
Macrophage Repolarization as a Therapeutic Strategy for Osteosarcoma



Abstract
:1. Introduction
2. Osteosarcoma Tumor-Microenvironment
3. Macrophages Involved in Bone Microenvironment
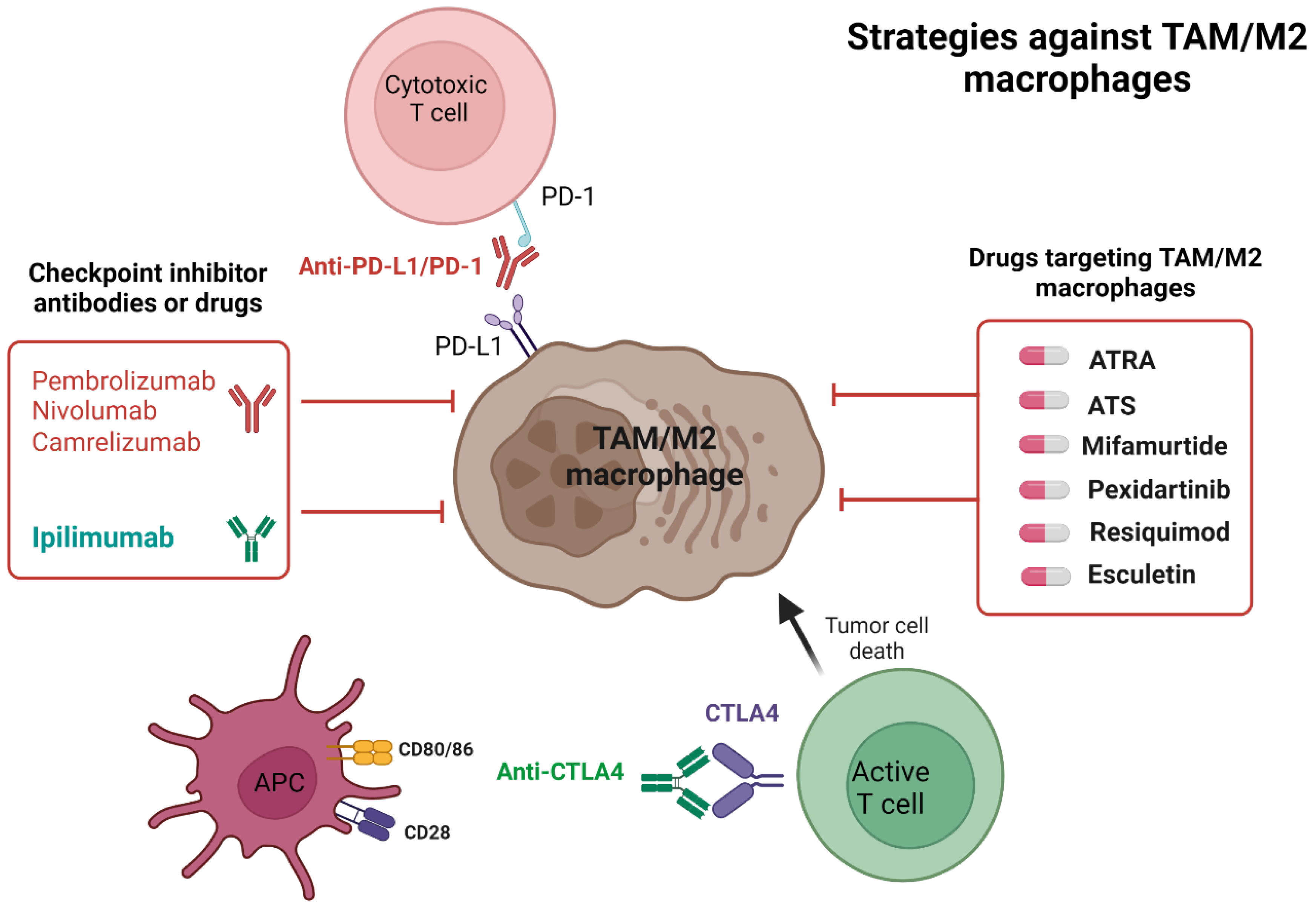
4. Checkpoint Inhibitors in Osteosarcoma
4.1. Checkpoint Inhibitor’s Mechanism of Action
4.2. The Rationale for Checkpoint Inhibitors in Osteosarcoma
4.3. Checkpoint Inhibitors Clinical Trials in Osteosarcoma

{kind=link}
{kind=link}
| Drug/NCT Number/n (Osteosarcoma Subgroup) | Target Molecule | Trial Design | Inclusion Criteria | Primary Outcome | Time to Event Outcomes |
|---|---|---|---|---|---|
| Pembrolizumab NCT03013127 [7] n = 12 | PD-1 | Single arm, phase 2 | ≥18 y.o, r/r osteosarcoma | CBR: 0% (0/12) | Estimated mPFS: 1.4 m.o Estimated mOS: 6.6 m.o |
| Pembrolizumab NCT02301039 [6] n = 86 (n=22) | PD-1 | Single-arm, phase 2 | ≥12 y.o, r/r locally advanced or metastatic sarcoma | ORR (bone sarcoma arm): 5% (2/40) ORR (osteosarcoma): 5% (1/22) | Bone sarcoma arm mPFS: 8 wk mOS: 52 wk |
| Atezolizumab NCT02541604 [56] n = 90 (n =12) | PD-L1 | Single arm, phase 1–2 | <30 y.o, r/r solid tumors and lymphomas | ORR (overall): 5% (4/87) ORR (osteosarcoma): 0% (0/10) | Overall mPFS: 1.3 m.o mOS: 7.4 m.o |
| Nivolumab + Ipilimumab NCT02500797 [58] n = 85 (n = 1) * | PD-1, CTLA-4 | Non-comparative, randomized, two-arm, phase 2 | ≥18 y.o, r/r advanced or metastatic sarcoma | ORR (monotherapy): 8% (3/38) ORR (combination): 15% (6/41) | Monotherapy mPFS: 1.7 m.o mOS: 10.7 m.o Combination mPFS: 4.1 mo. mOS: 14.3 m.o |
| Durvalumab + Tremelimumab NCT02815995 [59] n = 57 (n = 5) | PD-L1, CTLA-4 | Single arm, phase 2 | ≥18 y.o, r/r metastatic sarcomas | 12 wk PFS rate (overall): 49% (28/57) 12 wk PFS rate (osteosarcoma): 20% (1/5) | Overall mPFS: 2.8 m.o mOS: 21.6 m.o Osteosarcoma mPFS: 1.81 m.o |
| Nivolumab + bempegaldesleukin NCT03282344 [60] n = 84 (n = 10) | PD-1, CD122 (IL-2) | Single arm, pilot study | ≥12 y.o, r/r locally advanced or metastatic sarcoma | ORR (osteosarcoma): 0% | Osteosarcoma mPFS: 2.0 m.o mOS: 6.3 m.o |
| Apatinib + Camrelizumab NCT03359018 [8] n = 43 | PD-1, VEGFR-2 | Single arm, phase 2 | ≥11 y.o, r/r locally advanced or metastatic osteosarcoma | mPFS: 6.2 m.o | mOS: 11.3 m.o |
| Pembrolizumab + Cyclophosphamide NCT02406781 [67] n = 15 | PD-1, alkylating agent (DNA damage) | Single arm, phase 2 | ≥18 y.o, r/r metastatic osteosarcoma | 6 m.o non-progression rate: 13.3% (2/15) | mPFS: 1.4 m.o mOS: 5.6 m.o |
5. Drugs Targeting Macrophage Repolarization in OS
5.1. ATRA
5.2. ATS
5.3. PTT
5.4. Mifamurtide
5.5. Pexidartinib
5.6. Resiquimod
5.7. Esculetin
6. Other Strategies to Target TAM/M2 Macrophages
6.1. CD47/SIRPα
6.2. CD40
6.3. SIGLEC-15
6.4. Complement
6.5. PI3K
6.6. ERK5
7. Conclusions
Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Raymond, A.K.; Jaffe, N. Osteosarcoma multidisciplinary approach to the management from the pathologist’s perspective. Pediatr. Adolesc. Osteosarcoma 2009, 152, 63–84. [Google Scholar]
- Hayden, J.B.; Hoang, B.H.J.O.C. Osteosarcoma: Basic science and clinical implications. Orthop. Clin. 2006, 37, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bacci, G.; Longhi, A.; Bertoni, F.; Bacchini, P.; Ruggeri, P.; Versari, M.; Picci, P. Primary high-grade osteosarcoma: Comparison between preadolescent and older patients. J. Pediatr. Hematol. /Oncol. 2005, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Altekruse, S.F.; Adamson, P.C.; Reaman, G.H.; Seibel, N.L. Declining childhood and adolescent cancer mortality. Cancer 2014, 120, 2497–2506. [Google Scholar] [CrossRef] [PubMed]
- O’Kane, G.; Cadoo, K.; Walsh, E.; Emerson, R.; Dervan, P.; O’keane, C.; Hurson, B.; O’toole, G.; Dudeney, S.; Kavanagh, E.; et al. Perioperative chemotherapy in the treatment of osteosarcoma: A 26-year single institution review. Clin. Sarcoma Res. 2015, 5, 1–8. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Boye, K.; Longhi, A.; Guren, T.; Lorenz, S.; Næss, S.; Pierini, M.; Taksdal, I.; Lobmaier, I.; Cesari, M.; Paioli, A.; et al. Pembrolizumab in advanced osteosarcoma: Results of a single-arm, open-label, phase 2 trial. Cancer Immunol. Immunother. 2021, 70, 2617–2624. [Google Scholar] [CrossRef]
- Xie, L.; Xu, J.; Sun, X.; Guo, W.; Gu, J.; Liu, K.; Zheng, B.; Ren, T.; Huang, Y.; Tang, X.; et al. Apatinib plus camrelizumab (anti-PD1 therapy, SHR-1210) for advanced osteosarcoma (APFAO) progressing after chemotherapy: A single-arm, open-label, phase 2 trial. J. Immunother. Cancer 2020, 8, e000798. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef]
- Overholtzer, M.; Rao, P.H.; Favis, R.; Lu, X.-Y.; Elowitz, M.B.; Barany, F.; Ladanyi, M.; Gorlick, R.; Levine, A.J. The presence of p53 mutations in human osteosarcomas correlates with high levels of genomic instability. Proc. Natl. Acad. Sci. USA 2003, 100, 11547–11552. [Google Scholar] [CrossRef]
- Molchadsky, A.; Shats, I.; Goldfinger, N.; Pevsner-Fischer, M.; Olson, M.; Rinon, A.; Tzahor, E.; Lozano, G.; Zipori, D.; Sarig, R.; et al. p53 plays a role in mesenchymal differentiation programs, in a cell fate dependent manner. PLoS ONE 2008, 3, e3707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tataria, M.; Quarto, N.; Longaker, M.T.; Sylvester, K.G. Absence of the p53 tumor suppressor gene promotes osteogenesis in mesenchymal stem cells. J. Pediatr. Surg. 2006, 41, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, G.M.; Kong, E.; Sabbagh, Y.; Brown, N.E.; Lee, J.-S.; Demay, M.B.; Thomas, D.M.; Hinds, P.W. Impaired bone development and increased mesenchymal progenitor cells in calvaria of RB1−/− mice. Proc. Natl. Acad. Sci. USA 2008, 105, 18402–18407. [Google Scholar] [CrossRef] [PubMed]
- Calo, E.; Quintero-Estades, J.A.; Danielian, P.S.; Nedelcu, S.; Berman, S.D.; Lees, J.A. Rb regulates fate choice and lineage commitment in vivo. Nature 2010, 466, 1110–1114. [Google Scholar] [CrossRef] [PubMed]
- TSUkAMOTO, S.; Honoki, K.; Fujii, H.; Tohma, Y.; Kido, A.; Mori, T.; Tsujiuchi, T.; Tanaka, Y. Mesenchymal stem cells promote tumor engraftment and metastatic colonization in rat osteosarcoma model. Int. J. Oncol. 2012, 40, 163–169. [Google Scholar] [PubMed]
- VanPutte, C.L.; Regan, J.L.; Russo, A.F.; Seeley, R.R.; Stephens, T.; Tate, P. Seeley’s Anatomy & Physiology; McGraw-Hill: New York, NY, USA, 2019. [Google Scholar]
- Tu, B.; Peng, Z.-X.; Fan, Q.-M.; Du, L.; Yan, W.; Tang, T.-T. Osteosarcoma cells promote the production of pro-tumor cytokines in mesenchymal stem cells by inhibiting their osteogenic differentiation through the TGF-β/Smad2/3 pathway. Exp. Cell Res. 2014, 320, 164–173. [Google Scholar] [CrossRef]
- Florencio-Silva, R.; Sasso, G.R.d.S.; Sasso-Cerri, E.; Simões, M.J.; Cerri, P.S. Biology of bone tissue: Structure, function, and factors that influence bone cells. BioMed Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef]
- Yu, F.; Shen, H.; Deng, H.-W. Systemic analysis of osteoblast-specific DNA methylation marks reveals novel epigenetic basis of osteoblast differentiation. Bone Rep. 2017, 6, 109–119. [Google Scholar] [CrossRef]
- Mutsaers, A.J.; Walkley, C.R.J.B. Cells of origin in osteosarcoma: Mesenchymal stem cells or osteoblast committed cells? Bone 2014, 62, 56–63. [Google Scholar] [CrossRef]
- Larionova, I.; Cherdyntseva, N.; Liu, T.; Patysheva, M.; Rakina, M.; Kzhyshkowska, J.J.O. Interaction of tumor-associated macrophages and cancer chemotherapy. Oncoimmunology 2019, 8, e1596004. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F.J.n. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The local tumor microenvironment. In General Principles of Tumor Immunotherapy; Springer: Berlin/Heidelberg, Germany, 2007; pp. 145–167. [Google Scholar]
- Heymann, M.-F.; Lézot, F.; Heymann, D.J.C.i. The contribution of immune infiltrates and the local microenvironment in the pathogenesis of osteosarcoma. Cell. Immunol. 2019, 343, 103711. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.K.; Raggatt, L.-J.; Alexander, K.A.; Kuliwaba, J.S.; Fazzalari, N.L.; Schroder, K.; Maylin, E.R.; Ripoll, V.M.; Hume, D.A.; Pettit, A.R. Osteal tissue macrophages are intercalated throughout human and mouse bone lining tissues and regulate osteoblast function in vitro and in vivo. J. Immunol. 2008, 181, 1232–1244. [Google Scholar] [CrossRef] [PubMed]
- Miron, R.J.; Bosshardt, D.D. OsteoMacs: Key players around bone biomaterials. Biomaterials 2016, 82, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Galán, L.; Olleros, M.L.; Vesin, D.; Garcia, I.J.F.i.i. Much more than M1 and M2 macrophages, there are also CD169+ and TCR+ macrophages. Front. Immunol. 2015, 6, 263. [Google Scholar] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Cassetta, L.; Kitamura, T. Targeting tumor-associated macrophages as a potential strategy to enhance the response to immune checkpoint inhibitors. Front. Cell Dev. Biol. 2018, 38. [Google Scholar] [CrossRef]
- Han, Y.; Guo, W.; Ren, T.; Huang, Y.; Wang, S.; Liu, K.; Zheng, B.; Yang, K.; Zhang, H.; Liang, X. Tumor-associated macrophages promote lung metastasis and induce epithelial-mesenchymal transition in osteosarcoma by activating the COX-2/STAT3 axis. Cancer Lett. 2019, 440, 116–125. [Google Scholar] [CrossRef]
- Wolf-Dennen, K.; Gordon, N.; Kleinerman, E.S. Exosomal communication by metastatic osteosarcoma cells modulates alveolar macrophages to an M2 tumor-promoting phenotype and inhibits tumoricidal functions. Oncoimmunology 2020, 9, 1747677. [Google Scholar] [CrossRef]
- Cersosimo, F.; Lonardi, S.; Bernardini, G.; Telfer, B.; Mandelli, G.E.; Santucci, A.; Vermi, W.; Giurisato, E. Tumor-associated macrophages in osteosarcoma: From mechanisms to therapy. Int. J. Mol. Sci. 2020, 21, 5207. [Google Scholar] [CrossRef]
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.W.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019, 572, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Rőszer, T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Su, Y.; He, H. Research Progress of M2 Macrophage. Int. J. Front. Med. 2022, 4, 60–64. [Google Scholar]
- Wang, L.x.; Zhang, S.x.; Wu, H.j.; Rong, X.l.; Guo, J.J.J.o.l.b. M2b macrophage polarization and its roles in diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef]
- Severn, C.E.; Oates, T.C.; Moura, P.L.; Cross, S.J.; Roberts, K.; Baum, H.E.; Haydn-Smith, K.; Wilson, M.C.; Heesom, K.J.; Toye, A.M. Characterizing the polarization continuum of macrophage subtypes M1, M2a and M2c. bioRxiv 2022. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Hughes, R.; Lewis, C.E. Tumor-associated macrophages: Effectors of angiogenesis and tumor progression. Biochim. Et Biophys. Acta (BBA)-Rev. Cancer 2009, 1796, 11–18. [Google Scholar] [CrossRef]
- Franklin, R.A.; Li, M.O. Ontogeny of tumor-associated macrophages and its implication in cancer regulation. Trends Cancer 2016, 2, 20–34. [Google Scholar] [CrossRef]
- Inagaki, Y.; Hookway, E.; Williams, K.; Hassan, A.; Oppermann, U.; Tanaka, Y.; Soilleux, E.; Athanasou, N. Dendritic and mast cell involvement in the inflammatory response to primary malignant bone tumours. Clin. Sarcoma Res. 2016, 6, 1–8. [Google Scholar] [CrossRef]
- Alvarez, C.; Monasterio, G.; Cavalla, F.; Córdova, L.A.; Hernández, M.; Heymann, D.; Garlet, G.P.; Sorsa, T.; Pärnänen, P.; Lee, H.-M.; et al. Osteoimmunology of oral and maxillofacial diseases: Translational applications based on biological mechanisms. Front. Immunol. 2019, 10, 1664. [Google Scholar] [CrossRef]
- Jia, X.-h.; Feng, G.-w.; Wang, Z.-l.; Du, Y.; Shen, C.; Hui, H.; Peng, D.; Li, Z.-j.; Kong, D.-l.; Tian, J. Activation of mesenchymal stem cells by macrophages promotes tumor progression through immune suppressive effects. Oncotarget 2016, 7, 20934. [Google Scholar] [CrossRef]
- Liu, W.; Xie, X.; Qi, Y.; Wu, J. Exploration of immune-related gene expression in osteosarcoma and association with outcomes. JAMA Netw. Open 2021, 4, e2119132. [Google Scholar] [CrossRef] [PubMed]
- Dumars, C.; Ngyuen, J.-M.; Gaultier, A.; Lanel, R.; Corradini, N.; Gouin, F.; Heymann, D.; Heymann, M.-F. Dysregulation of macrophage polarization is associated with the metastatic process in osteosarcoma. Oncotarget 2016, 7, 78343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ségaliny, A.I.; Mohamadi, A.; Dizier, B.; Lokajczyk, A.; Brion, R.; Lanel, R.; Amiaud, J.; Charrier, C.; Boisson-Vidal, C.; Heymann, D. Interleukin-34 promotes tumor progression and metastatic process in osteosarcoma through induction of angiogenesis and macrophage recruitment. Int. J. Cancer 2015, 137, 73–85. [Google Scholar] [CrossRef]
- Pham, T.; Roth, S.; Kong, J.; Guerra, G.; Narasimhan, V.; Pereira, L.; Desai, J.; Heriot, A.; Ramsay, R. An update on immunotherapy for solid tumors: A review. Ann. Surg. Oncol. 2018, 25, 3404–3412. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727. [Google Scholar] [PubMed]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Chikuma, S. CTLA-4, an essential immune-checkpoint for T-cell activation. In Emerging Concepts Targeting Immune Checkpoints in Cancer and Autoimmunity; Springer: Berlin/Heidelberg, Germany, 2017; pp. 99–126. [Google Scholar]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef]
- Hashimoto, K.; Nishimura, S.; Akagi, M. Characterization of PD-1/PD-L1 immune checkpoint expression in osteosarcoma. Diagnostics 2020, 10, 528. [Google Scholar] [CrossRef] [PubMed]
- Lussier, D.M.; O’Neill, L.; Nieves, L.M.; McAfee, M.S.; Holechek, S.A.; Collins, A.W.; Dickman, P.; Jacobsen, J.; Hingorani, P.; Blattman, J.N. Enhanced T-cell immunity to osteosarcoma through antibody blockade of PD-1/PD-L1 interactions. J. Immunother. 2015, 38, 96. [Google Scholar] [CrossRef]
- Koirala, P.; Roth, M.E.; Gill, J.; Piperdi, S.; Chinai, J.M.; Geller, D.S.; Hoang, B.H.; Park, A.; Fremed, M.A.; Zang, X.; et al. Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. Sci. Rep. 2016, 6, 30093. [Google Scholar] [CrossRef]
- Zheng, W.; Xiao, H.; Liu, H.; Zhou, Y. Expression of programmed death 1 is correlated with progression of osteosarcoma. Apmis 2015, 123, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Jin, Z.; Zhang, M.; Tang, Y.; Yang, G.; Yuan, X.; Yao, J.; Sun, D. Prognostic value of programmed death-ligand 1 in sarcoma: A meta-analysis. Oncotarget 2017, 8, 59570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geoerger, B.; Zwaan, C.M.; Marshall, L.V.; Michon, J.; Bourdeaut, F.; Casanova, M.; Corradini, N.; Rossato, G.; Farid-Kapadia, M.; Shemesh, C.S.; et al. Atezolizumab for children and young adults with previously treated solid tumours, non-Hodgkin lymphoma, and Hodgkin lymphoma (iMATRIX): A multicentre phase 1–2 study. Lancet Oncol. 2020, 21, 134–144. [Google Scholar] [CrossRef]
- Vafaei, S.; Zekiy, A.O.; Khanamir, R.A.; Zaman, B.A.; Ghayourvahdat, A.; Azimizonuzi, H.; Zamani, M. Combination therapy with immune checkpoint inhibitors (ICIs); a new frontier. Cancer Cell Int. 2022, 22, 1–27. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Mahoney, M.R.; Van Tine, B.A.; Atkins, J.; Milhem, M.M.; Jahagirdar, B.N.; Antonescu, C.R.; Horvath, E.; Tap, W.D.; Schwartz, G.K. A non-comparative multi-center randomized phase II study of nivolumab+/− ipilimumab for patients with metastatic sarcoma (Alliance A091401). Lancet. Oncol. 2018, 19, 416. [Google Scholar] [CrossRef] [PubMed]
- Somaiah, N.; Conley, A.P.; Parra, E.R.; Lin, H.; Amini, B.; Soto, L.S.; Salazar, R.; Barreto, C.; Chen, H.; Gite, S.; et al. Durvalumab plus tremelimumab in advanced or metastatic soft tissue and bone sarcomas: A single-centre phase 2 trial. Lancet Oncol. 2022, 23, 1156–1166. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Richards, A.L.; Conley, A.P.; Woo, H.J.; Dickson, M.A.; Gounder, M.; Kelly, C.; Keohan, M.L.; Movva, S.; Thornton, K.; et al. Pilot study of bempegaldesleukin in combination with nivolumab in patients with metastatic sarcoma. Nat. Commun. 2022, 13, 1–11. [Google Scholar] [CrossRef]
- Davis, L.E.; Bolejack, V.; Ryan, C.W.; Ganjoo, K.N.; Loggers, E.T.; Chawla, S.; Agulnik, M.; Livingston, M.B.; Reed, D.; Keedy, V.; et al. Randomized double-blind phase II study of regorafenib in patients with metastatic osteosarcoma. J. Clin. Oncol. 2019, 37, 1424. [Google Scholar] [CrossRef]
- Grignani, G.; Palmerini, E.; Dileo, P.; Asaftei, S.; D’Ambrosio, L.; Pignochino, Y.; Mercuri, M.; Picci, P.; Fagioli, F.; Casali, P.; et al. A phase II trial of sorafenib in relapsed and unresectable high-grade osteosarcoma after failure of standard multimodal therapy: An Italian Sarcoma Group study. Ann. Oncol. 2012, 23, 508–516. [Google Scholar] [CrossRef]
- Keremu, A.; Aimaiti, A.; Liang, Z.; Zou, X. Role of the HDAC6/STAT3 pathway in regulating PD-L1 expression in osteosarcoma cell lines. Cancer Chemother. Pharmacol. 2019, 83, 255–264. [Google Scholar] [CrossRef]
- Duan, X.L.; Guo, J.P.; Li, F.; Xiu, C.; Wang, H. Sunitinib inhibits PD-L1 expression in osteosarcoma by targeting STAT3 and remodels the immune system in tumor-bearing mice. Future Oncol. 2020, 16, 1815–1824. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wen, J.; Wu, C.; Hu, C.; Wang, J.; Bao, Q.; Wang, H.; Wang, J.; Zhou, Q.; Wei, L.; et al. MicroRNA-200a induces immunosuppression by promoting PTEN-mediated PD-L1 upregulation in osteosarcoma. Aging 2020, 12, 1213. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Ren, T.; Huang, Y.; Guo, W. Apatinib inhibits migration and invasion as well as PD-L1 expression in osteosarcoma by targeting STAT3. Biochem. Biophys. Res. Commun. 2018, 495, 1695–1701. [Google Scholar] [CrossRef]
- Le Cesne, A.; Marec-Berard, P.; Blay, J.-Y.; Gaspar, N.; Bertucci, F.; Penel, N.; Bompas, E.; Cousin, S.; Toulmonde, M.; Bessede, A.; et al. Programmed cell death 1 (PD-1) targeting in patients with advanced osteosarcomas: Results from the PEMBROSARC study. Eur. J. Cancer 2019, 119, 151–157. [Google Scholar] [CrossRef]
- Zhou, Q.; Xian, M.; Xiang, S.; Xiang, D.; Shao, X.; Wang, J.; Cao, J.; Yang, X.; Yang, B.; Ying, M.; et al. All-trans retinoic acid prevents osteosarcoma metastasis by inhibiting M2 polarization of tumor-associated macrophages. Cancer Immunol. Res. 2017, 5, 547–559. [Google Scholar] [CrossRef]
- Li, D.-k.; Wang, G.-h. Asiaticoside reverses M2 phenotype macrophage polarization-evoked osteosarcoma cell malignant behaviour by TRAF6/NF-κB inhibition. Pharm. Biol. 2022, 60, 1635–1645. [Google Scholar] [CrossRef]
- Deng, X.; Liang, H.; Yang, W.; Shao, Z. Polarization and function of tumor-associated macrophages mediate graphene oxide-induced photothermal cancer therapy. J. Photochem. Photobiol. B Biol. 2020, 208, 111913. [Google Scholar] [CrossRef]
- Punzo, F.; Bellini, G.; Tortora, C.; Di Pinto, D.; Argenziano, M.; Pota, E.; Di Paola, A.; Di Martino, M.; Rossi, F. Mifamurtide and TAM-like macrophages: Effect on proliferation, migration and differentiation of osteosarcoma cells. Oncotarget 2020, 11, 687–698. [Google Scholar] [CrossRef]
- Fujiwara, T.; Yakoub, M.A.; Chandler, A.; Christ, A.B.; Yang, G.; Ouerfelli, O.; Rajasekhar, V.K.; Yoshida, A.; Kondo, H.; Hata, T. CSF1/CSF1R signaling inhibitor pexidartinib (PLX3397) reprograms tumor-associated macrophages and stimulates T-cell infiltration in the sarcoma microenvironment. Mol. Cancer Ther. 2021, 20, 1388–1399. [Google Scholar] [CrossRef]
- Zhang, Y.; Yuan, T.; Li, Z.; Luo, C.; Wu, Y.; Zhang, J.; Zhang, X.; Fan, W. Hyaluronate-based self-stabilized nanoparticles for immunosuppression reversion and immunochemotherapy in osteosarcoma treatment. ACS Biomater. Sci. Eng. 2021, 7, 1515–1525. [Google Scholar] [CrossRef]
- Kimura, Y.; Sumiyoshi, M. Antitumor and antimetastatic actions of dihydroxycoumarins (esculetin or fraxetin) through the inhibition of M2 macrophage differentiation in tumor-associated macrophages and/or G1 arrest in tumor cells. Eur. J. Pharmacol. 2015, 746, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.d.M.; Teixeira, F.M.E.; Sato, M.N. Impact of retinoic acid on immune cells and inflammatory diseases. Mediat. Inflamm. 2018, 2018, 3067126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alatshan, A.; Kovács, G.E.; Aladdin, A.; Czimmerer, Z.; Tar, K.; Benkő, S. All-trans retinoic acid enhances both the signaling for priming and the glycolysis for activation of NLRP3 inflammasome in human macrophage. Cells 2020, 9, 1591. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.-j.; Xiang, S.-f.; Chen, Y.-q.; Zhang, N.; Cao, J.; Zhu, H.; Yang, B.; Zhou, Q.; Ying, M.-d.; He, Q.-j. Inhibition of M2-like macrophages by all-trans retinoic acid prevents cancer initiation and stemness in osteosarcoma cells. Acta Pharmacol. Sin. 2019, 40, 1343–1350. [Google Scholar] [CrossRef]
- Todesco, A.; Carli, M.; Iacona, I.; Frascella, E.; Ninfo, V.; Rosolen, A. All-trans retinoic acid and interferon-α in the treatment of a patient with resistant metastatic osteosarcoma: A case report. Cancer 2000, 89, 2661–2666. [Google Scholar] [CrossRef]
- Cox-Georgian, D.; Ramadoss, N.; Dona, C.; Basu, C. Therapeutic and medicinal uses of terpenes. In Medicinal Plants; Springer: Berlin/Heidelberg, Germany, 2019; pp. 333–359. [Google Scholar]
- Zhi, D.; Yang, T.; O’hagan, J.; Zhang, S.; Donnelly, R.F. Photothermal therapy. J. Control. Release 2020, 325, 52–71. [Google Scholar] [CrossRef]
- Pan, S.; Xing, H.; Fu, X.; Yu, H.; Yang, Z.; Yang, Y.; Sun, W. The effect of photothermal therapy on osteosarcoma with polyacrylic acid–coated gold nanorods. Dose-Response 2018, 16, 1559325818789841. [Google Scholar] [CrossRef]
- Ando, K.; Mori, K.; Corradini, N.; Redini, F.; Heymann, D. Mifamurtide for the treatment of nonmetastatic osteosarcoma. Expert Opin. Pharmacother. 2011, 12, 285–292. [Google Scholar] [CrossRef]
- Tacyildiz, N.; Incesoy Ozdemir, S.; Unal, E.; Berber, M.; Dincaslan, H.; Yavuz, G. The efficiency and toxicity of mifamurtide in childhood osteosarcoma. J. Pediatr. Hematol./Oncol. 2018, 40, e373–e376. [Google Scholar] [CrossRef]
- Creaven, P.J.; Cowens, J.; Brenner, D.E.; Dadey, B.M.; Han, T.; Huben, R.; Karakousis, C.; Frost, H.; LeSher, D.; Hanagan, J. Initial clinical trial of the macrophage activator muramyl tripeptide-phosphatidylethanolamine encapsulated in liposomes in patients with advanced cancer. J. Biol. Response Modif. 1990, 9, 492–498. [Google Scholar]
- Kleinerman, E.S.; Gano, J.B.; Johnston, D.A.; Benjamin, R.S.; Jaffe, N. Efficacy of liposomal muramyl tripeptide (CGP 19835A) in the treatment of relapsed osteosarcoma. Am. J. Clin. Oncol. 1995, 18, 93–99. [Google Scholar] [CrossRef]
- Múdry, P.; Kýr, M.; Rohleder, O.; Mahdal, M.; Zambo, I.S.; Ježová, M.; Tomáš, T.; Štěrba, J. Improved osteosarcoma survival with addition of mifamurtide to conventional chemotherapy–Observational prospective single institution analysis. J. Bone Oncol. 2021, 28, 100362. [Google Scholar] [CrossRef] [PubMed]
- Benner, B.; Good, L.; Quiroga, D.; Schultz, T.E.; Kassem, M.; Carson, W.E.; Cherian, M.A.; Sardesai, S.; Wesolowski, R. Pexidartinib, a novel small molecule CSF-1R inhibitor in use for tenosynovial giant cell tumor: A systematic review of pre-clinical and clinical development. Drug Des. Dev. Ther. 2020, 14, 1693. [Google Scholar] [CrossRef] [PubMed]
- Manji, G.A.; Van Tine, B.A.; Lee, S.M.; Raufi, A.G.; Pellicciotta, I.; Hirbe, A.C.; Pradhan, J.; Chen, A.; Rabadan, R.; Schwartz, G.K. A phase I study of the combination of pexidartinib and sirolimus to target tumor-associated macrophages in unresectable sarcoma and malignant peripheral nerve sheath tumors. Clin. Cancer Res. 2021, 27, 5519–5527. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Sumiyoshi, M.; Sakanaka, M.; Taniguchi, M.; Baba, K. In vitro and in vivo antiproliferative effect of a combination of ultraviolet-A and alkoxy furocoumarins isolated from umbelliferae medicinal plants, in melanoma cells. Photochem. Photobiol. 2013, 89, 1216–1225. [Google Scholar] [CrossRef]
- Sumiyoshi, M.; Sakanaka, M.; Taniguchi, M.; Baba, K.; Kimura, Y. Anti-tumor effects of various furocoumarins isolated from the roots, seeds and fruits of Angelica and Cnidium species under ultraviolet A irradiation. J. Nat. Med. 2014, 68, 83–94. [Google Scholar] [CrossRef]
- Willingham, S.B.; Volkmer, J.-P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef]
- Veillette, A.; Chen, J. SIRPα–CD47 immune checkpoint blockade in anticancer therapy. Trends Immunol. 2018, 39, 173–184. [Google Scholar] [CrossRef]
- Cao, F.; Nguyen, P.; Hong, B.; DeRenzo, C.; Rainusso, N.C.; Rodriguez Cruz, T.; Wu, M.F.; Liu, H.; Song, X.T.; Suzuki, M.; et al. Engineering oncolytic vaccinia virus to redirect macrophages to tumor cells. Adv. Cell Gene Ther. 2021, 4, e99. [Google Scholar] [CrossRef]
- Mohanty, S.; Yerneni, K.; Theruvath, J.L.; Graef, C.M.; Nejadnik, H.; Lenkov, O.; Pisani, L.; Rosenberg, J.; Mitra, S.; Cordero, A.S.; et al. Nanoparticle enhanced MRI can monitor macrophage response to CD47 mAb immunotherapy in osteosarcoma. Cell Death Dis. 2019, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, S.; Aghighi, M.; Yerneni, K.; Theruvath, J.L.; Daldrup-Link, H.E. Improving the efficacy of osteosarcoma therapy: Combining drugs that turn cancer cell ‘don’t eat me’signals off and ‘eat me’signals on. Mol. Oncol. 2019, 13, 2049–2061. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, B.; Li, S.; Lin, W.; Wang, Z.; Wang, S.; Chen, W.; Shi, W.; Chen, T.; Zhou, H. Metabolic control of CD47 expression through LAT2-mediated amino acid uptake promotes tumor immune evasion. Nat. Commun. 2022, 13, 6308. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, Y.; Yang, S.; Zhang, L.; Wang, W. Anti-CD40 mAb enhanced efficacy of anti-PD1 against osteosarcoma. J. Bone Oncol. 2019, 17, 100245. [Google Scholar] [CrossRef]
- Hiruma, Y.; Hirai, T.; Tsuda, E. Siglec-15, a member of the sialic acid-binding lectin, is a novel regulator for osteoclast differentiation. Biochem. Biophys. Res. Commun. 2011, 409, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Ishida-Kitagawa, N.; Tanaka, K.; Bao, X.; Kimura, T.; Miura, T.; Kitaoka, Y.; Hayashi, K.; Sato, M.; Maruoka, M.; Ogawa, T.; et al. Siglec-15 protein regulates formation of functional osteoclasts in concert with DNAX-activating protein of 12 kDa (DAP12). J. Biol. Chem. 2012, 287, 17493–17502. [Google Scholar] [CrossRef]
- Fan, M.-k.; Zhang, G.-c.; Chen, W.; Qi, L.-l.; Xie, M.-f.; Zhang, Y.-y.; Wang, L.; Zhang, Q. Siglec-15 promotes tumor progression in osteosarcoma via DUSP1/MAPK pathway. Front. Oncol. 2021, 11, 710689. [Google Scholar] [CrossRef]
- Abraham, S.; Clark, A. Dual-specificity phosphatase 1: A critical regulator of innate immune responses. Biochem. Soc. Trans. 2006, 34, 1018–1023. [Google Scholar] [CrossRef]
- Walport, M.J. Complement first of two parts. N. Engl. J. Med. 2001, 344, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Ignatius, A.; Schoengraf, P.; Kreja, L.; Liedert, A.; Recknagel, S.; Kandert, S.; Brenner, R.E.; Schneider, M.; Lambris, J.D.; Huber-Lang, M. Complement C3a and C5a modulate osteoclast formation and inflammatory response of osteoblasts in synergism with IL-1β. J. Cell. Biochem. 2011, 112, 2594–2605. [Google Scholar] [CrossRef]
- Rutkowski, M.J.; Sughrue, M.E.; Kane, A.J.; Mills, S.A.; Parsa, A.T. Cancer and the Complement CascadeCancer and the Complement Cascade. Mol. Cancer Res. 2010, 8, 1453–1465. [Google Scholar] [CrossRef]
- Jeon, H.; Han, S.R.; Lee, S.; Park, S.J.; Kim, J.H.; Yoo, S.-M.; Lee, M.-S. Activation of the complement system in an osteosarcoma cell line promotes angiogenesis through enhanced production of growth factors. Sci. Rep. 2018, 8, 5415. [Google Scholar] [CrossRef] [PubMed]
- Astarita, J.L.; Acton, S.E.; Turley, S.J. Podoplanin: Emerging functions in development, the immune system, and cancer. Front. Immunol. 2012, 3, 283. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, W.; Bi, J.; Wang, J.; Ni, L.; Shi, Q.; Meng, Q. Association of high PDPN expression with pulmonary metastasis of osteosarcoma and patient prognosis. Oncol. Lett. 2019, 18, 6323–6330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemoto, A.; Takagi, S.; Ukaji, T.; Gyobu, N.; Kakino, M.; Takami, M.; Kobayashi, A.; Lebel, M.; Kawaguchi, T.; Sugawara, M.; et al. Targeting podoplanin for the treatment of osteosarcoma. Clin. Cancer Res. 2022, 28, OF1–OF13. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K pathway in human disease. Cell 2017, 170, 605–635. [Google Scholar]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef]
- De Henau, O.; Rausch, M.; Winkler, D.; Campesato, L.F.; Liu, C.; Cymerman, D.H.; Budhu, S.; Ghosh, A.; Pink, M.; Tchaicha, J.; et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature 2016, 539, 443–447. [Google Scholar] [CrossRef]
- Zhang, J.; Yu, X.-H.; Yan, Y.-G.; Wang, C.; Wang, W.-J. PI3K/Akt signaling in osteosarcoma. Clin. Chim. Acta 2015, 444, 182–192. [Google Scholar] [CrossRef]
- Zhang, Y.; Weng, Q.; Han, J.; Chen, J. Alantolactone suppresses human osteosarcoma through the PI3K/AKT signaling pathway. Mol. Med. Rep. 2020, 21, 675–684. [Google Scholar] [CrossRef]
- Kato, Y.; Chao, T.-H.; Hayashi, M.; Tapping, R.I.; Lee, J.-D. Role of BMK1 in regulation of growth factor-induced cellular responses. Immunol. Res. 2000, 21, 233–237. [Google Scholar] [CrossRef]
- Gomez, N.; Erazo, T.; Lizcano, J.M. ERK5 and cell proliferation: Nuclear localization is what matters. Front. Cell Dev. Biol. 2016, 4, 105. [Google Scholar] [CrossRef] [PubMed]
- Giurisato, E.; Xu, Q.; Lonardi, S.; Telfer, B.; Russo, I.; Pearson, A.; Finegan, K.G.; Wang, W.; Wang, J.; Gray, N.S.; et al. Myeloid ERK5 deficiency suppresses tumor growth by blocking protumor macrophage polarization via STAT3 inhibition. Proc. Natl. Acad. Sci. USA 2018, 115, E2801–E2810. [Google Scholar] [CrossRef] [PubMed]
- Giurisato, E.; Lonardi, S.; Telfer, B.; Lussoso, S.; Risa-Ebrí, B.; Zhang, J.; Russo, I.; Wang, J.; Santucci, A.; Finegan, K.G. Extracellular-Regulated Protein Kinase 5-Mediated Control of p21 Expression Promotes Macrophage Proliferation Associated with Tumor Growth and MetastasisTargeting ERK5 Blocks TAM Proliferation and Tumor Malignancy. Cancer Res. 2020, 80, 3319–3330. [Google Scholar] [CrossRef] [PubMed]
- Green, D.; Eyre, H.; Singh, A.; Taylor, J.T.; Chu, J.; Jeys, L.; Sumathi, V.; Coonar, A.; Rassl, D.; Babur, M. Targeting the MAPK7/MMP9 axis for metastasis in primary bone cancer. Oncogene 2020, 39, 5553–5569. [Google Scholar] [CrossRef] [PubMed]


| Drug | Target Cell Type | Markers Used for Flow Cytometry/IHC/RT-PCR | Inhibition In Vitro Drug Concentration | Target Cells Type: In Vitro/ Primary Tumor/Pulmonary Metastasis/In Vivo | Mechanism |
|---|---|---|---|---|---|
| All-Trans Retinoic Acid (ATRA) [68] | TAM/M2 | F4/80, CD206+ CD209, CD86 CD14 | Pretreatment of mice for 7 days at 20 mg/kg and post injection 40 mg/kg for 4 weeks | ATRA reduced TAM macrophage polarization in vitro. Secondary lung macroscopic metastatic reduction was seen to 60% and 95% after 1- and 2-weeks treatment respectively. | MMP12 Inhibition from M2 macrophages to suppress metastasis |
| Asiaticoside (ATS) [69] | M2 | CD206, CD14, CD86, Ki67, Bcl-2, Bax, VEGF | 40 µM invitro and 10 mg/kg in vivo every 2nd day for 30 days | ATS restrained the M2 phenotype and helped reduce the tumor weight by 3-fold and suppressed OS progression. | TRAF6/NF-kB inhibition |
| Graphene Oxide (GO) mediated Photothermal therapy (PTT) [70] | M2 | CD206, CD209, Arg-1 | 0.05 mg/mL in vitro and 808 nm light (0.7 W/cm2, 1.5 min in vivo, temperature ≥45 °C | Low-temperature PPT helped polarize to M1 phenotype and show antitumor effects. | Suppression of IL-2 induced M2 repolarization |
| Mifamurtide [71] | M2 | CD11b, CD3, CD45.2, Ly6.G, MMP2/ MMP9, TNF-Ƴ, TRPV1 | 100 µM in vitro and 5 mg/mL in vivo | Treatment showed a reduction in osteoblast markers. M1 treated cells showed increased iron transporter expression of DMT1. | Inhibition of STAT3 pathway/anti RANKL therapy |
| Pexidartinib (PLX3397) [72] | TAM/M2 | CD206, CD86, iNOS, IL-1beta, CD80, CD206, CCL2 | 10 mmol/L in vitro In vivo 5 and 10 mg/kg | Treatment showed suppression of TAM phenotype and increased chemotaxis. The mouse model showed suppressed primary tumor and metastasis and possibility of transition into immunotherapy. | Inhibition of CSF1/CSF1R signaling |
| Resiquimod cisplatin loaded nanoparticle (CDDPNPR848) [73] | TAM/M2 | CD86, CD206 CD44, CD62L | 10 µg/mL | Treatment effectively suppressed the tumor growth in vivo and stimulated the induction of immune memory response in spleen. | D88-dependent signaling pathway |
| Esculetin and fraxetin [74] | M2 | Cyclin D1 and CDK4 | In vitro- 10–100 µM In vivo- 3 or 10 mg/kg for 35 days | Esculetin showed cell cycle arrest at S phase and differentiation of M2 macrophages. Esculetin and fraxetin showed antitumor activity against primary and secondary metastatic cancer. | Inhibition of M2 macrophage differentiation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anand, N.; Peh, K.H.; Kolesar, J.M. Macrophage Repolarization as a Therapeutic Strategy for Osteosarcoma. Int. J. Mol. Sci. 2023, 24, 2858. https://doi.org/10.3390/ijms24032858
Anand N, Peh KH, Kolesar JM. Macrophage Repolarization as a Therapeutic Strategy for Osteosarcoma. International Journal of Molecular Sciences. 2023; 24(3):2858. https://doi.org/10.3390/ijms24032858
Chicago/Turabian StyleAnand, Namrata, Keng Hee Peh, and Jill M. Kolesar. 2023. "Macrophage Repolarization as a Therapeutic Strategy for Osteosarcoma" International Journal of Molecular Sciences 24, no. 3: 2858. https://doi.org/10.3390/ijms24032858
APA StyleAnand, N., Peh, K. H., & Kolesar, J. M. (2023). Macrophage Repolarization as a Therapeutic Strategy for Osteosarcoma. International Journal of Molecular Sciences, 24(3), 2858. https://doi.org/10.3390/ijms24032858