
Genetics and Molecular Basis of Congenital Heart Defects in Down Syndrome: Role of Extracellular Matrix Regulation
 , ,
, ,
Abstract
:1. Introduction

2. Candidate Genes for CHD in DS
2.1. Genes Mapping to Hsa21
2.2. Genes Mapping to Other Chromosomes
2.3. Role of ncRNAs in the Pathophysiology of CHD in DS
3. Role of ECM Regulation in the Development of CHD in DS
4. The Hsa21 Transcription Factor RUNX1 Regulates the Expression of ECM Components
5. Conclusions

Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Der Linde, D.; Konings, E.E.M.; Slager, M.A.; Witsenburg, M.; Helbing, W.A.; Takkenberg, J.J.M.; Roos-Hesselink, J.W. Birth Prevalence of Congenital Heart Disease Worldwide: A Systematic Review and Meta-Analysis. J. Am. Coll. Cardiol. 2011, 58, 2241–2247. [Google Scholar] [CrossRef]
- Fahed, A.C.; Gelb, B.D.; Seidman, J.G.; Seidman, C.E. Genetics of Congenital Heart Disease: The Glass Half Empty. Circ. Res. 2013, 112, 707–720. [Google Scholar] [CrossRef]
- Urbano, R. Health Issues in Persons with Down Syndrome; Academic Press: Cambridge, MA, USA, 2012; Volume 39, ISBN 9780123744777. [Google Scholar]
- Stoll, C.; Dott, B.; Alembik, Y.; Roth, M.-P. Associated congenital anomalies among cases with Down syndrome. Eur. J. Med. Genet. 2015, 58, 674–680. [Google Scholar] [CrossRef]
- Maslen, C.L.; Babcock, D.; Robinson, S.W.; Bean, L.J.H.; Dooley, K.J.; Willour, V.L.; Sherman, S.L. CRELD1 mutations contribute to the occurrence of cardiac atrioventricular septal defects in Down syndrome. Am. J. Med. Genet. Part A 2006, 140, 2501–2505. [Google Scholar] [CrossRef]
- Asim, A.; Agarwal, S. Congenital heart defects among Down’s syndrome cases: An updated review from basic research to an emerging diagnostics technology and genetic counselling. J. Genet. 2021, 100, 45. [Google Scholar] [CrossRef]
- Rehman, Y.; Wazir, H.D.; Akbar, A.; Khan, A.M.; Hussain, I.; Afridi, A.; Gul, H.; Sadia, H. Congenital Heart Disease and Its Association in Children with Down Syndrome. Cureus 2022, 14, e29176. [Google Scholar] [CrossRef]
- Rahmani, Z.; Blouin, J.L.; Creau-Goldberg, N.; Watkins, P.C.; Mattei, J.F.; Poissonnier, M.; Prieur, M.; Chettouh, Z.; Nicole, A.; Aurias, A. Critical role of the D21S55 region on chromosome 21 in the pathogenesis of Down syndrome. Proc. Natl. Acad. Sci. USA 1989, 86, 5958–5962. [Google Scholar] [CrossRef]
- McCormick, M.K.; Schinzel, A.; Petersen, M.B.; Stetten, G.; Driscoll, D.J.; Cantu, E.S.; Tranebjaerg, L.; Mikkelsen, M.; Watkins, P.C.; Antonarakis, S.E. Molecular genetic approach to the characterization of the “Down syndrome region” of chromosome 21. Genomics 1989, 5, 325–331. [Google Scholar] [CrossRef]
- Korenberg, J.R.; Kawashima, H.; Pulst, S.M.; Ikeuchi, T.; Ogasawara, N.; Yamamoto, K.; Schonberg, S.A.; West, R.; Allen, L.; Magenis, E.; et al. Molecular definition of a region of chromosome 21 that causes features of the Down syndrome phenotype. Am. J. Hum. Genet. 1990, 47, 236–246. [Google Scholar]
- Korenberg, J.R.; Bradley, C.; Disteche, C.M. Down syndrome: Molecular mapping of the congenital heart disease and duodenal stenosis. Am. J. Hum. Genet. 1992, 50, 294–302. [Google Scholar]
- Delabar, J.-M.; Theophile, D.; Rahmani, Z.; Chettouh, Z.; Blouin, J.-L.; Prieur, M.; Noel, B.; Sinet, P.-M. Molecular Mapping of Twenty-Four Features of Down Syndrome on Chromosome 21. Eur. J. Hum. Genet. 1993, 1, 114–124. [Google Scholar] [CrossRef]
- Korenberg, J.R.; Chen, X.N.; Schipper, R.; Sun, Z.; Gonsky, R.; Gerwehr, S.; Carpenter, N.; Daumer, C.; Dignan, P.; Disteche, C. Down syndrome phenotypes: The consequences of chromosomal imbalance. Proc. Natl. Acad. Sci. USA 1994, 91, 4997–5001. [Google Scholar] [CrossRef] [Green Version]
- Barlow, G.M.; Chen, X.-N.; Shi, Z.Y.; Lyons, G.E.; Kurnit, D.M.; Celle, L.; Spinner, N.B.; Zackai, E.; Pettenati, M.J.; Van Riper, A.J.; et al. Down syndrome congenital heart disease: A narrowed region and a candidate gene. Anesth. Analg. 2001, 3, 91–101. [Google Scholar] [CrossRef]
- Korbel, J.O.; Tirosh-Wagner, T.; Urban, A.E.; Chen, X.N.; Kasowski, M.; Dai, L.; Grubert, F.; Erdman, C.; Gao, M.C.; Lange, K.; et al. The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc. Natl. Acad Sci. USA 2009, 106, 12031–12036. [Google Scholar] [CrossRef]
- Pelleri, M.C.; Gennari, E.; Locatelli, C.; Piovesan, A.; Caracausi, M.; Antonaros, F.; Rocca, A.; Donati, C.M.; Conti, L.; Strippoli, P.; et al. Genotype-phenotype correlation for congenital heart disease in Down syndrome through analysis of partial trisomy 21 cases. Genomics 2017, 109, 391–400. [Google Scholar] [CrossRef]
- Lyle, R.; Béna, F.; Gagos, S.; Gehrig, C.; Lopez, G.; Schinzel, A.; Lespinasse, J.; Bottani, A.; Dahoun, S.; Taine, L.; et al. Genotype–phenotype correlations in Down syndrome identified by array CGH in 30 cases of partial trisomy and partial monosomy chromosome 21. Eur. J. Hum. Genet. 2009, 17, 454–466. [Google Scholar] [CrossRef]
- Yu, T.; Li, Z.; Jia, Z.; Clapcote, S.J.; Liu, C.; Li, S.; Asrar, S.; Pao, A.; Chen, R.; Fan, N.; et al. A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Hum. Mol. Genet. 2010, 19, 2780–2791. [Google Scholar] [CrossRef]
- Liu, C.; Belichenko, P.V.; Zhang, L.; Fu, D.; Kleschevnikov, A.M.; Baldini, A.; Antonarakis, S.E.; Mobley, W.C.; Yu, Y.E. Mouse Models for Down Syndrome-Associated Developmental Cognitive Disabilities. Dev. Neurosci. 2011, 33, 404–413. [Google Scholar] [CrossRef]
- Liu, C.; Morishima, M.; Jiang, X.; Yu, T.; Meng, K.; Ray, D.; Pao, A.; Ye, P.; Parmacek, M.S.; Yu, Y.E. Engineered chromosome-based genetic mapping establishes a 3.7 Mb critical genomic region for Down syndrome-associated heart defects in mice. Hum. Genet. 2014, 133, 743–753. [Google Scholar] [CrossRef]
- Lana-Elola, E.; Watson-Scales, S.; Slender, A.; Gibbins, D.; Martineau, A.; Douglas, C.; Mohun, T.; MC Fisher, E.; Tybulewicz, V.L. Genetic dissection of Down syndrome-associated congenital heart defects using a new mouse mapping panel. eLife 2016, 5, e11614. [Google Scholar] [CrossRef]
- Venegas-Zamora, L.; Bravo-Acuña, F.; Sigcho, F.; Gomez, W.; Bustamante-Salazar, J.; Pedrozo, Z.; Parra, V. New Molecular and Organelle Alterations Linked to Down Syndrome Heart Disease. Front. Genet. 2022, 12, 2734. [Google Scholar] [CrossRef]
- Conti, A.; Fabbrini, F.; D’Agostino, P.; Negri, R.; Greco, D.; Genesio, R.; D’Armiento, M.; Olla, C.; Paladini, D.; Zannini, M.; et al. Altered expression of mitochondrial and extracellular matrix genes in the heart of human fetuses with chromosome 21 trisomy. BMC Genom. 2007, 8, 268. [Google Scholar] [CrossRef] [Green Version]
- Wright, T.C.; Orkin, R.W.; Destrempes, M.; Kurnit, D.M. Increased adhesiveness of Down syndrome fetal fibroblasts in vitro. Proc. Natl. Acad. Sci. USA 1984, 81, 2426–2430. [Google Scholar] [CrossRef]
- Kurnit, D.M.; Aldridge, J.F.; Matsuoka, R.; Matthysse, S.; Opitz, J.M.; Reynolds, J.F. Increased adhesiveness of trisomy 21 cells and atrioventricular canal malformations in down syndrome: A stochastic model. Am. J. Med. Genet. 1985, 20, 385–399. [Google Scholar] [CrossRef]
- Agarwala, K.L.; Ganesh, S.; Amano, K.; Suzuki, T.; Yamakawa, K. DSCAM, a Highly Conserved Gene in Mammals, Expressed in Differentiating Mouse Brain. Biochem. Biophys. Res. Commun. 2001, 281, 697–705. [Google Scholar] [CrossRef]
- Dunlevy, L.; Bennett, M.; Slender, A.; Lana-Elola, E.; Tybulewicz, V.L.; Fisher, E.M.; Mohun, T. Down’s syndrome-like cardiac developmental defects in embryos of the transchromosomic Tc1 mouse. Cardiovasc. Res. 2010, 88, 287–295. [Google Scholar] [CrossRef]
- Davies, G.E.; Howard, C.M.; Farrer, M.J.; Coleman, M.M.; Bennett, L.B.; Cullen, L.M.; Wyse, R.K.H.; Burn, J.; Williamson, R.; Kessling, A.M. Genetic variation in the COL6A1 region is associated with congenital heart defects in trisomy 21 (Down’s syndrome). Ann. Hum. Genet. 1995, 59, 253–269. [Google Scholar] [CrossRef]
- Groot, A.C.G.-D.; Bartram, U.; Oosthoek, P.W.; Bartelings, M.M.; Hogers, B.; Poelmann, R.E.; Jongewaard, I.N.; Klewer, S.E. Collagen type VI expression during cardiac development and in human fetuses with trisomy 21. Anat. Rec. 2003, 275, 1109–1116. [Google Scholar] [CrossRef]
- Vogel, W.; Gish, G.D.; Alves, F.; Pawson, T. The Discoidin Domain Receptor Tyrosine Kinases Are Activated by Collagen. Mol. Cell 1997, 1, 13–23. [Google Scholar] [CrossRef]
- Faraci, E.; Eck, M.; Gerstmayer, B.; Bosio, A.; Vogel, W.F. An extracellular matrix-specific microarray allowed the identification of target genes downstream of discoidin domain receptors. Matrix Biol. 2003, 22, 373–381. [Google Scholar] [CrossRef]
- Morales, M.O.; Price, R.L.; Goldsmith, E.C. Expression of Discoidin Domain Receptor 2 (DDR2) in the Developing Heart. Microsc. Microanal. 2005, 11, 260–267. [Google Scholar] [CrossRef]
- Grossman, T.R.; Gamliel, A.; Wessells, R.J.; Taghli-Lamallem, O.; Jepsen, K.; Ocorr, K.; Korenberg, J.R.; Peterson, K.L.; Rosenfeld, M.G.; Bodmer, R.; et al. Over-Expression of DSCAM and COL6A2 Cooperatively Generates Congenital Heart Defects. PLoS Genet. 2011, 7, e1002344. [Google Scholar] [CrossRef] [Green Version]
- Kosaki, R.; Kosaki, K.; Matsushima, K.; Mitsui, N.; Matsumoto, N.; Ohashi, H. Refining chromosomal region critical for Down syndrome-related heart defects with a case of cryptic 21q22.2 duplication. Congenit. Anomalies 2005, 45, 62–64. [Google Scholar] [CrossRef]
- Arron, J.R.; Winslow, M.M.; Polleri, A.; Chang, C.-P.; Wu, H.; Gao, X.; Neilson, J.R.; Chen, L.; Heit, J.J.; Kim, S.K.; et al. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 2006, 441, 595–600. [Google Scholar] [CrossRef]
- Graef, I.A.; Chen, F.; Chen, L.; Kuo, A.; Crabtree, G.R. Signals Transduced by Ca2+/Calcineurin and NFATc3/c4 Pattern the Developing Vasculature. Cell 2001, 105, 863–875. [Google Scholar] [CrossRef]
- Fuentes, J.J.; Genescà, L.; Kingsbury, T.J.; Cunningham, K.W.; Pérez-Riba, M.; Estivill, X.; de la Luna, S. DSCR1, overexpressed in Down syndrome, is an inhibitor of calcineurin-mediated signaling pathways. Hum. Mol. Genet. 2000, 9, 1681–1690. [Google Scholar] [CrossRef]
- Klee, C.B.; Ren, H.; Wang, X. Regulation of the Calmodulin-stimulated Protein Phosphatase, Calcineurin. J. Biol. Chem. 1998, 273, 13367–13370. [Google Scholar] [CrossRef]
- Flanagan, W.M.; Corthésy, B.; Bram, R.J.; Crabtree, G.R. Nuclear association of a T-cell transcription factor blocked by FK-506 and cyclosporin A. Nature 1991, 352, 803–807. [Google Scholar] [CrossRef]
- Crabtree, G.R.; Olson, E.N. NFAT Signaling: Choreographing the Social Lives of Cells. Cell 2002, 109, S67–S79. [Google Scholar] [CrossRef]
- De La Pompa, J.L.; Timmerman, L.A.; Takimoto, H.; Yoshida, H.; Elia, A.J.; Samper, E.; Potter, J.; Wakeham, A.; Marengere, L.; Langille, B.L.; et al. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature 1998, 392, 182–186. [Google Scholar] [CrossRef]
- Ranger, A.M.; Grusby, M.J.; Hodge, M.R.; Gravallese, E.M.; De La Brousse, F.C.; Hoey, T.; Mickanin, C.; Baldwin, H.S.; Glimcher, L.H. The transcription factor NF-ATc is essential for cardiac valve formation. Nature 1998, 392, 186–190. [Google Scholar] [CrossRef]
- Chang, C.-P.; Neilson, J.R.; Bayle, J.H.; Gestwicki, J.E.; Kuo, A.; Stankunas, K.; Graef, I.A.; Crabtree, G.R. A Field of Myocardial-Endocardial NFAT Signaling Underlies Heart Valve Morphogenesis. Cell 2004, 118, 649–663. [Google Scholar] [CrossRef] [Green Version]
- Casas, C.; Martínez, S.; Pritchard, M.; Fuentes, J.; Nadal, M.; Guimerà, J.; Arbonés, M.; Flórez, J.; Soriano, E.; Estivill, X.; et al. Dscr1, a novel endogenous inhibitor of calcineurin signaling, is expressed in the primitive ventricle of the heart and during neurogenesis. Mech. Dev. 2001, 101, 289–292. [Google Scholar] [CrossRef]
- Lange, A.W.; Molkentin, J.; E Yutzey, K. DSCR1 gene expression is dependent on NFATc1 during cardiac valve formation and colocalizes with anomalous organ development in trisomy 16 mice. Dev. Biol. 2004, 266, 346–360. [Google Scholar] [CrossRef]
- Adayev, T.; Chen-Hwang, M.-C.; Murakami, N.; Wang, R.; Hwang, Y.-W. MNB/DYRK1A phosphorylation regulates the interactions of synaptojanin 1 with endocytic accessory proteins. Biochem. Biophys. Res. Commun. 2006, 351, 1060–1065. [Google Scholar] [CrossRef]
- Cousin, M.; Robinson, P.J. The dephosphins: Dephosphorylation by calcineurin triggers synaptic vesicle endocytosis. Trends Neurosci. 2001, 24, 659–665. [Google Scholar] [CrossRef]
- Fasano, D.; Parisi, S.; Pierantoni, G.M.; De Rosa, A.; Picillo, M.; Amodio, G.; Pellecchia, M.T.; Barone, P.; Moltedo, O.; Bonifati, V.; et al. Alteration of endosomal trafficking is associated with early-onset parkinsonism caused by SYNJ1 mutations. Cell Death Dis. 2018, 9, 385. [Google Scholar] [CrossRef]
- Amodio, G.; Moltedo, O.; Fasano, D.; Zerillo, L.; Oliveti, M.; Di Pietro, P.; Faraonio, R.; Barone, P.; Pellecchia, M.T.; De Rosa, A.; et al. PERK-Mediated Unfolded Protein Response Activation and Oxidative Stress in PARK20 Fibroblasts. Front. Neurosci. 2019, 13, 673. [Google Scholar] [CrossRef]
- Choudhry, H.; Aggarwal, M.; Pan, P.-Y. Mini-review: Synaptojanin 1 and its implications in membrane trafficking. Neurosci. Lett. 2021, 765, 136288. [Google Scholar] [CrossRef]
- Cossec, J.-C.; Lavaur, J.; Berman, D.E.; Rivals, I.; Hoischen, A.; Stora, S.; Ripoll, C.; Mircher, C.; Grattau, Y.; OlivoMarin, J.-C.; et al. Trisomy for Synaptojanin1 in Down syndrome is functionally linked to the enlargement of early endosomes. Hum. Mol. Genet. 2012, 21, 3156–3172. [Google Scholar] [CrossRef]
- De Rosa, L.; Fasano, D.; Zerillo, L.; Valente, V.; Izzo, A.; Mollo, N.; Amodio, G.; Polishchuk, E.; Polishchuk, R.; Melone, M.A.B.; et al. Down Syndrome Fetal Fibroblasts Display Alterations of Endosomal Trafficking Possibly due to SYNJ1 Overexpression. Front. Genet. 2022, 13, 867989. [Google Scholar] [CrossRef]
- Curran, J.; Makara, M.A.; Mohler, P.J. Endosome-based protein trafficking and Ca2+ homeostasis in the heart. Front. Physiol. 2015, 6, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardel, N.; Zolles, G.; Fakler, B.; Klöcker, N. Recycling endosomes supply cardiac pacemaker channels for regulated surface expression. Cardiovasc. Res. 2008, 79, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Turan, N.N.; Moshal, K.S.; Roder, K.; Baggett, B.C.; Kabakov, A.Y.; Dhakal, S.; Teramoto, R.; Chiang, D.Y.-E.; Zhong, M.; Xie, A.; et al. The endosomal trafficking regulator LITAF controls the cardiac Nav1.5 channel via the ubiquitin ligase NEDD4-2. J. Biol. Chem. 2020, 295, 18148–18159. [Google Scholar] [CrossRef] [PubMed]
- Sailani, M.R.; Makrythanasis, P.; Valsesia, A.; Santoni, F.A.; Deutsch, S.; Popadin, K.; Borel, C.; Migliavacca, E.; Sharp, A.J.; Sail, G.D.; et al. The complex SNP and CNV genetic architecture of the increased risk of congenital heart defects in Down syndrome. Genome Res. 2013, 23, 1410–1421. [Google Scholar] [CrossRef]
- Calcagni, G.; Pugnaloni, F.; Digilio, M.; Unolt, M.; Putotto, C.; Niceta, M.; Baban, A.; Sparascio, F.P.; Drago, F.; De Luca, A.; et al. Cardiac Defects and Genetic Syndromes: Old Uncertainties and New Insights. Genes 2021, 12, 1047. [Google Scholar] [CrossRef]
- Balistreri, C.R.; Ammoscato, C.L.; Scola, L.; Fragapane, T.; Giarratana, R.M.; Lio, D.; Piccione, M. Susceptibility to Heart Defects in Down Syndrome Is Associated with Single Nucleotide Polymorphisms in HAS 21 Interferon Receptor Cluster and VEGFA Genes. Genes 2020, 11, 1428. [Google Scholar] [CrossRef]
- Robinson, S.W.; Morris, C.D.; Goldmuntz, E.; Reller, M.D.; Jones, M.A.; Steiner, R.D.; Maslen, C.L. Missense Mutations in CRELD1 Are Associated with Cardiac Atrioventricular Septal Defects. Am. J. Hum. Genet. 2003, 72, 1047–1052. [Google Scholar] [CrossRef]
- Ackerman, C.; Locke, A.E.; Feingold, E.; Reshey, B.; Espana, K.; Thusberg, J.; Mooney, S.; Bean, L.J.; Dooley, K.J.; Cua, C.L.; et al. An Excess of Deleterious Variants in VEGF-A Pathway Genes in Down-Syndrome-Associated Atrioventricular Septal Defects. Am. J. Hum. Genet. 2012, 91, 646–659. [Google Scholar] [CrossRef]
- Asim, A.; Agarwal, S.; Panigrahi, I.; Sarangi, A.N.; Muthuswamy, S.; Kapoor, A. CRELD1 gene variants and atrioventricular septal defects in Down syndrome. Gene 2018, 641, 180–185. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, L.; Tian, J. Molecular mechanisms of congenital heart disease in down syndrome. Genes Dis. 2019, 6, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Reiter, T.A.; Abraham, R.T.; Choi, M.; Rusnak, F. Redox regulation of calcineurin in T-lymphocytes. JBIC J. Biol. Inorg. Chem. 1999, 4, 632–644. [Google Scholar] [CrossRef]
- Weston, G.; Haviv, I.; Rogers, P. Microarray analysis of VEGF-responsive genes in myometrial endothelial cells. Mol. Hum. Reprod. 2002, 8, 855–863. [Google Scholar] [CrossRef]
- Law, E.W.L.; Cheung, A.K.L.; Kashuba, V.I.; Pavlova, T.V.; Zabarovsky, E.R.; Lung, H.L.; Cheng, Y.; Chua, D.; Kwong, D.L.-W.; Tsao, S.W.; et al. Anti-angiogenic and tumor-suppressive roles of candidate tumor-suppressor gene, Fibulin-2, in nasopharyngeal carcinoma. Oncogene 2012, 31, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Person, A.D.; Garriock, R.J.; Krieg, P.A.; Runyan, R.B.; Klewer, S.E. Frzb modulates Wnt-9a-mediated β-catenin signaling during avian atrioventricular cardiac cushion development. Dev. Biol. 2005, 278, 35–48. [Google Scholar] [CrossRef]
- Armstrong, E.J.; Bischoff, J. Heart Valve Development: Endothelial Cell Signaling and Differentiation. Circ. Res. 2004, 95, 459–470. [Google Scholar] [CrossRef]
- Lambrechts, D.; Carmeliet, P. Sculpting Heart Valves with NFATc and VEGF. Cell 2004, 118, 532–534. [Google Scholar] [CrossRef] [PubMed]
- Dor, Y.; Porat, R.; Keshet, E.; Godoy, A.; Montecinos, V.P.; Gray, D.R.; Sotomayor, P.; Yau, J.M.; Vethanayagam, R.R.; Singh, S.; et al. Vascular endothelial growth factor and vascular adjustments to perturbations in oxygen homeostasis. Am. J. Physiol. Physiol. 2001, 280, C1367–C1374. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.D.; Yong, S.-K.; Dheen, S.T.; Bay, B.-H.; Tay, S.S.-W. Cardiac Malformations Are Associated with Altered Expression of Vascular Endothelial Growth Factor and Endothelial Nitric Oxide Synthase Genes in Embryos of Diabetic Mice. Exp. Biol. Med. 2008, 233, 1421–1432. [Google Scholar] [CrossRef]
- Hallaq, H.; Pinter, E.; Enciso, J.; McGrath, J.; Zeiss, C.; Brueckner, M.; Madri, J.; Jacobs, H.C.; Wilson, C.M.; Vasavada, H.; et al. A null mutation of Hhex results in abnormal cardiac development, defective vasculogenesis and elevated Vegfa levels. Development 2004, 131, 5197–5209. [Google Scholar] [CrossRef]
- Montano, M.M.; Doughman, Y.Q.; Deng, H.; Chaplin, L.; Yang, J.; Wang, N.; Zhou, Q.; Ward, N.L.; Watanabe, M. Mutation of the HEXIM1 Gene Results in Defects During Heart and Vascular Development Partly Through Downregulation of Vascular Endothelial Growth Factor. Circ. Res. 2008, 102, 415–422. [Google Scholar] [CrossRef]
- Miquerol, L.; Langille, B.; Nagy, A. Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development 2000, 127, 3941–3946. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Cherry, S.; Klinedinst, D.; DeLeon, V.; Redig, J.; Reshey, B.; Chin, M.T.; Sherman, S.L.; Maslen, C.L.; Reeves, R.H. Genetic Modifiers Predisposing to Congenital Heart Disease in the Sensitized Down Syndrome Population. Circ. Cardiovasc. Genet. 2012, 5, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, K.M.; Al-Mazroea, A.H.; Abdallah, A.M.; Almohammadi, Y.; Carlus, S.J.; Basit, S. Targeted Next-Generation Sequencing of 406 Genes Identified Genetic Defects Underlying Congenital Heart Disease in Down Syndrome Patients. Pediatr. Cardiol. 2018, 39, 1676–1680. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, C.; Izzo, A.; Scrima, R.; Bonfiglio, F.; Manco, R.; Negri, R.; Quarato, G.; Cela, O.; Ripoli, M.; Prisco, M.; et al. Chronic pro-oxidative state and mitochondrial dysfunctions are more pronounced in fibroblasts from Down syndrome foeti with congenital heart defects. Hum. Mol. Genet. 2013, 22, 1218–1232. [Google Scholar] [CrossRef]
- Ong, S.-B.; Hausenloy, D.J. Mitochondrial morphology and cardiovascular disease. Cardiovasc. Res. 2010, 88, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Huang, Y.; Xu, C.; An, P.; Luo, Y.; Jiao, L.; Luo, J.; Li, Y. Mitochondrial Dysfunction and Therapeutic Perspectives in Cardiovascular Diseases. Int. J. Mol. Sci. 2022, 23, 16053. [Google Scholar] [CrossRef]
- Izzo, A.; Manco, R.; Bonfiglio, F.; Calì, G.; De Cristofaro, T.; Patergnani, S.; Cicatiello, R.; Scrima, R.; Zannini, M.; Pinton, P.; et al. NRIP1/RIP140 siRNA-mediated attenuation counteracts mitochondrial dysfunction in Down syndrome. Hum. Mol. Genet. 2014, 23, 4406–4419. [Google Scholar] [CrossRef]
- Izzo, A.; Nitti, M.; Mollo, N.; Paladino, S.; Procaccini, C.; Faicchia, D.; Calì, G.; Genesio, R.; Bonfiglio, F.; Cicatiello, R.; et al. Metformin restores the mitochondrial network and reverses mitochondrial dysfunction in Down syndrome cells. Hum. Mol. Genet. 2017, 26, 1056–1069. [Google Scholar] [CrossRef]
- Mollo, N.; Nitti, M.; Zerillo, L.; Faicchia, D.; Micillo, T.; Accarino, R.; Secondo, A.; Petrozziello, T.; Calì, G.; Cicatiello, R.; et al. Pioglitazone Improves Mitochondrial Organization and Bioenergetics in Down Syndrome Cells. Front. Genet. 2019, 10, 606. [Google Scholar] [CrossRef]
- Mollo, N.; Cicatiello, R.; Aurilia, M.; Scognamiglio, R.; Genesio, R.; Charalambous, M.; Paladino, S.; Conti, A.; Nitsch, L.; Izzo, A. Targeting Mitochondrial Network Architecture in Down Syndrome and Aging. Int. J. Mol. Sci. 2020, 21, 3134. [Google Scholar] [CrossRef]
- Izzo, A.; Mollo, N.; Nitti, M.; Paladino, S.; Calì, G.; Genesio, R.; Bonfiglio, F.; Cicatiello, R.; Barbato, M.; Sarnataro, V.; et al. Mitochondrial dysfunction in down syndrome: Molecular mechanisms and therapeutic targets. Mol. Med. 2018, 24, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Stennard, F.A.; Costa, M.W.; Elliott, D.A.; Rankin, S.; Haast, S.J.P.; Lai, D.; McDonald, L.P.A.; Niederreither, K.; Dolle, P.; Bruneau, B.G.; et al. Cardiac T-box factor Tbx20 directly interacts with Nkx2-5, GATA4, and GATA5 in regulation of gene expression in the developing heart. Dev. Biol. 2003, 262, 206–224. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Nomura-Kitabayashi, A.; Cai, W.; Yan, J.; Christoffels, V.M.; Cai, C.-L. Myocardial Tbx20 regulates early atrioventricular canal formation and endocardial epithelial–mesenchymal transition via Bmp2. Dev. Biol. 2011, 360, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Samal, E.; Srivastava, D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature 2005, 436, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.; Shetty, P.M.V.; Frey, N.; Rangrez, A.Y. SRF: A seriously responsible factor in cardiac development and disease. J. Biomed. Sci. 2022, 29, 38. [Google Scholar] [CrossRef]
- Coppola, A.; Romito, A.; Borel, C.; Gehrig, C.; Gagnebin, M.; Falconnet, E.; Izzo, A.; Altucci, L.; Banfi, S.; Antonarakis, S.; et al. Cardiomyogenesis is controlled by the miR-99a/let-7c cluster and epigenetic modifications. Stem Cell Res. 2014, 12, 323–337. [Google Scholar] [CrossRef]
- Brás, A.; Rodrigues, A.S.; Gomes, B.; Rueff, J. Down syndrome and microRNAs (Review). Biomed. Rep. 2018, 8, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Latronico, M.V.G.; Catalucci, D.; Condorelli, G. MicroRNA and Cardiac Pathologies. Physiol. Genom. 2008, 34, 239–242. [Google Scholar] [CrossRef]
- Izzo, A.; Manco, R.; de Cristofaro, T.; Bonfiglio, F.; Cicatiello, R.; Mollo, N.; De Martino, M.; Genesio, R.; Zannini, M.; Conti, A.; et al. Overexpression of Chromosome 21 miRNAs May Affect Mitochondrial Function in the Hearts of Down Syndrome Fetuses. Int. J. Genom. 2017, 2017, 8737649. [Google Scholar] [CrossRef]
- Mollo, N.; Izzo, A.; Nitti, M.; Paladino, S.; Calì, G.; Genesio, R.; Bonfiglio, F.; Cicatiello, R.; Conti, A.; Nitsch, L. Mitochondria in Down Syndrome: From Organelle Abnormalities to Clinical Phenotype. In Down Syndrome; SM Group: Manila, Philippines, 2017. [Google Scholar]
- Quiñones-Lombraña, A.; Blanco, J.G. Chromosome 21-derived hsa-miR-155-5p regulates mitochondrial biogenesis by targeting Mitochondrial Transcription Factor A (TFAM). Biochim. et Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 1420–1427. [Google Scholar] [CrossRef]
- Ono, K.; Kuwabara, Y.; Han, J. MicroRNAs and cardiovascular diseases. FEBS J. 2011, 278, 1619–1633. [Google Scholar] [CrossRef] [Green Version]
- Sayed, D.; Hong, C.; Chen, I.-Y.; Lypowy, J.; Abdellatif, M. MicroRNAs Play an Essential Role in the Development of Cardiac Hypertrophy. Circ. Res. 2007, 100, 416–424. [Google Scholar] [CrossRef]
- Ramachandran, D.; Zeng, Z.; Locke, A.E.; Mulle, J.G.; Bean, L.J.; Rosser, T.C.; Dooley, K.J.; Cua, C.L.; Capone, G.T.; Reeves, R.H.; et al. Genome-Wide Association Study of Down Syndrome-Associated Atrioventricular Septal Defects. G3 Genes Genomes Genet. 2015, 5, 1961–1971. [Google Scholar] [CrossRef]
- Just, S.; Hirth, S.; Berger, I.M.; Fishman, M.C.; Rottbauer, W. The mediator complex subunit Med10 regulates heart valve formation in zebrafish by controlling Tbx2b-mediated Has2 expression and cardiac jelly formation. Biochem. Biophys. Res. Commun. 2016, 477, 581–588. [Google Scholar] [CrossRef]
- Koch, C.D.; Lee, C.M.; Apte, S.S. Aggrecan in Cardiovascular Development and Disease. J. Histochem. Cytochem. 2020, 68, 777–795. [Google Scholar] [CrossRef]
- Bloksgaard, M.; Lindsey, M.; Martinez-Lemus, L.A. Extracellular matrix in cardiovascular pathophysiology. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1687–H1690. [Google Scholar] [CrossRef]
- Stevens, M.V.; Parker, P.; Vaillancourt, R.R.; Camenisch, T.D. MEKK4 regulates developmental EMT in the embryonic heart. Dev. Dyn. 2006, 235, 2761–2770. [Google Scholar] [CrossRef]
- Shelton, E.L.; Yutzey, K.E. Tbx20 regulation of endocardial cushion cell proliferation and extracellular matrix gene expression. Dev. Biol. 2007, 302, 376–388. [Google Scholar] [CrossRef]
- Vilardell, M.; Rasche, A.; Thormann, A.; Maschke-Dutz, E.; Pérez-Jurado, L.A.; Lehrach, H.; Herwig, R. Meta-analysis of heterogeneous Down Syndrome data reveals consistent genome-wide dosage effects related to neurological processes. BMC Genom. 2011, 12, 229. [Google Scholar] [CrossRef]
- Vilardell, M.; Civit, S.; Herwig, R. An integrative computational analysis provides evidence for FBN1-associated network deregulation in trisomy 21. Biol. Open 2013, 2, 771–778. [Google Scholar] [CrossRef]
- De Toma, I.; Sierra, C.; Dierssen, M. Meta-analysis of transcriptomic data reveals clusters of consistently deregulated gene and disease ontologies in Down syndrome. PLoS Comput. Biol. 2021, 17, e1009317. [Google Scholar] [CrossRef]
- Twal, W.O.; Czirok, A.; Hegedus, B.; Knaak, C.; Chintalapudi, M.R.; Okagawa, H.; Sugi, Y.; Argraves, W.S. Fibulin-1 suppression of fibronectin-regulated cell adhesion and motility. J. Cell Sci. 2001, 114, 4587–4598. [Google Scholar] [CrossRef]
- Kern, C.B.; Twal, W.O.; Mjaatvedt, C.H.; Fairey, S.E.; Toole, B.P.; Iruela-Arispe, M.L.; Argraves, W.S. Proteolytic cleavage of versican during cardiac cushion morphogenesis. Dev. Dyn. 2006, 235, 2238–2247. [Google Scholar] [CrossRef]
- Kern, C.B.; Norris, R.A.; Thompson, R.P.; Argraves, W.S.; Fairey, S.E.; Reyes, L.; Hoffman, S.; Markwald, R.R.; Mjaatvedt, C.H. Versican proteolysis mediates myocardial regression during outflow tract development. Dev. Dyn. 2007, 236, 671–683. [Google Scholar] [CrossRef]
- Stankunas, K.; Hang, C.T.; Tsun, Z.-Y.; Chen, H.; Lee, N.V.; Wu, J.I.; Shang, C.; Bayle, J.H.; Shou, W.; Iruela-Arispe, M.L.; et al. Endocardial Brg1 Represses ADAMTS1 to Maintain the Microenvironment for Myocardial Morphogenesis. Dev. Cell 2008, 14, 298–311. [Google Scholar] [CrossRef]
- Lockhart, M.; Wirrig, E.; Phelps, A.; Wessels, A. Extracellular matrix and heart development. Birth Defects Res. Part A Clin. Mol. Teratol. 2011, 91, 535–550. [Google Scholar] [CrossRef]
- McCulloch, D.R.; Le Goff, C.; Bhatt, S.; Dixon, L.J.; Sandy, J.D.; Apte, S.S. Adamts5, the gene encoding a proteoglycan-degrading metalloprotease, is expressed by specific cell lineages during mouse embryonic development and in adult tissues. Gene Expr. Patterns 2009, 9, 314–323. [Google Scholar] [CrossRef]
- Fuentes, J.; Pritchard, M.; Estivill, X. Genomic Organization, Alternative Splicing, and Expression Patterns of the DSCR1 (Down Syndrome Candidate Region 1) Gene. Genomics 1997, 44, 358–361. [Google Scholar] [CrossRef]
- Bonaldo, P.; Braghetta, P.; Zanetti, M.; Piccolo, S.; Volpin, D.; Bressan, G.M. Collagen VI deficiency induces early onset myopathy in the mouse: An animal model for Bethlem myopathy. Hum. Mol. Genet. 1998, 7, 2135–2140. [Google Scholar] [CrossRef]
- Klewer, S.E.; Krob, S.L.; Kolker, S.J.; Kitten, G.T. Expression of Type VI Collagen in the Developing Mouse Heart. Dev. Dyn. 1998, 211, 248–255. [Google Scholar] [CrossRef]
- Kruithof, B.P.; Krawitz, S.A.; Gaussin, V. Atrioventricular valve development during late embryonic and postnatal stages involves condensation and extracellular matrix remodeling. Dev. Biol. 2007, 302, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Utriainen, A.; Sormunen, R.; Kettunen, M.; Carvalhaes, L.S.; Sajanti, E.; Eklund, L.; Kauppinen, R.; Kitten, G.T.; Pihlajaniemi, T. Structurally altered basement membranes and hydrocephalus in a type XVIII collagen deficient mouse line. Hum. Mol. Genet. 2004, 13, 2089–2099. [Google Scholar] [CrossRef]
- Carvalhaes, L.S.; Gervásio, O.L.; Guatimosim, C.; Heljasvaara, R.; Sormunen, R.; Pihlajaniemi, T.; Kitten, G.T. Collagen XVIII/endostatin is associated with the epithelial-mesenchymal transformation in the atrioventricular valves during cardiac development. Dev. Dyn. 2006, 235, 132–142. [Google Scholar] [CrossRef]
- Reynolds, L.E.; Watson, A.R.; Baker, M.; Jones, T.A.; D’Amico, G.; Robinson, S.D.; Joffre, C.; Garrido-Urbani, S.; Rodriguez-Manzaneque, J.C.; Martino-Echarri, E.; et al. Tumour angiogenesis is reduced in the Tc1 mouse model of Down’s syndrome. Nature 2010, 465, 813–817. [Google Scholar] [CrossRef]
- Li, H.; Edie, S.; Klinedinst, D.; Jeong, J.S.; Blackshaw, S.; Maslen, C.L.; Reeves, R.H. Penetrance of Congenital Heart Disease in a Mouse Model of Down Syndrome Depends on a Trisomic Potentiator of a Disomic Modifier. Genetics 2016, 203, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Levanon, D.; Brenner, O.; Negreanu, V.; Bettoun, D.; Woolf, E.; Eilam, R.; Lotem, J.; Gat, U.; Otto, F.; Speck, N.; et al. Spatial and temporal expression pattern of Runx3 (Aml2) and Runx1 (Aml1) indicates non-redundant functions during mouse embryogenesis. Mech. Dev. 2001, 109, 413–417. [Google Scholar] [CrossRef]
- Gattenlöhner, S.; Waller, C.; Ertl, G.; Bültmann, B.-D.; Müller-Hermelink, H.-K.; Marx, A. NCAM(CD56) and RUNX1(AML1) Are Up-Regulated in Human Ischemic Cardiomyopathy and a Rat Model of Chronic Cardiac Ischemia. Am. J. Pathol. 2003, 163, 1081–1090. [Google Scholar] [CrossRef]
- Kubin, T.; Pöling, J.; Kostin, S.; Gajawada, P.; Hein, S.; Rees, W.; Wietelmann, A.; Tanaka, M.; Lörchner, H.; Schimanski, S.; et al. Oncostatin M Is a Major Mediator of Cardiomyocyte Dedifferentiation and Remodeling. Cell Stem Cell 2011, 9, 420–432. [Google Scholar] [CrossRef]
- Eulalio, A.; Mano, M.; Ferro, M.D.; Zentilin, L.; Sinagra, G.; Zacchigna, S.; Giacca, M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 2012, 492, 376–381. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, S.; Wang, Q.; Hu, W.; Wang, D.; Li, X.; Su, T.; Qin, X.; Zhang, X.; Ma, K.; et al. Effects of cannabinoid receptor type 2 on endogenous myocardial regeneration by activating cardiac progenitor cells in mouse infarcted heart. Sci. China Life Sci. 2014, 57, 201–208. [Google Scholar] [CrossRef]
- Lluri, G.; Huang, V.; Touma, M.; Liu, X.; Harmon, A.W.; Nakano, A. Hematopoietic progenitors are required for proper development of coronary vasculature. J. Mol. Cell. Cardiol. 2015, 86, 199–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Górnikiewicz, B.; Ronowicz, A.; Krzemiński, M.; Sachadyn, P. Changes in gene methylation patterns in neonatal murine hearts: Implications for the regenerative potential. BMC Genom. 2016, 17, 231. [Google Scholar] [CrossRef] [PubMed]
- McCarroll, C.S.; He, W.; Foote, K.; Bradley, A.; Mcglynn, K.; Vidler, F.; Nixon, C.; Nather, K.; Fattah, C.; Riddell, A.; et al. Runx1 Deficiency Protects Against Adverse Cardiac Remodeling After Myocardial Infarction. Circulation 2018, 137, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, J.; Alfieri, C.M.; Yutzey, K.E. Development of heart valve leaflets and supporting apparatus in chicken and mouse embryos. Dev. Dyn. 2004, 230, 239–250. [Google Scholar] [CrossRef]
- Liu, X.; Wu, H.; Byrne, M.; Krane, S.; Jaenisch, R. Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 1852–1856. [Google Scholar] [CrossRef]
- Tan, M.; Wang, X.; Liu, H.; Peng, X.; Yang, Y.; Yu, H.; Xu, L.; Li, J.; Cao, H. Genetic Diagnostic Yield and Novel Causal Genes of Congenital Heart Disease. Front. Genet. 2022, 13, 1693. [Google Scholar] [CrossRef]
- Wenstrup, R.J.; Florer, J.B.; Brunskill, E.W.; Bell, S.M.; Chervoneva, I.; Birk, D.E. Type V Collagen Controls the Initiation of Collagen Fibril Assembly. J. Biol. Chem. 2004, 279, 53331–53337. [Google Scholar] [CrossRef]
- Eklund, L.; Piuhola, J.; Komulainen, J.; Sormunen, R.; Ongvarrasopone, C.; Fässler, R.; Muona, A.; Ilves, M.; Ruskoaho, H.; Takala, T.E.S.; et al. Lack of type XV collagen causes a skeletal myopathy and cardiovascular defects in mice. Proc. Natl. Acad. Sci. USA 2001, 98, 1194–1199. [Google Scholar] [CrossRef]
- Muona, A.; Eklund, L.; Väisänen, T.; Pihlajaniemi, T. Developmentally regulated expression of type XV collagen correlates with abnormalities in Col15a1−/− mice. Matrix Biol. 2002, 21, 89–102. [Google Scholar] [CrossRef]
- Rasi, K.; Piuhola, J.; Czabanka, M.; Sormunen, R.; Ilves, M.; Leskinen, H.; Rysä, J.; Kerkelä, R.; Janmey, P.; Heljasvaara, R.; et al. Collagen XV Is Necessary for Modeling of the Extracellular Matrix and Its Deficiency Predisposes to Cardiomyopathy. Circ. Res. 2010, 107, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Spence, S.; Argraves, W.; Walters, L.; Hungerford, J.E.; Little, C.D. Fibulin is localized at sites of epithelial-mesenchymal transitions in the early avian embryo. Dev. Biol. 1992, 151, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Cooley, M.A.; Kern, C.B.; Fresco, V.M.; Wessels, A.; Thompson, R.P.; McQuinn, T.C.; Twal, W.O.; Mjaatvedt, C.H.; Drake, C.J.; Argraves, W.S. Fibulin-1 is required for morphogenesis of neural crest-derived structures. Dev. Biol. 2008, 319, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Mjaatvedt, C.; Lepera, R.; Markwald, R. Myocardial specificity for initiating endothelial-mesenchymal cell transition in embryonic chick heart correlates with a particulate distribution of fibronectin. Dev. Biol. 1987, 119, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Ffrench-Constant, C.; Hynes, R. Alternative splicing of fibronectin is temporally and spatially regulated in the chicken embryo. Development 1989, 106, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Icardo, J.M.; Nakamura, A.; Fernandez-Teran, M.A.; Manasek, F.J. Effects of injecting fibronectin and antifibronectin antibodies on cushion mesenchyme formation in the chick. Anat. Embryol. 1992, 185, 239–247. [Google Scholar] [CrossRef]
- Astrof, S.; Crowley, D.; Hynes, R.O. Multiple cardiovascular defects caused by the absence of alternatively spliced segments of fibronectin. Dev. Biol. 2007, 311, 11–24. [Google Scholar] [CrossRef]
- Chan, C.K.; Rolle, M.W.; Potter-Perigo, S.; Braun, K.R.; Van Biber, B.P.; Laflamme, M.A.; Murry, C.E.; Wight, T.N. Differentiation of cardiomyocytes from human embryonic stem cells is accompanied by changes in the extracellular matrix production of versican and hyaluronan. J. Cell. Biochem. 2010, 111, 585–596. [Google Scholar] [CrossRef]
- Delom, F.; Burt, E.; Hoischen, A.; Veltman, J.; Groet, J.; Cotter, F.E.; Nizetic, D. Transchromosomic cell model of Down syndrome shows aberrant migration, adhesion and proteome response to extracellular matrix. Proteome Sci. 2009, 7, 31. [Google Scholar] [CrossRef]
- Marino, B. Congenital heart disease in patients with Down’s Syndrome: Anatomic and genetic aspects. Biomed. Pharmacother. 1993, 47, 197–200. [Google Scholar] [CrossRef]
- Jongewaard, I.N.; Lauer, R.M.; Behrendt, D.A.; Patil, S.; Klewer, S.E. Beta 1 integrin activation mediates adhesive differences between trisomy 21 and non-trisomic fibroblasts on type VI collagen. Am. J. Med. Genet. 2002, 109, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Aumailley, M.; Specks, U.; Timpl, R. Cell Adhesion to Type-VI Collagen. Biochem. Soc. Trans. 1991, 19, 843–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankov, R.; Yamada, K.M. Fibronectin at a glance. J. Cell Sci. 2002, 115, 3861–3863. [Google Scholar] [CrossRef]
- George, E.; Georges-Labouesse, E.; Patel-King, R.; Rayburn, H.; Hynes, R. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development 1993, 119, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.V.; Rodriguez-Manzaneque, J.C.; Thai, S.N.-M.; Twal, W.O.; Luque, A.; Lyons, K.M.; Argraves, W.S.; Iruela-Arispe, M.L. Fibulin-1 Acts as a Cofactor for the Matrix Metalloprotease ADAMTS-1. J. Biol. Chem. 2005, 280, 34796–34804. [Google Scholar] [CrossRef] [PubMed]
- Mollo, N.; Aurilia, M.; Scognamiglio, R.; Zerillo, L.; Cicatiello, R.; Bonfiglio, F.; Pagano, P.; Paladino, S.; Conti, A.; Nitsch, L.; et al. Overexpression of the Hsa21 Transcription Factor RUNX1 Modulates the Extracellular Matrix in Trisomy 21 Cells. Front. Genet. 2022, 13, 824922. [Google Scholar] [CrossRef]
- De Cegli, R.; Romito, A.; Iacobacci, S.; Mao, L.; Lauria, M.; Fedele, A.O.; Klose, J.; Borel, C.; Descombes, P.; Antonarakis, S.E.; et al. A mouse embryonic stem cell bank for inducible overexpression of human chromosome 21 genes. Genome Biol. 2010, 11, R64. [Google Scholar] [CrossRef]
- Levanon, D.; Groner, Y. Structure and regulated expression of mammalian RUNX genes. Oncogene 2004, 23, 4211–4219. [Google Scholar] [CrossRef]
- Lie-A-Ling, M.; Marinopoulou, E.; Li, Y.; Patel, R.; Stefanska, M.; Bonifer, C.; Miller, C.; Kouskoff, V.; Lacaud, G. RUNX1 positively regulates a cell adhesion and migration program in murine hemogenic endothelium prior to blood emergence. Blood 2014, 124, e11–e20. [Google Scholar] [CrossRef]
- Michaud, J.; Simpson, K.M.; Escher, R.; Buchet-Poyau, K.; Beissbarth, T.; Carmichael, C.; Ritchie, M.E.; Schütz, F.; Cannon, P.; Liu, M.; et al. Integrative analysis of RUNX1 downstream pathways and target genes. BMC Genom. 2008, 9, 363. [Google Scholar] [CrossRef] [PubMed]
- Wotton, S.; Terry, A.; Kilbey, A.; Jenkins, A.; Herzyk, P.; Cameron, E.; Neil, J.C. Gene array analysis reveals a common Runx transcriptional programme controlling cell adhesion and survival. Oncogene 2008, 27, 5856–5866. [Google Scholar] [CrossRef] [PubMed]
- Barutcu, A.R.; Hong, D.; Lajoie, B.R.; McCord, R.P.; van Wijnen, A.J.; Lian, J.B.; Stein, J.L.; Dekker, J.; Imbalzano, A.N.; Stein, G.S. RUNX1 contributes to higher-order chromatin organization and gene regulation in breast cancer cells. Biochim. et Biophys. Acta (BBA)-Gene Regul. Mech. 2016, 1859, 1389–1397. [Google Scholar] [CrossRef] [Green Version]
- Sobol, M.; Klar, J.; Laan, L.; Shahsavani, M.; Schuster, J.; Annerén, G.; Konzer, A.; Mi, J.; Bergquist, J.; Nordlund, J.; et al. Transcriptome and Proteome Profiling of Neural Induced Pluripotent Stem Cells from Individuals with Down Syndrome Disclose Dynamic Dysregulations of Key Pathways and Cellular Functions. Mol. Neurobiol. 2019, 56, 7113–7127. [Google Scholar] [CrossRef]
- Zheng, Q.; Zhou, G.; Morello, R.; Chen, Y.; Garcia-Rojas, X.; Lee, B. Type X collagen gene regulation by Runx2 contributes directly to its hypertrophic chondrocyte–specific expression in vivo. J. Cell Biol. 2003, 162, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Higashikawa, A.; Saito, T.; Ikeda, T.; Kamekura, S.; Kawamura, N.; Kan, A.; Oshima, Y.; Ohba, S.; Ogata, N.; Takeshita, K.; et al. Identification of the core element responsive to runt-related transcription factor 2 in the promoter of human type x collagen gene. Arthritis Rheum. 2009, 60, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Shimazu, J.; Makinistoglu, M.P.; Maurizi, A.; Kajimura, D.; Zong, H.; Takarada, T.; Iezaki, T.; Pessin, J.E.; Hinoi, E.; et al. Glucose Uptake and Runx2 Synergize to Orchestrate Osteoblast Differentiation and Bone Formation. Cell 2015, 161, 1576–1591. [Google Scholar] [CrossRef]
- Wang, T.; Jin, H.; Hu, J.; Li, X.; Ruan, H.; Xu, H.; Wei, L.; Dong, W.; Teng, F.; Gu, J.; et al. COL4A1 promotes the growth and metastasis of hepatocellular carcinoma cells by activating FAK-Src signaling. J. Exp. Clin. Cancer Res. 2020, 39, 148. [Google Scholar] [CrossRef]
- Sarohi, V.; Chakraborty, S.; Basak, T. Exploring the cardiac ECM during fibrosis: A new era with next-gen proteomics. Front. Mol. Biosci. 2022, 9, 1030226. [Google Scholar] [CrossRef]
- Li, Q.; Lai, Q.; He, C.; Fang, Y.; Yan, Q.; Zhang, Y.; Wang, X.; Gu, C.; Wang, Y.; Ye, L.; et al. RUNX1 promotes tumour metastasis by activating the Wnt/β-catenin signalling pathway and EMT in colorectal cancer. J. Exp. Clin. Cancer Res. 2019, 38, 334. [Google Scholar] [CrossRef]
- Keita, M.; Bachvarova, M.; Morin, C.; Plante, M.; Gregoire, J.; Renaud, M.-C.; Sebastianelli, A.; Trinh, X.B.; Bachvarov, D. The RUNX1 transcription factor is expressed in serous epithelial ovarian carcinoma and contributes to cell proliferation, migration and invasion. Cell Cycle 2013, 12, 972–986. [Google Scholar] [CrossRef]
- Planagumà, J.; Liljeström, M.; Alameda, F.; Butzow, R.; Virtanen, I.; Reventós, J.; Hukkanen, M. Matrix metalloproteinase-2 and matrix metalloproteinase-9 codistribute with transcription factors RUNX1/AML1 and ETV5/ERM at the invasive front of endometrial and ovarian carcinoma. Hum. Pathol. 2011, 42, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Sangpairoj, K.; Vivithanaporn, P.; Apisawetakan, S.; Chongthammakun, S.; Sobhon, P.; Chaithirayanon, K. RUNX1 Regulates Migration, Invasion, and Angiogenesis via p38 MAPK Pathway in Human Glioblastoma. Cell. Mol. Neurobiol. 2017, 37, 1243–1255. [Google Scholar] [CrossRef]
- Laufer, B.I.; Hwang, H.; Jianu, J.M.; Mordaunt, C.E.; Korf, I.F.; Hertz-Picciotto, I.; LaSalle, J.M. Low-pass whole genome bisulfite sequencing of neonatal dried blood spots identifies a role for RUNX1 in Down syndrome DNA methylation profiles. Hum. Mol. Genet. 2020, 29, 3465–3476. [Google Scholar] [CrossRef] [PubMed]
- Dobosz, A.; Grabowska, A.; Bik-Multanowski, M. Hypermethylation of NRG1 gene correlates with the presence of heart defects in Down’s syndrome. J. Genet. 2019, 98, 110. [Google Scholar] [CrossRef] [PubMed]
- Mollo, N.; Esposito, M.; Aurilia, M.; Scognamiglio, R.; Accarino, R.; Bonfiglio, F.; Cicatiello, R.; Charalambous, M.; Procaccini, C.; Micillo, T.; et al. Human Trisomic iPSCs from Down Syndrome Fibroblasts Manifest Mitochondrial Alterations Early during Neuronal Differentiation. Biology 2021, 10, 609. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Yamada, K.M.; Collins, J.W.; Walma, D.A.C.; Doyle, A.D.; Morales, S.G.; Lu, J.; Matsumoto, K.; Nazari, S.S.; Sekiguchi, R.; Shinsato, Y.; et al. Extracellular matrix dynamics in cell migration, invasion and tissue morphogenesis. Int. J. Exp. Pathol. 2019, 100, 144–152. [Google Scholar] [CrossRef]
- Garcia, O.; Torres, M.; Helguera, P.; Coskun, P.; Busciglio, J. A Role for Thrombospondin-1 Deficits in Astrocyte-Mediated Spine and Synaptic Pathology in Down’s Syndrome. PLoS ONE 2010, 5, e14200. [Google Scholar] [CrossRef]
- Danopoulos, S.; Bhattacharya, S.; Deutsch, G.; Nih, L.R.; Slaunwhite, C.; Mariani, T.J.; Al Alam, D. Prenatal histological, cellular, and molecular anomalies in Trisomy 21 lung. J. Pathol. 2021, 255, 41–51. [Google Scholar] [CrossRef]
- Von Kaisenberg, C.S.; Krenn, V.; Ludwig, M.; Nicolaides, K.; Brand-Saberi, B. Morphological classification of nuchal skin in human fetuses with trisomy 21, 18, and 13 at 12–18 weeks and in a trisomy 16 mouse. Anat. Embryol. 1998, 197, 105–124. [Google Scholar] [CrossRef]
- Wirrig, E.E.; Snarr, B.S.; Chintalapudi, M.R.; O’Neal, J.L.; Phelps, A.L.; Barth, J.L.; Fresco, V.M.; Kern, C.B.; Mjaatvedt, C.H.; Toole, B.P.; et al. Cartilage link protein 1 (Crtl1), an extracellular matrix component playing an important role in heart development. Dev. Biol. 2007, 310, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franz, M.; Rodriguez, H.; Lopes, C.; Zuberi, K.; Montojo, J.; Bader, G.D.; Morris, Q. GeneMANIA update 2018. Nucleic Acids Res. 2018, 46, W60–W64. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
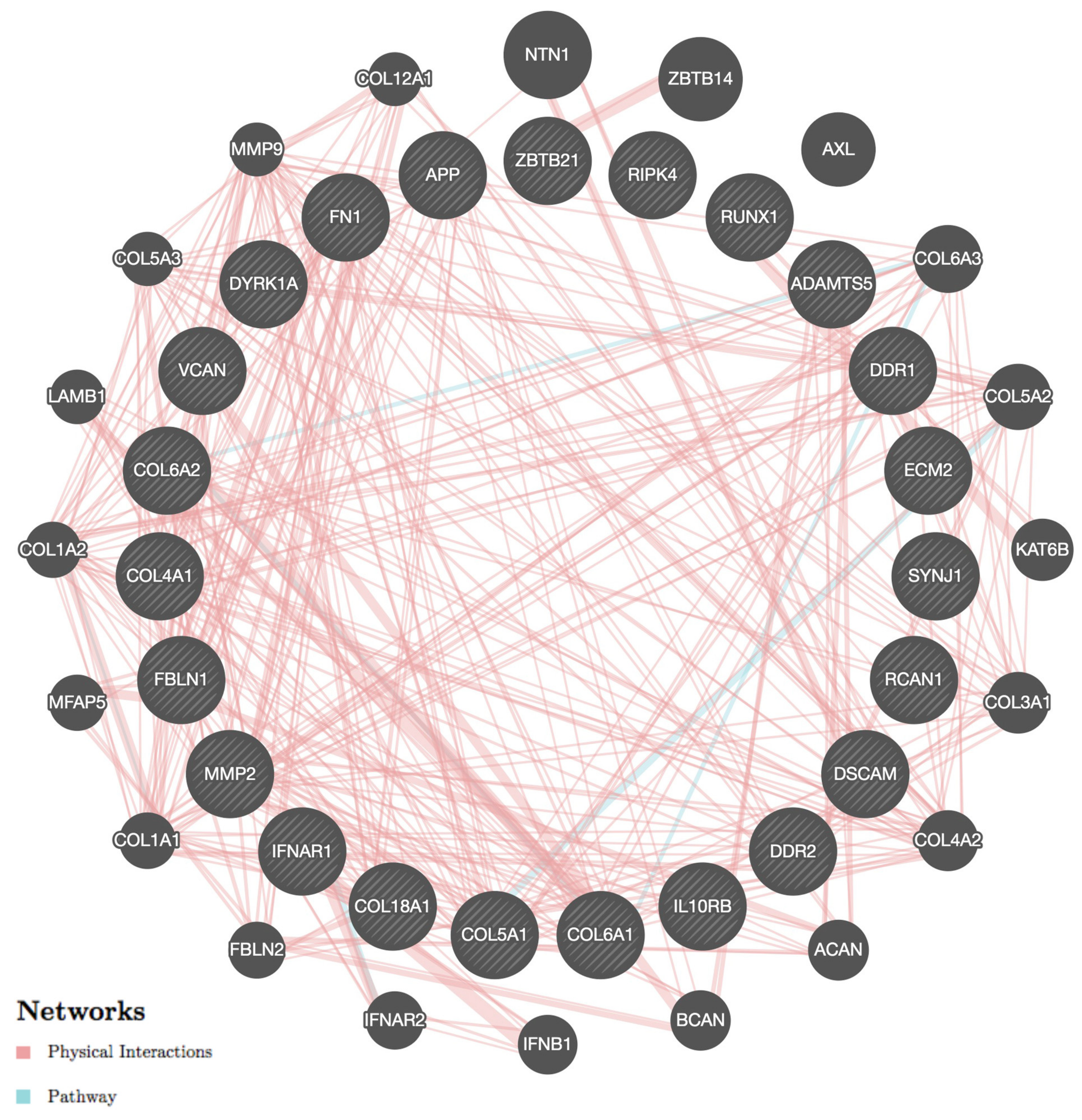{kind=link}
| Gene Name | Location | Gene Product | Potential Role in CHD |
|---|---|---|---|
| ADAMTS1 (ADAM Metallopeptidase with Thrombospondin Type 1 Motif 1) | 21q21.3 | Cleaves aggrecan, a cartilage proteoglycan, at the ‘1938-Glu-|-Leu-1939’ site (within the chondroitin sulfate attachment domain) and may be involved in its turnover. Has angiogenic inhibitor activity. | [23,105,106,107,108,109] |
| ADAMTS5 (ADAM Metallopeptidase with Thrombospondin Type 1 Motif 5) | 21q21.3 | ECM-degrading enzyme that shows proteolytic activity towards the hyalectan group of chondroitin sulfate proteoglycans, including ACAN, VCAN, BCAN and NCAN. Plays an important role in connective tissue organization, development, inflammation and cell migration. | [23,109,110] |
| COL6A1 (Collagen Type VI Alpha 1 Chain)/COL6A2 (Collagen Type VI Alpha 2 Chain) | 21q22.3/21q22.3 | Collagen VI acts as a cell-binding protein. Collagen VI is a ubiquitous nonfibrillar collagen composed of three chains (α1(VI), α2(VI), and α3(VI)) organized into a network of microfibrils important in anchoring the basement membrane to the ECM. Each chain contains a comparatively short triple helical domain with repeating Gly-X-Y subunits flanked by large globular von Willebrand factor type A domains. | [23,28,29,33,109,111,112,113,114] |
| COL18A1 (Collagen Type XVIII Alpha 1 Chain) | 21q22.3 | Regulates ECM-dependent motility and morphogenesis of endothelial and non-endothelial cells. Inhibits angiogenesis by binding to the heparan sulfate proteoglycans involved in growth factor signaling. Inhibits VEGFA-induced endothelial cell proliferation and migration. Modulates endothelial cell migration in an integrin-dependent manner. | [23,109,115,116] |
| DSCAM (DS Cell Adhesion Molecule) | 21q22.2 | Plays a role in neuronal self-avoidance, promotes lamina-specific synaptic connections in the retina and mediates homophilic intercellular adhesion. | [14,27,33] |
| JAM2 (Junctional Adhesion Molecule 2) | 21q21.3 | Mediates heterotypic cell–cell interactions with its cognate receptor JAM3 to regulate different cellular processes.Plays a role in homing and mobilization of hematopoietic stem and progenitor cells within the bone marrow. Plays a central role in leukocytes extravasation by facilitating not only transmigration, but also tethering and rolling of leukocytes along the endothelium. During myogenesis, it is involved in myocyte fusion. | [23,117,118] |
| RUNX1 (Runt-Related Transcription Factor 1) | 21q22.12 | Required for the development of normal hematopoiesis. Forms the heterodimeric complex core-binding factor with CBFB. RUNX members modulate the transcription of their target genes. The heterodimers bind to the core site of several enhancers and promoters. Several studies have ascribed to RUNX1 an important role in regulating ECM genes, cell adhesion and migration. | [119,120,121,122,123,124,125,126] |
| COL1A1 (Collagen Type I Alpha 1 Chain)/COL1A2 (Collagen Type I Alpha 2 Chain) | 17q21.33/7q21.3 | Type I collagen is a member of group I collagen (fibrillar forming collagen). COL1A1 and COL1A2 genes provide instructions for making part of type I collagen. Type I collagen is the most abundant form of collagen in the human body. | [23,109,114,127] |
| COL3A1 (Collagen Type III Alpha 1 Chain) | 2q32.2 | Involved in regulation of cortical development, it is the major ligand of ADGRG1 in the developing brain. Binding to ADGRG1 inhibits neuronal migration and activates the RhoA pathway by coupling ADGRG1 to GNA13 and possibly GNA12. | [23,109,128,129] |
| COL5A1 (Collagen Type V Alpha 2 Chain)/COL5A2 (Collagen Type V Alpha 2 Chain) | 9q34.3/2q32.2 | Type V collagen is a member of group I collagen (fibrillar-forming collagen). It is a minor connective tissue component of nearly ubiquitous distribution. Type V collagen binds to DNA, heparan sulfate, thrombospondin, heparin and insulin. Type V collagen is a key determinant in the assembly of tissue-specific matrices. | [23,109,114,130] |
| COL15A1 (Collagen Type XV Alpha 1 Chain) | 9q22.33 | Structural protein that stabilizes microvessels and muscle cells, both in heart and in skeletal muscle. | [23,109,131,132,133] |
| DDR1 (Discoidin Domain Receptor Tyrosine Kinase 1) | 6p21.33 | Functions as cell surface receptor for fibrillar collagen and regulates cell attachment to the ECM, remodeling of the ECM, cell migration, differentiation, survival and cell proliferation. Regulates remodeling of the ECM by upregulation of the metalloproteinases MMP2, MMP7 and MMP9, and thereby facilitates cell migration and wound healing. | [21] |
| DDR2 (Discoidin Domain Receptor Tyrosine Kinase 2) | 1q23.3 | Required for normal bone development. Functions as cell surface receptor for fibrillar collagen and regulates cell differentiation, remodeling of the ECM, cell migration and cell proliferation. Regulates remodeling of the ECM by upregulation of the collagenases MMP1, MMP2 and MMP13, and thereby facilitates cell migration and tumor cell invasion. Promotes fibroblast migration and proliferation, and thereby contributes to cutaneous wound healing. | [23,32] |
| FBLN1 (Fibulin 1) | 22q13.31 | Incorporated into fibronectin-containing matrix fibers. Plays a role in cell adhesion and migration along protein fibers within the ECM and contributes to the supramolecular organization of ECM architecture. Has been implicated in a role in cellular transformation and tumor invasion. Plays a role in hemostasis and thrombosis owing to its ability to bind fibrinogen and incorporate into clots. | [105,106,109,134,135] |
| FN1 (Fibronectin 1) | 2q35 | Fibronectins bind cell surfaces and various compounds including collagen, fibrin, heparin, DNA and actin. Fibronectins are involved in cell adhesion, cell motility, opsonization, wound healing and maintenance of cell shape. Participates in the regulation of type I collagen deposition by osteoblasts. | [109,136,137,138,139] |
| VCAN (Versican) | 5q14.2–q14.3 | Plays a role in intercellular signaling and in connecting cells with the ECM. Takes part in the regulation of cell motility, growth and differentiation. Binds hyaluronic acid. | [106,107,109,140] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mollo, N.; Scognamiglio, R.; Conti, A.; Paladino, S.; Nitsch, L.; Izzo, A. Genetics and Molecular Basis of Congenital Heart Defects in Down Syndrome: Role of Extracellular Matrix Regulation. Int. J. Mol. Sci. 2023, 24, 2918. https://doi.org/10.3390/ijms24032918
Mollo N, Scognamiglio R, Conti A, Paladino S, Nitsch L, Izzo A. Genetics and Molecular Basis of Congenital Heart Defects in Down Syndrome: Role of Extracellular Matrix Regulation. International Journal of Molecular Sciences. 2023; 24(3):2918. https://doi.org/10.3390/ijms24032918
Chicago/Turabian StyleMollo, Nunzia, Roberta Scognamiglio, Anna Conti, Simona Paladino, Lucio Nitsch, and Antonella Izzo. 2023. "Genetics and Molecular Basis of Congenital Heart Defects in Down Syndrome: Role of Extracellular Matrix Regulation" International Journal of Molecular Sciences 24, no. 3: 2918. https://doi.org/10.3390/ijms24032918
APA StyleMollo, N., Scognamiglio, R., Conti, A., Paladino, S., Nitsch, L., & Izzo, A. (2023). Genetics and Molecular Basis of Congenital Heart Defects in Down Syndrome: Role of Extracellular Matrix Regulation. International Journal of Molecular Sciences, 24(3), 2918. https://doi.org/10.3390/ijms24032918