286, 1237–1247. [Google Scholar] [CrossRef] [Green Version]
- Vidal-Puig, A.J.; Considine, R.V.; Jimenez-Linan, M.; Werman, A.; Pories, W.J.; Caro, J.F.; Flier, J.S. Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids. J. Clin. Investig. 1997, 99, 2416–2422. [Google Scholar] [CrossRef]
- Pan, X.; Wang, P.; Luo, J.; Wang, Z.; Song, Y.; Ye, J.; Hou, X. Adipogenic changes of hepatocytes in a high-fat diet-induced fatty liver mice model and non-alcoholic fatty liver disease patients. Endocrine 2015, 48, 834–847. [Google Scholar] [CrossRef] [PubMed]
- Wolf Greenstein, A.; Majumdar, N.; Yang, P.; Subbaiah, P.V.; Kineman, R.D.; Cordoba-Chacon, J. Hepatocyte-specific, PPARgamma-regulated mechanisms to promote steatosis in adult mice. J. Endocrinol. 2017, 232, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Wahli, W.; Michalik, L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Moore, R.; Negishi, M.; Sueyoshi, T. Nuclear pregnane X receptor cross-talk with FoxA2 to mediate drug-induced regulation of lipid metabolism in fasting mouse liver. J. Biol. Chem. 2007, 282, 9768–9776. [Google Scholar] [CrossRef] [Green Version]
- Roth, A.; Looser, R.; Kaufmann, M.; Blattler, S.M.; Rencurel, F.; Huang, W.; Moore, D.D.; Meyer, U.A. Regulatory cross-talk between drug metabolism and lipid homeostasis: Constitutive androstane receptor and pregnane X receptor increase Insig-1 expression. Mol. Pharmacol. 2008, 73, 1282–1289. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Xu, C.X.; Krager, S.L.; Bottum, K.M.; Liao, D.F.; Tischkau, S.A. Aryl hydrocarbon receptor deficiency enhances insulin sensitivity and reduces PPAR-α pathway activity in mice. Environ. Health Perspect. 2011, 119, 1739–1744. [Google Scholar] [CrossRef] [Green Version]
- Vitellius, G.; Lombes, M. Genetics in Endocrinology: Glucocorticoid resistance syndrome. Eur. J. Endocrinol. 2020, 182, R15–R27. [Google Scholar] [CrossRef] [Green Version]
- Lazar, M.A. Thyroid hormone receptors: Multiple forms, multiple possibilities. Endocr. Rev. 1993, 14, 184–193. [Google Scholar] [CrossRef]
- Singh, B.K.; Sinha, R.A.; Zhou, J.; Tripathi, M.; Ohba, K.; Wang, M.E.; Astapova, I.; Ghosh, S.; Hollenberg, A.N.; Gauthier, K.; et al. Hepatic FOXO1 Target Genes Are Co-regulated by Thyroid Hormone via RICTOR Protein Deacetylation and MTORC2-AKT Protein Inhibition. J. Biol. Chem. 2016, 291, 198–214. [Google Scholar] [CrossRef] [Green Version]
- Shukla, P.K.; Meena, A.S.; Dalal, K.; Canelas, C.; Samak, G.; Pierre, J.F.; Rao, R. Chronic stress and corticosterone exacerbate alcohol-induced tissue injury in the gut-liver-brain axis. Sci. Rep. 2021, 11, 826. [Google Scholar] [CrossRef]
- Regnier, M.; Polizzi, A.; Lippi, Y.; Fouche, E.; Michel, G.; Lukowicz, C.; Smati, S.; Marrot, A.; Lasserre, F.; Naylies, C.; et al. Insights into the role of hepatocyte PPARα activity in response to fasting. Mol. Cell. Endocrinol. 2018, 471, 75–88. [Google Scholar] [CrossRef]
- Montagner, A.; Polizzi, A.; Fouche, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Regnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Grygiel-Gorniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsouris, D.; Mandard, S.; Voshol, P.J.; Escher, P.; Tan, N.S.; Havekes, L.M.; Koenig, W.; Marz, W.; Tafuri, S.; Wahli, W.; et al. PPARα governs glycerol metabolism. J. Clin. Investig. 2004, 114, 94–103. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Wagner, M.; Xiao, R.; Kim, K.H.; Feng, D.; Lazar, M.A.; Moore, D.D. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014, 516, 112–115. [Google Scholar] [CrossRef] [Green Version]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Mutemberezi, V.; Guillemot-Legris, O.; Muccioli, G.G. Oxysterols: From cholesterol metabolites to key mediators. Prog. Lipid Res. 2016, 64, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Bjorkhem, I. Do oxysterols control cholesterol homeostasis? J. Clin. Investig. 2002, 110, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Repa, J.J.; Liang, G.; Ou, J.; Bashmakov, Y.; Lobaccaro, J.M.; Shimomura, I.; Shan, B.; Brown, M.S.; Goldstein, J.L.; Mangelsdorf, D.J. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRα and LXRβ. Genes Dev. 2000, 14, 2819–2830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef] [PubMed]
- Claudel, T.; Staels, B.; Kuipers, F. The Farnesoid X receptor: A molecular link between bile acid and lipid and glucose metabolism. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2020–2030. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsukuma, K.E.; Bennett, M.K.; Huang, J.; Wang, L.; Gil, G.; Osborne, T.F. Coordinated control of bile acids and lipogenesis through FXR-dependent regulation of fatty acid synthase. J. Lipid Res. 2006, 47, 2754–2761. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Horai, Y.; Houten, S.M.; Morimoto, K.; Sugizaki, T.; Arita, E.; Mataki, C.; Sato, H.; Tanigawara, Y.; Schoonjans, K.; et al. Lowering bile acid pool size with a synthetic farnesoid X receptor (FXR) agonist induces obesity and diabetes through reduced energy expenditure. J. Biol. Chem. 2011, 286, 26913–26920. [Google Scholar] [CrossRef] [Green Version]
- Yamagata, K.; Daitoku, H.; Shimamoto, Y.; Matsuzaki, H.; Hirota, K.; Ishida, J.; Fukamizu, A. Bile acids regulate gluconeogenic gene expression via small heterodimer partner-mediated repression of hepatocyte nuclear factor 4 and Foxo1. J. Biol. Chem. 2004, 279, 23158–23165. [Google Scholar] [CrossRef] [Green Version]
- Stayrook, K.R.; Bramlett, K.S.; Savkur, R.S.; Ficorilli, J.; Cook, T.; Christe, M.E.; Michael, L.F.; Burris, T.P. Regulation of carbohydrate metabolism by the farnesoid X receptor. Endocrinology 2005, 146, 984–991. [Google Scholar] [CrossRef] [Green Version]
- Han, C.Y. Update on FXR Biology: Promising Therapeutic Target? Int. J. Mol. Sci. 2018, 19, 2069. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.C.; Turner, N. FOX01 Is the Headline Akt Regulating Hepatic Glucose Metabolism. Endocrinology 2017, 158, 2436–2438. [Google Scholar] [CrossRef] [PubMed]
- Coleman, D.L. Obese and diabetes: Two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia 1978, 14, 141–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, F.K.; Levi, J.; Denroche, H.C.; Gray, S.L.; Voshol, P.J.; Neumann, U.H.; Speck, M.; Chua, S.C.; Covey, S.D.; Kieffer, T.J. Disruption of hepatic leptin signaling protects mice from age- and diet-related glucose intolerance. Diabetes 2010, 59, 3032–3040. [Google Scholar] [CrossRef]
- Huynh, F.K.; Neumann, U.H.; Wang, Y.; Rodrigues, B.; Kieffer, T.J.; Covey, S.D. A role for hepatic leptin signaling in lipid metabolism via altered very low density lipoprotein composition and liver lipase activity in mice. Hepatology 2013, 57, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.J.; Han, H.S.; Kim, M.J.; Koo, S.H. CREB and FoxO1: Two transcription factors for the regulation of hepatic gluconeogenesis. BMB Rep. 2013, 46, 567–574. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Xu, G.; Qin, Y.; Zhang, C.; Tang, H.; Yin, Y.; Xiang, X.; Li, Y.; Zhao, J.; Mulholland, M.; et al. Ghrelin promotes hepatic lipogenesis by activation of mTOR-PPARγ signaling pathway. Proc. Natl. Acad. Sci. USA 2014, 111, 13163–13168. [Google Scholar] [CrossRef] [Green Version]
- Altosaar, K.; Balaji, P.; Bond, R.A.; Bylund, D.B.; Cotecchia, S.; Devost, D.; Doze, V.A.; Eikenburg, D.C.; Gora, S.; Goupil, E.; et al. Adrenoceptors (version 2021.3). IUPHAR/BPS Guide Pharmacol. CITE 2021, 2021. [Google Scholar] [CrossRef]
- Bylund, D.B. Adrenergic Receptors. In Encyclopedia of Biological Chemistry, 2nd ed.; Lennarz, W.J., Lane, M.D., Eds.; Academic Press: Cambridge, MA, USA, 2013; pp. 57–60. [Google Scholar]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Iwabu, M.; Okada-Iwabu, M.; Kadowaki, T. Adiponectin receptors: A review of their structure, function and how they work. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 15–23. [Google Scholar] [CrossRef]
- Roberts, L.D.; Bostrom, P.; O’Sullivan, J.F.; Schinzel, R.T.; Lewis, G.D.; Dejam, A.; Lee, Y.K.; Palma, M.J.; Calhoun, S.; Georgiadi, A.; et al. β-Aminoisobutyric acid induces browning of white fat and hepatic β-oxidation and is inversely correlated with cardiometabolic risk factors. Cell Metab. 2014, 19, 96–108. [Google Scholar] [CrossRef] [Green Version]
- Tanianskii, D.A.; Jarzebska, N.; Birkenfeld, A.L.; O’Sullivan, J.F.; Rodionov, R.N. Beta-Aminoisobutyric Acid as a Novel Regulator of Carbohydrate and Lipid Metabolism. Nutrients 2019, 11, 524. [Google Scholar] [CrossRef] [Green Version]
- Mathews, S.T.; Chellam, N.; Srinivas, P.R.; Cintron, V.J.; Leon, M.A.; Goustin, A.S.; Grunberger, G. Alpha2-HSG, a specific inhibitor of insulin receptor autophosphorylation, interacts with the insulin receptor. Mol. Cell. Endocrinol. 2000, 164, 87–98. [Google Scholar] [CrossRef]
- Takata, H.; Ikeda, Y.; Suehiro, T.; Ishibashi, A.; Inoue, M.; Kumon, Y.; Terada, Y. High glucose induces transactivation of the α2-HS glycoprotein gene through the ERK1/2 signaling pathway. J. Atheroscler. Thromb. 2009, 16, 448–456. [Google Scholar] [CrossRef]
- Iroz, A.; Couty, J.P.; Postic, C. Hepatokines: Unlocking the multi-organ network in metabolic diseases. Diabetologia 2015, 58, 1699–1703. [Google Scholar] [CrossRef] [PubMed]
- Bourebaba, L.; Marycz, K. Pathophysiological Implication of Fetuin-A Glycoprotein in the Development of Metabolic Disorders: A Concise Review. J. Clin. Med. 2019, 8, 2033. [Google Scholar] [CrossRef] [Green Version]
- Kliewer, S.A.; Mangelsdorf, D.J. A Dozen Years of Discovery: Insights into the Physiology and Pharmacology of FGF21. Cell Metab. 2019, 29, 246–253. [Google Scholar] [CrossRef] [Green Version]
- Markan, K.R.; Naber, M.C.; Ameka, M.K.; Anderegg, M.D.; Mangelsdorf, D.J.; Kliewer, S.A.; Mohammadi, M.; Potthoff, M.J. Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes 2014, 63, 4057–4063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.J.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A.; et al. FGF-21 as a novel metabolic regulator. J. Clin. Investig. 2005, 115, 1627–1635. [Google Scholar] [CrossRef] [Green Version]
- BonDurant, L.D.; Potthoff, M.J. Fibroblast Growth Factor 21: A Versatile Regulator of Metabolic Homeostasis. Annu. Rev. Nutr. 2018, 38, 173–196. [Google Scholar] [CrossRef] [PubMed]
- Laeger, T.; Henagan, T.M.; Albarado, D.C.; Redman, L.M.; Bray, G.A.; Noland, R.C.; Munzberg, H.; Hutson, S.M.; Gettys, T.W.; Schwartz, M.W.; et al. FGF21 is an endocrine signal of protein restriction. J. Clin. Investig. 2014, 124, 3913–3922. [Google Scholar] [CrossRef] [Green Version]
- Misu, H.; Takamura, T.; Takayama, H.; Hayashi, H.; Matsuzawa-Nagata, N.; Kurita, S.; Ishikura, K.; Ando, H.; Takeshita, Y.; Ota, T.; et al. A liver-derived secretory protein, selenoprotein P, causes insulin resistance. Cell Metab. 2010, 12, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Speckmann, B.; Walter, P.L.; Alili, L.; Reinehr, R.; Sies, H.; Klotz, L.O.; Steinbrenner, H. Selenoprotein P expression is controlled through interaction of the coactivator PGC-1α with FoxO1a and hepatocyte nuclear factor 4α transcription factors. Hepatology 2008, 48, 1998–2006. [Google Scholar] [CrossRef]
- Mao, J.; Teng, W. The relationship between selenoprotein P and glucose metabolism in experimental studies. Nutrients 2013, 5, 1937–1948. [Google Scholar] [CrossRef]
- Koishi, R.; Ando, Y.; Ono, M.; Shimamura, M.; Yasumo, H.; Fujiwara, T.; Horikoshi, H.; Furukawa, H. Angptl3 regulates lipid metabolism in mice. Nat. Genet. 2002, 30, 151–157. [Google Scholar] [CrossRef]
- Mattijssen, F.; Kersten, S. Regulation of triglyceride metabolism by Angiopoietin-like proteins. Biochim. Biophys. Acta 2012, 1821, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Inaba, T.; Matsuda, M.; Shimamura, M.; Takei, N.; Terasaka, N.; Ando, Y.; Yasumo, H.; Koishi, R.; Makishima, M.; Shimomura, I. Angiopoietin-like protein 3 mediates hypertriglyceridemia induced by the liver X receptor. J. Biol. Chem. 2003, 278, 21344–21351. [Google Scholar] [CrossRef] [Green Version]
- Matsusue, K.; Miyoshi, A.; Yamano, S.; Gonzalez, F.J. Ligand-activated PPARβ efficiently represses the induction of LXR-dependent promoter activity through competition with RXR. Mol. Cell. Endocrinol. 2006, 256, 23–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oike, Y.; Akao, M.; Yasunaga, K.; Yamauchi, T.; Morisada, T.; Ito, Y.; Urano, T.; Kimura, Y.; Kubota, Y.; Maekawa, H.; et al. Angiopoietin-related growth factor antagonizes obesity and insulin resistance. Nat. Med. 2005, 11, 400–408. [Google Scholar] [CrossRef]
- Boulware, S.D.; Tamborlane, W.V.; Rennert, N.J.; Gesundheit, N.; Sherwin, R.S. Comparison of the metabolic effects of recombinant human insulin-like growth factor-I and insulin. Dose-response relationships in healthy young and middle-aged adults. J. Clin. Investig. 1994, 93, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Ohlsson, C.; Mohan, S.; Sjogren, K.; Tivesten, A.; Isgaard, J.; Isaksson, O.; Jansson, J.O.; Svensson, J. The role of liver-derived insulin-like growth factor-I. Endocr. Rev. 2009, 30, 494–535. [Google Scholar] [CrossRef] [Green Version]
- Fusco, A.; Miele, L.; D’Uonnolo, A.; Forgione, A.; Riccardi, L.; Cefalo, C.; Barini, A.; Bianchi, A.; Giampietro, A.; Cimino, V.; et al. Nonalcoholic fatty liver disease is associated with increased GHBP and reduced GH/IGF-I levels. Clin. Endocrinol. 2012, 77, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Adamek, A.; Kasprzak, A. Insulin-Like Growth Factor (IGF) System in Liver Diseases. Int. J. Mol. Sci. 2018, 19, 1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saez-Lopez, C.; Salcedo-Allende, M.T.; Hernandez, C.; Simo-Servat, O.; Simo, R.; Selva, D.M. Sex Hormone-Binding Globulin Expression Correlates With Acetyl-Coenzyme A Carboxylase and Triglyceride Content in Human Liver. J. Clin. Endocrinol. Metab. 2019, 104, 1500–1507. [Google Scholar] [CrossRef]
- Le, T.N.; Nestler, J.E.; Strauss, J.F., 3rd; Wickham, E.P., 3rd. Sex hormone-binding globulin and type 2 diabetes mellitus. Trends Endocrinol. Metab. 2012, 23, 32–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, M.; Halilbasic, E.; Marschall, H.U.; Zollner, G.; Fickert, P.; Langner, C.; Zatloukal, K.; Denk, H.; Trauner, M. CAR and PXR agonists stimulate hepatic bile acid and bilirubin detoxification and elimination pathways in mice. Hepatology 2005, 42, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Pan, S.; Guo, Y.; Xu, P.; Zhai, Y. PPARs as Nuclear Receptors for Nutrient and Energy Metabolism. Molecules 2019, 24, 2545. [Google Scholar] [CrossRef] [Green Version]
- Janowski, B.A.; Willy, P.J.; Devi, T.R.; Falck, J.R.; Mangelsdorf, D.J. An oxysterol signalling pathway mediated by the nuclear receptor LXRα. Nature 1996, 383, 728–731. [Google Scholar] [CrossRef]
- Ritter, M.J.; Amano, I.; Hollenberg, A.N. Thyroid Hormone Signaling and the Liver. Hepatology 2020, 72, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Staiger, H.; Keuper, M.; Berti, L.; Hrabe de Angelis, M.; Haring, H.U. Fibroblast Growth Factor 21-Metabolic Role in Mice and Men. Endocr. Rev. 2017, 38, 468–488. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A.; Birnbaum, M.J. Glucagon: Acute actions on hepatic metabolism. Diabetologia 2016, 59, 1376–1381. [Google Scholar] [CrossRef] [Green Version]
- Titchenell, P.M.; Lazar, M.A.; Birnbaum, M.J. Unraveling the Regulation of Hepatic Metabolism by Insulin. Trends Endocrinol. Metab. 2017, 28, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M. Adverse Outcome Pathways as Tools to Assess Drug-Induced Toxicity. Methods Mol. Biol. 2016, 1425, 325–337. [Google Scholar] [CrossRef] [Green Version]
- Mellor, C.L.; Steinmetz, F.P.; Cronin, M.T. The identification of nuclear receptors associated with hepatic steatosis to develop and extend adverse outcome pathways. Crit. Rev. Toxicol. 2016, 46, 138–152. [Google Scholar] [CrossRef]
- Gijbels, E.; Vilas-Boas, V.; Annaert, P.; Vanhaecke, T.; Devisscher, L.; Vinken, M. Robustness testing and optimization of an adverse outcome pathway on cholestatic liver injury. Arch. Toxicol. 2020, 94, 1151–1172. [Google Scholar] [CrossRef]
- Cicatiello, A.G.; Di Girolamo, D.; Dentice, M. Metabolic Effects of the Intracellular Regulation of Thyroid Hormone: Old Players, New Concepts. Front. Endocrinol. 2018, 9, 474. [Google Scholar] [CrossRef] [Green Version]
- Brenta, G. Why can insulin resistance be a natural consequence of thyroid dysfunction? J. Thyroid Res. 2011, 2011, 152850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legeay, S.; Faure, S. Is bisphenol A an environmental obesogen? Fundam. Clin. Pharmacol. 2017, 31, 594–609. [Google Scholar] [CrossRef] [Green Version]
- Huc, L.; Lemarie, A.; Gueraud, F.; Helies-Toussaint, C. Low concentrations of bisphenol A induce lipid accumulation mediated by the production of reactive oxygen species in the mitochondria of HepG2 cells. Toxicol. In Vitro 2012, 26, 709–717. [Google Scholar] [CrossRef]
- Marmugi, A.; Ducheix, S.; Lasserre, F.; Polizzi, A.; Paris, A.; Priymenko, N.; Bertrand-Michel, J.; Pineau, T.; Guillou, H.; Martin, P.G.; et al. Low doses of bisphenol A induce gene expression related to lipid synthesis and trigger triglyceride accumulation in adult mouse liver. Hepatology 2012, 55, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhang, A.; Li, T.; Gao, R.; Peng, C.; Liu, L.; Cheng, Q.; Mei, M.; Song, Y.; Xiang, X.; et al. Dysregulated Autophagy in Hepatocytes Promotes Bisphenol A-Induced Hepatic Lipid Accumulation in Male Mice. Endocrinology 2017, 158, 2799–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Arevalo, M.; Alonso-Magdalena, P.; Rebelo Dos Santos, J.; Quesada, I.; Carneiro, E.M.; Nadal, A. Exposure to bisphenol-A during pregnancy partially mimics the effects of a high-fat diet altering glucose homeostasis and gene expression in adult male mice. PLoS ONE 2014, 9, e100214. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Xia, W.; Wang, D.Q.; Wan, Y.J.; Xu, B.; Chen, X.; Li, Y.Y.; Xu, S.Q. Hepatic DNA methylation modifications in early development of rats resulting from perinatal BPA exposure contribute to insulin resistance in adulthood. Diabetologia 2013, 56, 2059–2067. [Google Scholar] [CrossRef]
- Meng, Z.; Wang, D.; Yan, S.; Li, R.; Yan, J.; Teng, M.; Zhou, Z.; Zhu, W. Effects of perinatal exposure to BPA and its alternatives (BPS, BPF and BPAF) on hepatic lipid and glucose homeostasis in female mice adolescent offspring. Chemosphere 2018, 212, 297–306. [Google Scholar] [CrossRef]
- Becher, R.; Wirsel, S.G. Fungal cytochrome P450 sterol 14α-demethylase (CYP51) and azole resistance in plant and human pathogens. Appl. Microbiol. Biotechnol. 2012, 95, 825–840. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, P.R.; Axelstad, M.; Boberg, J.; Isling, L.K.; Christiansen, S.; Mandrup, K.R.; Berthelsen, L.O.; Vinggaard, A.M.; Hass, U. Persistent developmental toxicity in rat offspring after low dose exposure to a mixture of endocrine disrupting pesticides. Reprod. Toxicol. 2012, 34, 237–250. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Plant Protection Products and their Residues (PPR Panel). Scientific Opinion on Risk Assessment for a Selected Group of Pesticides from the Triazole Group to Test Possible Methodologies to Assess Cumulative Effects from Exposure through Food from these Pesticides on Human Health. EFSA J. 2009, 7, 1167. [Google Scholar] [CrossRef]
- Marx-Stoelting, P.; Ganzenberg, K.; Knebel, C.; Schmidt, F.; Rieke, S.; Hammer, H.; Schmidt, F.; Potz, O.; Schwarz, M.; Braeuning, A. Hepatotoxic effects of cyproconazole and prochloraz in wild-type and hCAR/hPXR mice. Arch. Toxicol. 2017, 91, 2895–2907. [Google Scholar] [CrossRef]
- Knebel, C.; Buhrke, T.; Sussmuth, R.; Lampen, A.; Marx-Stoelting, P.; Braeuning, A. Pregnane X receptor mediates steatotic effects of propiconazole and tebuconazole in human liver cell lines. Arch. Toxicol. 2019, 93, 1311–1322. [Google Scholar] [CrossRef]
- Lichtenstein, D.; Luckert, C.; Alarcan, J.; de Sousa, G.; Gioutlakis, M.; Katsanou, E.S.; Konstantinidou, P.; Machera, K.; Milani, E.S.; Peijnenburg, A.; et al. An adverse outcome pathway-based approach to assess steatotic mixture effects of hepatotoxic pesticides in vitro. Food Chem. Toxicol. 2020, 139, 111283. [Google Scholar] [CrossRef]
- Marx-Stoelting, P.; Knebel, C.; Braeuning, A. The Connection of Azole Fungicides with Xeno-Sensing Nuclear Receptors, Drug Metabolism and Hepatotoxicity. Cells 2020, 9, 1192. [Google Scholar] [CrossRef]
- Sunderland, E.M.; Hu, X.C.; Dassuncao, C.; Tokranov, A.K.; Wagner, C.C.; Allen, J.G. A review of the pathways of human exposure to poly- and perfluoroalkyl substances (PFASs) and present understanding of health effects. J. Expo. Sci. Environ. Epidemiol. 2019, 29, 131–147. [Google Scholar] [CrossRef] [Green Version]
- Stockholm Convention. Stockholm Convention (Decision SC-4/17). Available online: http://chm.pops.int/Implementation/IndustrialPOPs/PFOS/Overview/tabid/5221/Default.aspx (accessed on 23 November 2022).
- EU. Commission Delegated Regulation (EU) 2020/784 of 8 April 2020 Amending Annex I to Regulation (EU) 2019/1021 of the European Parliament and of the Council as Regards the Listing of Perfluorooctanoic acid (PFOA), Its Salts and PFOA-Related Compounds. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32020R0784&from=EN (accessed on 14 December 2022).
- Wang, Y.; Chang, W.; Wang, L.; Zhang, Y.; Zhang, Y.; Wang, M.; Wang, Y.; Li, P. A review of sources, multimedia distribution and health risks of novel fluorinated alternatives. Ecotoxicol. Environ. Saf. 2019, 182, 109402. [Google Scholar] [CrossRef]
- Buhrke, T.; Kibellus, A.; Lampen, A. In vitro toxicological characterization of perfluorinated carboxylic acids with different carbon chain lengths. Toxicol. Lett. 2013, 218, 97–104. [Google Scholar] [CrossRef]
- Wan, H.T.; Zhao, Y.G.; Wei, X.; Hui, K.Y.; Giesy, J.P.; Wong, C.K. PFOS-induced hepatic steatosis, the mechanistic actions on beta-oxidation and lipid transport. Biochim. Biophys. Acta 2012, 1820, 1092–1101. [Google Scholar] [CrossRef]
- Das, K.P.; Wood, C.R.; Lin, M.T.; Starkov, A.A.; Lau, C.; Wallace, K.B.; Corton, J.C.; Abbott, B.D. Perfluoroalkyl acids-induced liver steatosis: Effects on genes controlling lipid homeostasis. Toxicology 2017, 378, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Behr, A.C.; Plinsch, C.; Braeuning, A.; Buhrke, T. Activation of human nuclear receptors by perfluoroalkylated substances (PFAS). Toxicol. In Vitro 2020, 62, 104700. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Liang, Y.; Li, J.; Liu, Y.; Zhang, J.; Zhang, A.; Fu, J.; Jiang, G. PFOS induced lipid metabolism disturbances in BALB/c mice through inhibition of low density lipoproteins excretion. Sci. Rep. 2014, 4, 4582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louisse, J.; Rijkers, D.; Stoopen, G.; Janssen, A.; Staats, M.; Hoogenboom, R.; Kersten, S.; Peijnenburg, A. Perfluorooctanoic acid (PFOA), perfluorooctane sulfonic acid (PFOS), and perfluorononanoic acid (PFNA) increase triglyceride levels and decrease cholesterogenic gene expression in human HepaRG liver cells. Arch. Toxicol. 2020, 94, 3137–3155. [Google Scholar] [CrossRef]
- Behr, A.C.; Kwiatkowski, A.; Stahlman, M.; Schmidt, F.F.; Luckert, C.; Braeuning, A.; Buhrke, T. Impairment of bile acid metabolism by perfluorooctanoic acid (PFOA) and perfluorooctanesulfonic acid (PFOS) in human HepaRG hepatoma cells. Arch. Toxicol. 2020, 94, 1673–1686. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Zhang, H.; Zheng, F.; Sheng, N.; Guo, X.; Dai, J. Perfluorooctanoic acid exposure for 28 days affects glucose homeostasis and induces insulin hypersensitivity in mice. Sci. Rep. 2015, 5, 11029. [Google Scholar] [CrossRef] [Green Version]
- Safe, S. Toxicology, structure-function relationship, and human and environmental health impacts of polychlorinated biphenyls: Progress and problems. Environ. Health Perspect. 1993, 100, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, M.; Birnbaum, L.S.; Denison, M.; De Vito, M.; Farland, W.; Feeley, M.; Fiedler, H.; Hakansson, H.; Hanberg, A.; Haws, L.; et al. The 2005 World Health Organization reevaluation of human and Mammalian toxic equivalency factors for dioxins and dioxin-like compounds. Toxicol. Sci. 2006, 93, 223–241. [Google Scholar] [CrossRef] [Green Version]
- Honkakoski, P.; Sueyoshi, T.; Negishi, M. Drug-activated nuclear receptors CAR and PXR. Ann. Med. 2003, 35, 172–182. [Google Scholar] [CrossRef]
- Kopec, A.K.; Burgoon, L.D.; Ibrahim-Aibo, D.; Mets, B.D.; Tashiro, C.; Potter, D.; Sharratt, B.; Harkema, J.R.; Zacharewski, T.R. PCB153-elicited hepatic responses in the immature, ovariectomized C57BL/6 mice: Comparative toxicogenomic effects of dioxin and non-dioxin-like ligands. Toxicol. Appl. Pharmacol. 2010, 243, 359–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadupudi, G.S.; Klaren, W.D.; Olivier, A.K.; Klingelhutz, A.J.; Robertson, L.W. PCB126-Induced Disruption in Gluconeogenesis and Fatty Acid Oxidation Precedes Fatty Liver in Male Rats. Toxicol. Sci. 2016, 149, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Jan, J.; Hardesty, J.E.; Falkner, K.C.; Prough, R.A.; Balamurugan, A.N.; Mokshagundam, S.P.; Chari, S.T.; Cave, M.C. Polychlorinated biphenyl exposures differentially regulate hepatic metabolism and pancreatic function: Implications for nonalcoholic steatohepatitis and diabetes. Toxicol. Appl. Pharmacol. 2019, 363, 22–33. [Google Scholar] [CrossRef]
- Yadetie, F.; Oveland, E.; Doskeland, A.; Berven, F.; Goksoyr, A.; Karlsen, O.A. Quantitative proteomics analysis reveals perturbation of lipid metabolic pathways in the liver of Atlantic cod (Gadus morhua) treated with PCB 153. Aquat. Toxicol. 2017, 185, 19–28. [Google Scholar] [CrossRef]
- Mesnier, A.; Champion, S.; Louis, L.; Sauzet, C.; May, P.; Portugal, H.; Benbrahim, K.; Abraldes, J.; Alessi, M.C.; Amiot-Carlin, M.J.; et al. The Transcriptional Effects of PCB118 and PCB153 on the Liver, Adipose Tissue, Muscle and Colon of Mice: Highlighting of Glut4 and Lipin1 as Main Target Genes for PCB Induced Metabolic Disorders. PLoS ONE 2015, 10, e0128847. [Google Scholar] [CrossRef] [Green Version]
- Wahlang, B.; Falkner, K.C.; Gregory, B.; Ansert, D.; Young, D.; Conklin, D.J.; Bhatnagar, A.; McClain, C.J.; Cave, M. Polychlorinated biphenyl 153 is a diet-dependent obesogen that worsens nonalcoholic fatty liver disease in male C57BL6/J mice. J. Nutr. Biochem. 2013, 24, 1587–1595. [Google Scholar] [CrossRef] [Green Version]
- Ganning, A.E.; Brunk, U.; Dallner, G. Phthalate esters and their effect on the liver. Hepatology 1984, 4, 541–547. [Google Scholar] [CrossRef]
- Zhang, W.; Shen, X.Y.; Zhang, W.W.; Chen, H.; Xu, W.P.; Wei, W. The effects of di 2-ethyl hexyl phthalate (DEHP) on cellular lipid accumulation in HepG2 cells and its potential mechanisms in the molecular level. Toxicol. Mech. Methods 2017, 27, 245–252. [Google Scholar] [CrossRef] [PubMed]
- DeKeyser, J.G.; Stagliano, M.C.; Auerbach, S.S.; Prabhu, K.S.; Jones, A.D.; Omiecinski, C.J. Di(2-ethylhexyl) phthalate is a highly potent agonist for the human constitutive androstane receptor splice variant CAR2. Mol. Pharmacol. 2009, 75, 1005–1013. [Google Scholar] [CrossRef] [Green Version]
- Feige, J.N.; Gerber, A.; Casals-Casas, C.; Yang, Q.; Winkler, C.; Bedu, E.; Bueno, M.; Gelman, L.; Auwerx, J.; Gonzalez, F.J.; et al. The pollutant diethylhexyl phthalate regulates hepatic energy metabolism via species-specific PPARα-dependent mechanisms. Environ. Health Perspect. 2010, 118, 234–241. [Google Scholar] [CrossRef]
- Okey, A.B. An aryl hydrocarbon receptor odyssey to the shores of toxicology: The Deichmann Lecture, International Congress of Toxicology-XI. Toxicol. Sci. 2007, 98, 5–38. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Shirakawa, H.; Tomita, S.; Ohsaki, Y.; Haketa, K.; Tooi, O.; Santo, N.; Tohkin, M.; Furukawa, Y.; Gonzalez, F.J.; et al. Low-dose dioxins alter gene expression related to cholesterol biosynthesis, lipogenesis, and glucose metabolism through the aryl hydrocarbon receptor-mediated pathway in mouse liver. Toxicol. Appl. Pharmacol. 2008, 229, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Wada, T.; Febbraio, M.; He, J.; Matsubara, T.; Lee, M.J.; Gonzalez, F.J.; Xie, W. A novel role for the dioxin receptor in fatty acid metabolism and hepatic steatosis. Gastroenterology 2010, 139, 653–663. [Google Scholar] [CrossRef] [Green Version]
- Roig, B.; Cadiere, A.; Bressieux, S.; Biau, S.; Faure, S.; de Santa Barbara, P. Environmental concentration of nonylphenol alters the development of urogenital and visceral organs in avian model. Environ. Int. 2014, 62, 78–85. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekar, G.; Arner, A.; Kitambi, S.S.; Dahlman-Wright, K.; Lendahl, M.A. Developmental toxicity of the environmental pollutant 4-nonylphenol in zebrafish. Neurotoxicol. Teratol. 2011, 33, 752–764. [Google Scholar] [CrossRef] [PubMed]
- Kourouma, A.; Keita, H.; Duan, P.; Quan, C.; Bilivogui, K.K.; Qi, S.; Christiane, N.A.; Osamuyimen, A.; Yang, K. Effects of 4-nonylphenol on oxidant/antioxidant balance system inducing hepatic steatosis in male rat. Toxicol. Rep. 2015, 2, 1423–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Yang, X.; Luo, Y.; Yang, X.; Yang, M.; Yang, J.; Zhou, J.; Gao, F.; He, L.; Xu, J. Adverse effects of chronic exposure to nonylphenol on non-alcoholic fatty liver disease in male rats. PLoS ONE 2017, 12, e0180218. [Google Scholar] [CrossRef] [Green Version]
- Tinkov, A.A.; Ajsuvakova, O.P.; Skalnaya, M.G.; Skalny, A.V.; Aschner, M.; Suliburska, J.; Aaseth, J. Organotins in obesity and associated metabolic disturbances. J. Inorg. Biochem. 2019, 191, 49–59. [Google Scholar] [CrossRef]
- Chamorro-Garcia, R.; Sahu, M.; Abbey, R.J.; Laude, J.; Pham, N.; Blumberg, B. Transgenerational inheritance of increased fat depot size, stem cell reprogramming, and hepatic steatosis elicited by prenatal exposure to the obesogen tributyltin in mice. Environ. Health Perspect. 2013, 121, 359–366. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Z.; Chen, S.; Wu, T.; Zhang, J.; Su, Y.; Chen, Y.; Wang, C. Tributyltin causes obesity and hepatic steatosis in male mice. Environ. Toxicol. 2011, 26, 79–85. [Google Scholar] [CrossRef]
- Grun, F.; Watanabe, H.; Zamanian, Z.; Maeda, L.; Arima, K.; Cubacha, R.; Gardiner, D.M.; Kanno, J.; Iguchi, T.; Blumberg, B. Endocrine-disrupting organotin compounds are potent inducers of adipogenesis in vertebrates. Mol. Endocrinol. 2006, 20, 2141–2155. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Okuhira, K.; Ohoka, N.; Naito, M.; Kagechika, H.; Hirose, A.; Nishimaki-Mogami, T. Tributyltin chloride induces ABCA1 expression and apolipoprotein A-I-mediated cellular cholesterol efflux by activating LXRalpha/RXR. Biochem. Pharmacol. 2011, 81, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Lyssimachou, A.; Santos, J.G.; Andre, A.; Soares, J.; Lima, D.; Guimaraes, L.; Almeida, C.M.; Teixeira, C.; Castro, L.F.; Santos, M.M. The Mammalian “Obesogen” Tributyltin Targets Hepatic Triglyceride Accumulation and the Transcriptional Regulation of Lipid Metabolism in the Liver and Brain of Zebrafish. PLoS ONE 2015, 10, e0143911. [Google Scholar] [CrossRef] [PubMed]
- Stossi, F.; Dandekar, R.D.; Johnson, H.; Lavere, P.; Foulds, C.E.; Mancini, M.G.; Mancini, M.A. Tributyltin chloride (TBT) induces RXRA down-regulation and lipid accumulation in human liver cells. PLoS ONE 2019, 14, e0224405. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Li, Y.; Mai, J.; Ji, X.; Li, Q. Effect of Dibutyltin Dilaurate on Triglyceride Metabolism through the Inhibition of the mTOR Pathway in Human HL7702 Liver Cells. Molecules 2018, 23, 1654. [Google Scholar] [CrossRef] [Green Version]
- Machala, M.; Vondracek, J.; Blaha, L.; Ciganek, M.; Neca, J.V. Aryl hydrocarbon receptor-mediated activity of mutagenic polycyclic aromatic hydrocarbons determined using in vitro reporter gene assay. Mutat. Res. 2001, 497, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Bucher, S.; Tete, A.; Podechard, N.; Liamin, M.; Le Guillou, D.; Chevanne, M.; Coulouarn, C.; Imran, M.; Gallais, I.; Fernier, M.; et al. Co-exposure to benzo[a]pyrene and ethanol induces a pathological progression of liver steatosis in vitro and in vivo. Sci. Rep. 2018, 8, 5963. [Google Scholar] [CrossRef] [Green Version]
- Bucher, S.; Le Guillou, D.; Allard, J.; Pinon, G.; Begriche, K.; Tete, A.; Sergent, O.; Lagadic-Gossmann, D.; Fromenty, B. Possible Involvement of Mitochondrial Dysfunction and Oxidative Stress in a Cellular Model of NAFLD Progression Induced by Benzo[a]pyrene/Ethanol CoExposure. Oxidative Med. Cell. Longev. 2018, 2018, 4396403. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, L.; Nakamura, B.; Li, X.; Blumberg, B.; Luderer, U. In utero exposure to benzo[a]pyrene increases adiposity and causes hepatic steatosis in female mice, and glutathione deficiency is protective. Toxicol. Lett. 2013, 223, 260–267. [Google Scholar] [CrossRef] [Green Version]
- Goedtke, L.; John, A.; Lampen, A.; Seidel, A.; Braeuning, A.; Hessel-Pras, S. Mixture effects of food-relevant polycyclic aromatic hydrocarbons on the activation of nuclear receptors and gene expression, benzo[a]pyrene metabolite profile and DNA damage in HepaRG cells. Food Chem. Toxicol. 2020, 147, 111884. [Google Scholar] [CrossRef]
- Newbold, R.R.; Padilla-Banks, E.; Jefferson, W.N. Environmental estrogens and obesity. Mol. Cell. Endocrinol. 2009, 304, 84–89. [Google Scholar] [CrossRef] [Green Version]
- Hatch, E.E.; Troisi, R.; Palmer, J.R.; Wise, L.A.; Titus, L.; Strohsnitter, W.C.; Ricker, W.; Hyer, M.; Hoover, R.N. Prenatal diethylstilbestrol exposure and risk of obesity in adult women. J. Dev. Orig. Health Dis. 2015, 6, 201–207. [Google Scholar] [CrossRef] [Green Version]
- Hao, C.J.; Cheng, X.J.; Xia, H.F.; Ma, X. The endocrine disruptor diethylstilbestrol induces adipocyte differentiation and promotes obesity in mice. Toxicol. Appl. Pharmacol. 2012, 263, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Vega, A.; Baptissart, M.; Martinot, E.; Saru, J.P.; Baron, S.; Schoonjans, K.; Volle, D.H. Hepatotoxicity induced by neonatal exposure to diethylstilbestrol is maintained throughout adulthood via the nuclear receptor SHP. Expert Opin. Ther. Targets 2014, 18, 1367–1376. [Google Scholar] [CrossRef]
- Haeno, S.; Maeda, N.; Yamaguchi, K.; Sato, M.; Uto, A.; Yokota, H. Adrenal steroidogenesis disruption caused by HDL/cholesterol suppression in diethylstilbestrol-treated adult male rat. Endocrine 2016, 52, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Tamburro, C.H.; Makk, L.; Popper, H. Early hepatic histologic alterations among chemical (vinyl monomer) workers. Hepatology 1984, 4, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Easter, M.D.; Von Burg, R. Vinyl chloride. J. Appl. Toxicol. 1994, 14, 301–307. [Google Scholar] [CrossRef] [PubMed]
- U.S. EPA. IRIS Toxicological Review of Vinyl Chloride (Final Report); U.S. Environmental Protection Agency: Washington, DC, USA, 2000.
- Lang, A.L.; Chen, L.; Poff, G.D.; Ding, W.X.; Barnett, R.A.; Arteel, G.E.; Beier, J.I. Vinyl chloride dysregulates metabolic homeostasis and enhances diet-induced liver injury in mice. Hepatol. Commun. 2018, 2, 270–284. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Lang, A.L.; Poff, G.D.; Ding, W.X.; Beier, J.I. Vinyl chloride-induced interaction of nonalcoholic and toxicant-associated steatohepatitis: Protection by the ALDH2 activator Alda-1. Redox Biol. 2019, 24, 101205. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, L.; Chen, S.Q.; Ma, W.Y.; Guo, Y.L.; Gao, Y.; Tian, F.J.; Qiu, Y.L. Role of endoplasmic reticulum stress and oxidative stress in vinyl chloride-induced hepatic steatosis in mice. Toxicol. Appl. Pharmacol. 2019, 381, 114730. [Google Scholar] [CrossRef]
- Kojima, H.; Takeuchi, S.; Itoh, T.; Iida, M.; Kobayashi, S.; Yoshida, T. In vitro endocrine disruption potential of organophosphate flame retardants via human nuclear receptors. Toxicology 2013, 314, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.; Zhang, Z.; Lu, D.; Ding, B.; Shu, L.; Zhang, Q.; Wang, C. Organophosphorus Flame Retardants Impair Intracellular Lipid Metabolic Function in Human Hepatocellular Cells. Chem. Res. Toxicol. 2019, 32, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Negi, C.K.; Bajard, L.; Kohoutek, J.; Blaha, L. An adverse outcome pathway based in vitro characterization of novel flame retardants-induced hepatic steatosis. Environ. Pollut. 2021, 289, 117855. [Google Scholar] [CrossRef] [PubMed]
- Lasram, M.M.; Dhouib, I.B.; Bouzid, K.; Lamine, A.J.; Annabi, A.; Belhadjhmida, N.; Ahmed, M.B.; Fazaa, S.E.; Abdelmoula, J.; Gharbi, N. Association of inflammatory response and oxidative injury in the pathogenesis of liver steatosis and insulin resistance following subchronic exposure to malathion in rats. Environ. Toxicol. Pharmacol. 2014, 38, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi, M.; Donyavi, M.; Pournourmohammadi, S.; Saadat, M. Hyperglycemia associated with increased hepatic glycogen phosphorylase and phosphoenolpyruvate carboxykinase in rats following subchronic exposure to malathion. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2004, 137, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Sabir, S.; Akash, M.S.H.; Fiayyaz, F.; Saleem, U.; Mehmood, M.H.; Rehman, K. Role of cadmium and arsenic as endocrine disruptors in the metabolism of carbohydrates: Inserting the association into perspectives. Biomed. Pharmacother. 2019, 114, 108802. [Google Scholar] [CrossRef]
- Singhal, R.L.; Merali, Z.; Kacew, S.; Sutherland, D.J. Persistence of cadmium-induced metabolic changes in liver and kidney. Science 1974, 183, 1094–1096. [Google Scholar] [CrossRef]
- Alshehri, A.S.; El-Kott, A.F.; El-Kenawy, A.E.; Khalifa, H.S.; AlRamlawy, A.M. Cadmium chloride induces non-alcoholic fatty liver disease in rats by stimulating miR-34a/SIRT1/FXR/p53 axis. Sci. Total Environ. 2021, 784, 147182. [Google Scholar] [CrossRef]
- Young, J.L.; Cave, M.C.; Xu, Q.; Kong, M.; Xu, J.; Lin, Q.; Tan, Y.; Cai, L. Whole life exposure to low dose cadmium alters diet-induced NAFLD. Toxicol. Appl. Pharmacol. 2022, 436, 115855. [Google Scholar] [CrossRef]
- Franco, M.E.; Fernandez-Luna, M.T.; Ramirez, A.J.; Lavado, R. Metabolomic-based assessment reveals dysregulation of lipid profiles in human liver cells exposed to environmental obesogens. Toxicol. Appl. Pharmacol. 2020, 398, 115009. [Google Scholar] [CrossRef]
- Park, J.H.; Mangal, D.; Frey, A.J.; Harvey, R.G.; Blair, I.A.; Penning, T.M. Aryl hydrocarbon receptor facilitates DNA strand breaks and 8-oxo-2’-deoxyguanosine formation by the aldo-keto reductase product benzo[a]pyrene-7,8-dione. J. Biol. Chem. 2009, 284, 29725–29734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Go, Y.M.; Sutliff, R.L.; Chandler, J.D.; Khalidur, R.; Kang, B.Y.; Anania, F.A.; Orr, M.; Hao, L.; Fowler, B.A.; Jones, D.P. Low-Dose Cadmium Causes Metabolic and Genetic Dysregulation Associated With Fatty Liver Disease in Mice. Toxicol. Sci. 2015, 147, 524–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niture, S.; Gadi, S.; Lin, M.; Qi, Q.; Niture, S.S.; Moore, J.T.; Bodnar, W.; Fernando, R.A.; Levine, K.E.; Kumar, D. Cadmium modulates steatosis, fibrosis, and oncogenic signaling in liver cancer cells by activating notch and AKT/mTOR pathways. Environ. Toxicol. 2023. [Google Scholar] [CrossRef] [PubMed]
- European Chemical Agency (ECHA) and European Food Safety Authority (EFSA) with the technical support of the Joint Research Centre (JRC); Andersson, N.; Arena, M.; Auteri, D.; Barmaz, S.; Grignard, E.; Kienzler, A.; Lepper, P.; Lostia, A.M.; Munn, S.; et al. Guidance for the identification of endocrine disruptors in the context of Regulations (EU) No 528/2012 and (EC) No 1107/2009. EFSA J. 2018, 16, e05311. [Google Scholar] [CrossRef]
- OECD. Test No. 408: Repeated Dose 90-Day Oral Toxicity Study in Rodents; OECD: Paris, France, 2018. [Google Scholar] [CrossRef]
- OECD. Test No. 407: Repeated Dose 28-Day Oral Toxicity Study in Rodents; OECD: Paris, France, 2008. [Google Scholar] [CrossRef]
- Greaves, P. Histopathology of Preclinical Toxicity Studies: Interpretation and Relevance in Drug Safety Evaluation, 4th ed.; Elsevier Academic Press Inc.: San Diego, CA, USA, 2012. [Google Scholar]
- Elcombe, C.R.; Peffer, R.C.; Wolf, D.C.; Bailey, J.; Bars, R.; Bell, D.; Cattley, R.C.; Ferguson, S.S.; Geter, D.; Goetz, A.; et al. Mode of action and human relevance analysis for nuclear receptor-mediated liver toxicity: A case study with phenobarbital as a model constitutive androstane receptor (CAR) activator. Crit. Rev. Toxicol. 2014, 44, 64–82. [Google Scholar] [CrossRef] [Green Version]
- Corton, J.C.; Cunningham, M.L.; Hummer, B.T.; Lau, C.; Meek, B.; Peters, J.M.; Popp, J.A.; Rhomberg, L.; Seed, J.; Klaunig, J.E. Mode of action framework analysis for receptor-mediated toxicity: The peroxisome proliferator-activated receptor alpha (PPARα) as a case study. Crit. Rev. Toxicol. 2014, 44, 1–49. [Google Scholar] [CrossRef]
- Sprenger, H.; Rasinger, J.D.; Hammer, H.; Naboulsi, W.; Zabinsky, E.; Planatscher, H.; Schwarz, M.; Poetz, O.; Braeuning, A. Proteomic analysis of hepatic effects of phenobarbital in mice with humanized liver. Arch. Toxicol. 2022, 96, 2739–2754. [Google Scholar] [CrossRef]
- Gu, S.; A, J.; Wang, G.; Zha, W.; Yan, B.; Zhang, Y.; Ren, H.; Cao, B.; Liu, L. Metabonomic profiling of liver metabolites by gas chromatography-mass spectrometry and its application to characterizing hyperlipidemia. Biomed. Chromatogr. 2010, 24, 245–252. [Google Scholar] [CrossRef]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Innovation: Metabolomics: The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Marx-Stoelting, P.; Braeuning, A.; Buhrke, T.; Lampen, A.; Niemann, L.; Oelgeschlaeger, M.; Rieke, S.; Schmidt, F.; Heise, T.; Pfeil, R.; et al. Application of omics data in regulatory toxicology: Report of an international BfR expert workshop. Arch. Toxicol. 2015, 89, 2177–2184. [Google Scholar] [CrossRef]
- Pedro, P.F.; Tsakmaki, A.; Bewick, G.A. The Glucose Tolerance Test in Mice. Methods Mol. Biol. 2020, 2128, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Vinue, A.; Gonzalez-Navarro, H. Glucose and Insulin Tolerance Tests in the Mouse. Methods Mol. Biol. 2015, 1339, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Society for Advancement of AOPs. AOP-Wiki. 2022. Available online: http://aopwiki.org (accessed on 23 November 2022).
- Luckert, C.; Braeuning, A.; de Sousa, G.; Durinck, S.; Katsanou, E.S.; Konstantinidou, P.; Machera, K.; Milani, E.S.; Peijnenburg, A.; Rahmani, R.; et al. Adverse Outcome Pathway-Driven Analysis of Liver Steatosis in Vitro: A Case Study with Cyproconazole. Chem. Res. Toxicol. 2018, 31, 784–798. [Google Scholar] [CrossRef]
- Lichtenstein, D.; Mentz, A.; Schmidt, F.F.; Luckert, C.; Buhrke, T.; Marx-Stoelting, P.; Kalinowski, J.; Albaum, S.P.; Joos, T.O.; Poetz, O.; et al. Transcript and protein marker patterns for the identification of steatotic compounds in human HepaRG cells. Food Chem. Toxicol. 2020, 145, 111690. [Google Scholar] [CrossRef]
- European Chemical Agency. Read-Across Assessment Framework (RAAF); ECHA-17-R-01-EN; European Chemical Agency: Kelsinki, Finland, 2017; ISBN 978-92-9495-758-0. [CrossRef]
- Clish, C.B. Metabolomics: An emerging but powerful tool for precision medicine. Cold Spring Harb. Mol. Case Stud. 2015, 1, a000588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, P.; Barney, J.; Petriello, M.C.; Morris, A.J.; Wahlang, B.; Hennig, B. Hepatic metabolomics reveals that liver injury increases PCB 126-induced oxidative stress and metabolic dysfunction. Chemosphere 2019, 217, 140–149. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fritsche, K.; Ziková-Kloas, A.; Marx-Stoelting, P.; Braeuning, A. Metabolism-Disrupting Chemicals Affecting the Liver: Screening, Testing, and Molecular Pathway Identification. Int. J. Mol. Sci. 2023, 24, 2686. https://doi.org/10.3390/ijms24032686
Fritsche K, Ziková-Kloas A, Marx-Stoelting P, Braeuning A. Metabolism-Disrupting Chemicals Affecting the Liver: Screening, Testing, and Molecular Pathway Identification. International Journal of Molecular Sciences. 2023; 24(3):2686. https://doi.org/10.3390/ijms24032686
Chicago/Turabian StyleFritsche, Kristin, Andrea Ziková-Kloas, Philip Marx-Stoelting, and Albert Braeuning. 2023. "Metabolism-Disrupting Chemicals Affecting the Liver: Screening, Testing, and Molecular Pathway Identification" International Journal of Molecular Sciences 24, no. 3: 2686. https://doi.org/10.3390/ijms24032686
APA StyleFritsche, K., Ziková-Kloas, A., Marx-Stoelting, P., & Braeuning, A. (2023). Metabolism-Disrupting Chemicals Affecting the Liver: Screening, Testing, and Molecular Pathway Identification. International Journal of Molecular Sciences, 24(3), 2686. https://doi.org/10.3390/ijms24032686