
WGS Data Collections: How Do Genomic Databases Transform Medicine?


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
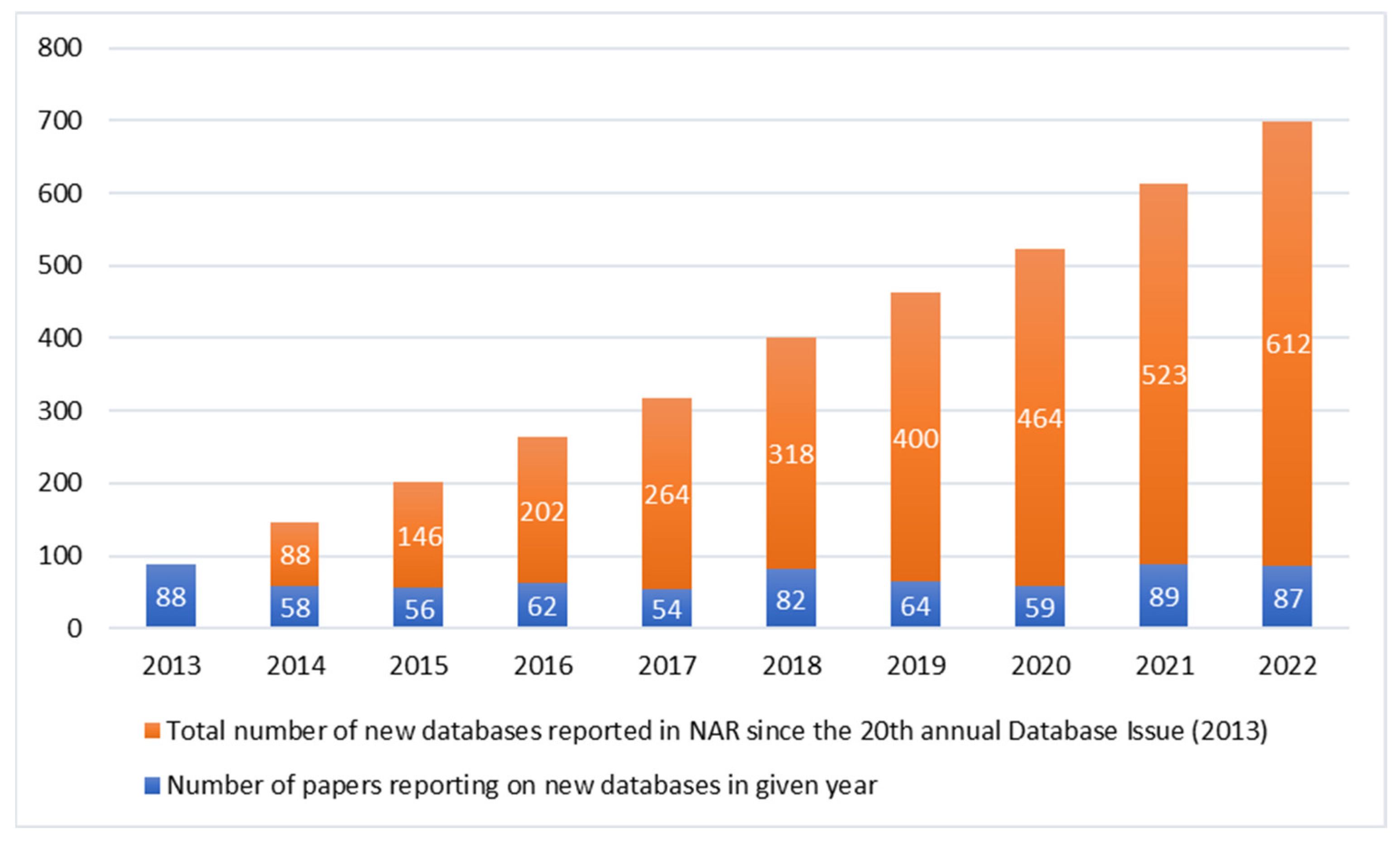
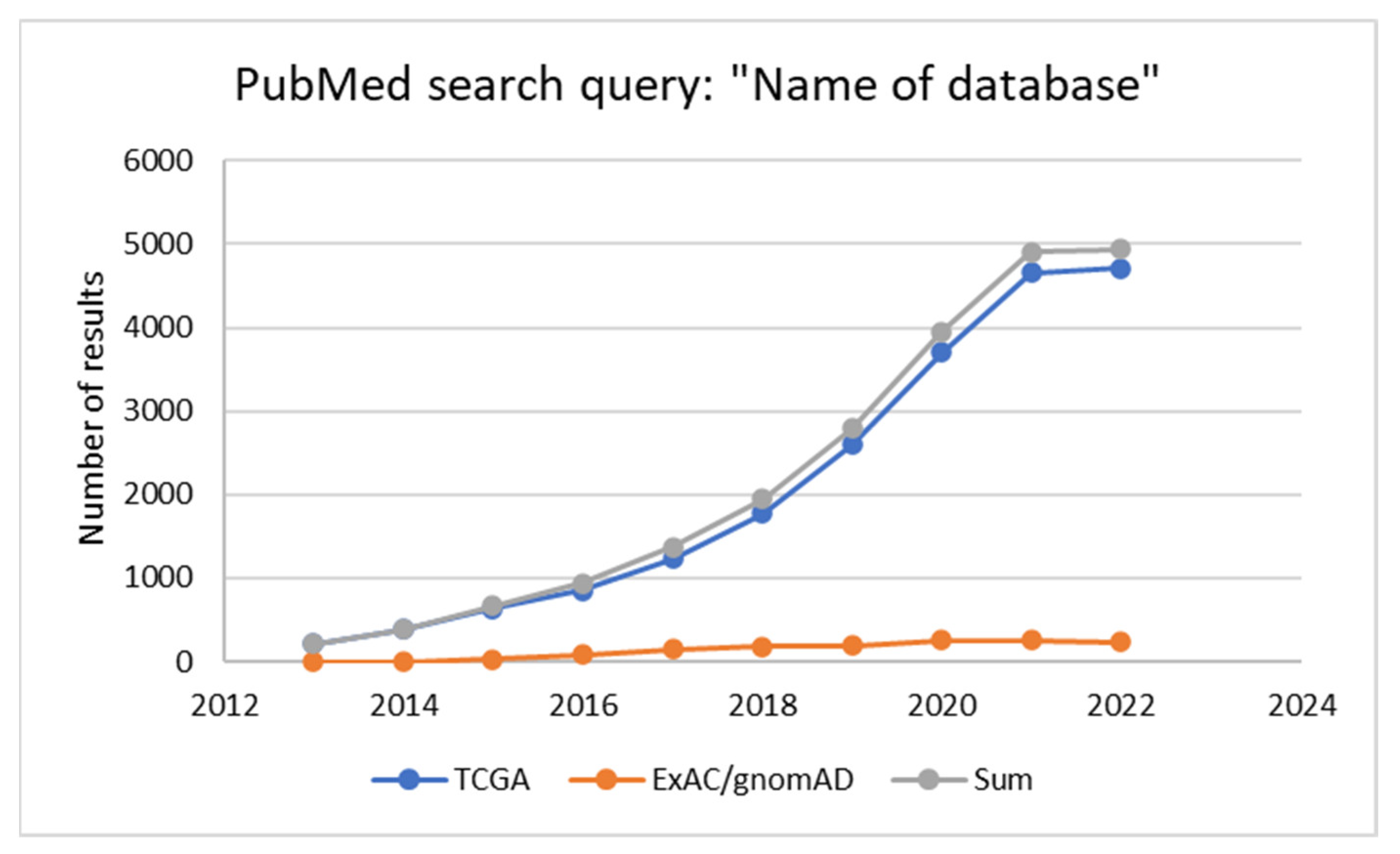
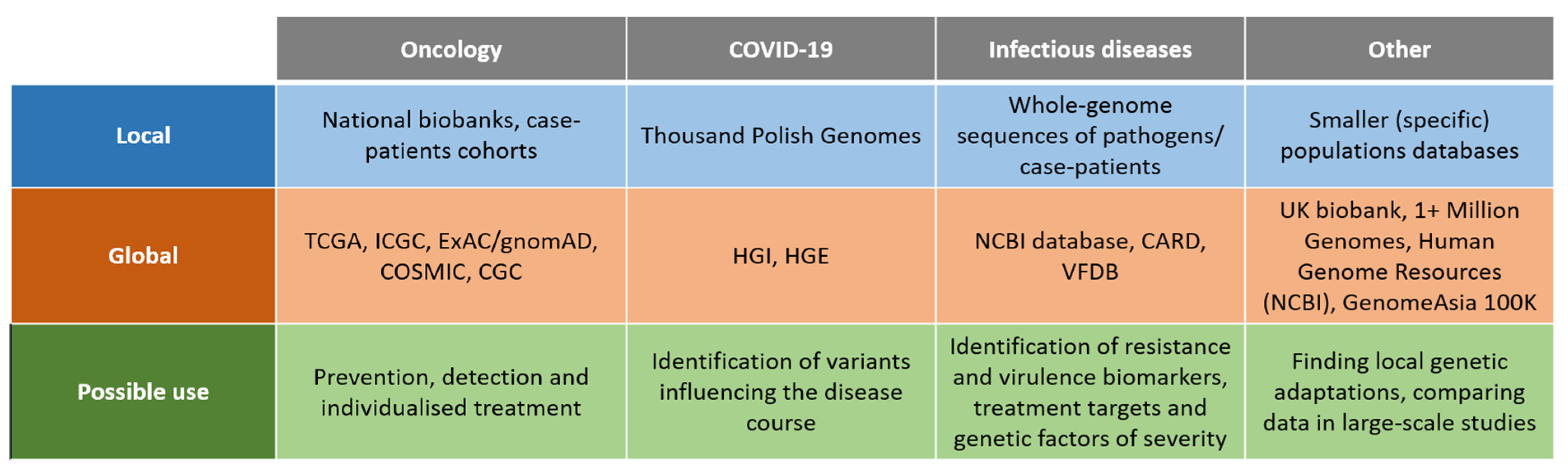
2. Genomic Databases: Global vs. Local Initiatives
3. Genomic Databases in Oncology
4. Genomic Databases in Infectious Diseases
5. Genomic Databases in Rare Diseases
6. Genomic Databases as a Fuel for AI-Driven Algorithms
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Lerner-Ellis, J.P. The clinical implementation of whole genome sequencing: A conversation with seven scientific experts. J. Inherit. Metab. Dis. 2012, 35, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Rossen, J.; Friedrich, A.; Moran-Gilad, J. Practical issues in implementing whole-genome-sequencing in routine diagnostic microbiology. Clin. Microbiol. Infect. 2018, 24, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Battke, F.; Schulte, B.; Schulze, M.; Biskup, S. The question of WGS’s clinical utility remains unanswered. Eur. J. Hum. Genet. 2021, 29, 722–723. [Google Scholar] [CrossRef]
- Dillon, O.J.; Melbourne Genomics Health Alliance; Lunke, S.; Stark, Z.; Yeung, A.; Thorne, N.; Gaff, C.; White, S.M.; Tan, T.Y. Exome sequencing has higher diagnostic yield compared to simulated disease-specific panels in children with suspected monogenic disorders. Eur. J. Hum. Genet. 2018, 26, 644–651. [Google Scholar] [CrossRef]
- Kerr, K.; McAneney, H.; Smyth, L.; Bailie, C.; McKee, S.; McKnight, A.J. A scoping review and proposed workflow for multi-omic rare disease research. Orphanet J. Rare Dis. 2020, 15, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Colin, E.; Duffourd, Y.; Tisserant, E.; Relator, R.; Bruel, A.-L.; Mau-Them, F.T.; Denommé-Pichon, A.-S.; Safraou, H.; Delanne, J.; Jean-Marçais, N.; et al. OMIXCARE: OMICS technologies solved about 33% of the patients with heterogeneous rare neuro-developmental disorders and negative exome sequencing results and identified 13% additional candidate variants. Front. Cell Dev. Biol. 2022, 10, 1021785. [Google Scholar] [CrossRef]
- Zhang, C.; Hansen, M.E.; Tishkoff, S.A. Advances in integrative African genomics. Trends Genet. 2021, 38, 152–168. [Google Scholar] [CrossRef]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef]
- Carey, N. Junk DNA: A Journey through the Dark Matter of the Genome; Columbia University Press: New York, NY, USA, 2015. [Google Scholar]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef]
- UCSF Health Center for Clinical Genetics and Genomics. UCSF 500 Cancer Gene Panel Test (UCSF500/UC500). Available online: https://genomics.ucsf.edu/content/ucsf-500-cancer-gene-panel-test-ucsf500-uc500 (accessed on 10 December 2022).
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Belkadi, A.; Bolze, A.; Itan, Y.; Cobat, A.; Vincent, Q.B.; Antipenko, A.; Shang, L.; Boisson, B.; Casanova, J.-L.; Abel, L. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. USA 2015, 112, 5473–5478. [Google Scholar] [CrossRef]
- Meienberg, J.; Bruggmann, R.; Oexle, K.; Matyas, G. Clinical sequencing: Is WGS the better WES? Hum. Genet. 2016, 135, 359–362. [Google Scholar] [CrossRef]
- Rigden, D.J.; Fernández, X.M. The 2022 Nucleic Acids Research database issue and the online molecular biology database collection. Nucleic Acids Res. 2021, 50, D1–D10. [Google Scholar] [CrossRef] [PubMed]
- Rigden, D.J.; Fernández, X.M. The 2021 Nucleic Acids Research database issue and the online molecular biology database collection. Nucleic Acids Res. 2020, 49, D1–D9. [Google Scholar] [CrossRef] [PubMed]
- Rigden, D.J.; Fernández, X.M. The 27th annual Nucleic Acids Research database issue and molecular biology database collection. Nucleic Acids Res. 2019, 48, D1–D8. [Google Scholar] [CrossRef]
- Rigden, D.J.; Fernández, X.M. The 26th annual Nucleic Acids Research database issue and Molecular Biology Database Collection. Nucleic Acids Res. 2018, 47, D1–D7. [Google Scholar] [CrossRef] [PubMed]
- Rigden, D.J.; Fernandez, X. The 2018 Nucleic Acids Research database issue and the online molecular biology database collection. Nucleic Acids Res. 2017, 46, D1–D7. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Fernández-Suárez, X.M.; Rigden, D.J. The 24th annual Nucleic Acids Research database issue: A look back and upcoming changes. Nucleic Acids Res. 2016, 45, D1–D11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigden, D.J.; Fernández-Suárez, X.M.; Galperin, M.Y. The 2016 database issue of Nucleic Acids Research and an updated molecular biology database collection. Nucleic Acids Res. 2015, 44, D1–D6. [Google Scholar] [CrossRef] [PubMed]
- Galperin, M.Y.; Rigden, D.J.; Fernández-Suárez, X.M. The 2015 Nucleic Acids Research Database Issue and Molecular Biology Database Collection. Nucleic Acids Res. 2015, 43, D1–D5. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Suárez, X.M.; Rigden, D.J.; Galperin, M.Y. The 2014 Nucleic Acids Research Database Issue and an updated NAR online Molecular Biology Database Collection. Nucleic Acids Res. 2013, 42, D1–D6. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, X.; Galperin, M.Y. The 2013 Nucleic Acids Research Database Issue and the online Molecular Biology Database Collection. Nucleic Acids Res. 2012, 41, D1–D7. [Google Scholar] [CrossRef]
- Ayuso, C.; Millán, J.M.; Mancheño, M.; Dal-Ré, R. Informed consent for whole-genome sequencing studies in the clinical setting. Proposed recommendations on essential content and process. Eur. J. Hum. Genet. 2013, 21, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Taliun, D.; NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium; Harris, D.N.; Kessler, M.D.; Carlson, J.; Szpiech, Z.A.; Torres, R.; Taliun, S.A.G.; Corvelo, A.; Gogarten, S.M.; et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 2021, 590, 290–299. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Halldorsson, B.V.; Eggertsson, H.P.; Moore, K.H.S.; Hauswedell, H.; Eiriksson, O.; Ulfarsson, M.O.; Palsson, G.; Hardarson, M.T.; Oddsson, A.; Jensson, B.O.; et al. The sequences of 150,119 genomes in the UK Biobank. Nature 2022, 607, 732–740. [Google Scholar] [CrossRef]
- Byrska-Bishop, M.; Evani, U.S.; Zhao, X.; Basile, A.O.; Abel, H.J.; Regier, A.A.; Corvelo, A.; Clarke, W.E.; Musunuri, R.; Nagulapalli, K.; et al. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 2022, 185, 3426–3440.e19. [Google Scholar] [CrossRef]
- European “1+ Million Genomes” Initiative|Shaping Europe’s Digital Future. Available online: https://digital-strategy.ec.europa.eu/en/policies/1-million-genomes (accessed on 28 December 2022).
- Human Genome Resources at NCBI. Available online: https://www.ncbi.nlm.nih.gov/genome/guide/human/ (accessed on 28 December 2022).
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.-H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Auto-antibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef] [PubMed]
- Du Bois, H.; Heim, T.A.; Rahman, S.A.; Yagnik, B.; Bastard, P.; Gervais, A.; Le Voyer, T.; Rosain, J.; Philippot, Q.; Manry, J.; et al. Autoantibodies neutralizing type I IFNs are present in ~4% of uninfected individuals over 70 years old and account for ~20% of COVID-19 deaths. Sci. Immunol. 2021, 6, eabl4340. [Google Scholar] [CrossRef]
- Need, A.C.; Goldstein, D.B. Next generation disparities in human genomics: Concerns and remedies. Trends Genet. 2009, 25, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Wonkam, A. Sequence three million genomes across Africa. Nature 2021, 590, 209–211. [Google Scholar] [CrossRef]
- GenomeAsia100K Consortium; Wall, J.D.; Stawiski, E.W.; Ratan, A.; Kim, H.L.; Kim, C.; Gupta, R.; Suryamohan, K.; Gusareva, E.S.; Purbojati, R.W.; et al. The GenomeAsia 100K Project enables genetic discoveries across Asia. Nature 2019, 576, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi-Kabata, Y.; Nariai, N.; Kawai, Y.; Sato, Y.; Kojima, K.; Tateno, M.; Katsuoka, F.; Yasuda, J.; Yamamoto, M.; Nagasaki, M. iJGVD: An integrative Japanese genome variation database based on whole-genome sequencing. Hum. Genome Var. 2015, 2, 15050. [Google Scholar] [CrossRef]
- Thareja, G.; Al-Sarraj, Y.; Belkadi, A.; Almotawa, M.; Ismail, S.; Al-Muftah, W.; Badji, R.; Mbarek, H.; Darwish, D.; Fadl, T.; et al. Whole genome sequencing in the Middle Eastern Qatari population identifies genetic associations with 45 clinically relevant traits. Nat. Commun. 2021, 12, 1–10. [Google Scholar] [CrossRef]
- Zhang, C.; Gao, Y.; Liu, J.; Xue, Z.; Lu, Y.; Deng, L.; Tian, L.; Feng, Q.; Xu, S. PGG.Population: A database for understanding the genomic diversity and genetic ancestry of human populations. Nucleic Acids Res. 2017, 46, D984–D993. [Google Scholar] [CrossRef]
- Gudbjartsson, D.F.; Helgason, H.; Gudjonsson, S.A.; Zink, F.; Oddsson, A.; Gylfason, A.; Besenbacher, S.; Magnusson, G.; Halldórsson, B.; Hjartarson, E.; et al. Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet. 2015, 47, 435–444. [Google Scholar] [CrossRef]
- Chheda, H.; Palta, P.; Pirinen, M.; McCarthy, S.; Walter, K.; Koskinen, S.; Salomaa, V.; Daly, M.; Durbin, R.; Palotie, A.; et al. Whole-genome view of the consequences of a population bottleneck using 2926 genome sequences from Finland and United Kingdom. Eur. J. Hum. Genet. 2017, 25, 477–484. [Google Scholar] [CrossRef] [Green Version]
- Leitsalu, L.; Haller, T.; Esko, T.; Tammesoo, M.-L.; Alavere, H.; Snieder, H.; Perola, M.; Ng, P.C.; Mägi, R.; Milani, L.; et al. Cohort Profile: Estonian Biobank of the Estonian Genome Center, University of Tartu. Leuk. Res. 2014, 44, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Smetana, J.; Brož, P. National Genome Initiatives in Europe and the United Kingdom in the Era of Whole-Genome Sequencing: A Comprehensive Review. Genes 2022, 13, 556. [Google Scholar] [CrossRef] [PubMed]
- Oleksyk, T.K.; Wolfsberger, W.W.; Schubelka, K.; Mangul, S.; O’Brien, S.J. The Pioneer Advantage: Filling the blank spots on the map of genome diversity in Europe. Gigascience 2022, 11, giac081. [Google Scholar] [CrossRef]
- Kaja, E.; Lejman, A.; Sielski, D.; Sypniewski, M.; Gambin, T.; Dawidziuk, M.; Suchocki, T.; Golik, P.; Wojtaszewska, M.; Mroczek, M.; et al. The Thousand Polish Genomes—A Database of Polish Variant Allele Frequencies. Int. J. Mol. Sci. 2022, 23, 4532. [Google Scholar] [CrossRef]
- Okada, Y.; Momozawa, Y.; Sakaue, S.; Kanai, M.; Ishigaki, K.; Akiyama, M.; Kishikawa, T.; Arai, Y.; Sasaki, T.; Kosaki, K.; et al. Deep whole-genome sequencing reveals recent selection signatures linked to evolution and disease risk of Japanese. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Lee, J.; Lee, J.; Jeon, S.; Lee, J.; Jang, I.; Yang, J.O.; Park, S.; Lee, B.; Choi, J.; Choi, B.-O.; et al. A database of 5305 healthy Korean individuals reveals genetic and clinical implications for an East Asian population. Exp. Mol. Med. 2022, 54, 1862–1871. [Google Scholar] [CrossRef]
- Udpa, N.; Ronen, R.; Zhou, D.; Liang, J.; Stobdan, T.; Appenzeller, O.; Yin, Y.; Du, Y.; Guo, L.; Cao, R.; et al. Whole genome sequencing of Ethiopian highlanders reveals conserved hypoxia tolerance genes. Genome Biol. 2014, 15, R36. [Google Scholar] [CrossRef] [PubMed]
- Fonseca-Montaño, M.A.; Blancas, S.; Herrera-Montalvo, L.A.; Hidalgo-Miranda, A. Cancer Genomics. Arch. Med Res. 2022, 53, 723–731. [Google Scholar] [CrossRef]
- Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabé, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; Gerhard, D.S.; et al. International Network of Cancer Genome Projects. Nature 2010, 464, 993–998. [Google Scholar] [CrossRef]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- About gnomAD. Available online: https://gnomad.broadinstitute.org/about (accessed on 10 December 2022).
- The Genome Aggregation Database (gnomAD). Available online: https://www.nature.com/immersive/d42859-020-00002-x/index.html (accessed on 12 December 2022).
- Peng, L.; Zhou, N.; Zhang, C.-Y.; Li, G.-C.; Yuan, X.-Q. eccDNAdb: A database of extrachromosomal circular DNA profiles in human cancers. Oncogene 2022, 41, 2696–2705. [Google Scholar] [CrossRef] [PubMed]
- Samy, M.D.; Yavorski, J.M.; Mauro, J.A.; Blanck, G. Impact of SNPs on CpG Islands in the MYC and HRAS oncogenes and in a wide variety of tumor suppressor genes: A multi-cancer approach. Cell Cycle 2016, 15, 1572–1578. [Google Scholar] [CrossRef] [PubMed]
- Manske, F.; Ogoniak, L.; Jürgens, L.; Grundmann, N.; Makałowski, W.; Wethmar, K. The new uORFdb: Integrating literature, sequence, and variation data in a central hub for uORF research. Nucleic Acids Res. 2022, 51, D328–D336. [Google Scholar] [CrossRef]
- Cantalupo, P.G.; Katz, J.P.; Pipas, J.M. Viral sequences in human cancer. Virology 2017, 513, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zheng, L.; Zhang, R.; Hou, P.; Wang, J.; Wu, L.; Li, J. Circ-ZEB1 promotes PIK3CA expression by silencing miR-199a-3p and affects the proliferation and apoptosis of hepatocellular carcinoma. Mol. Cancer 2022, 21, 1–15. [Google Scholar] [CrossRef]
- Kang, N.; Ou, Y.; Wang, G.; Chen, J.; Li, D.; Zhan, Q. miR-875-5p exerts tumor-promoting function via down-regulation of CAPZA1 in esophageal squamous cell carcinoma. Peerj 2021, 9, e10020. [Google Scholar] [CrossRef]
- Xu, M.; Li, Y.; Li, W.; Zhao, Q.; Zhang, Q.; Le, K.; Huang, Z.; Yi, P. Immune and Stroma Related Genes in Breast Cancer: A Comprehensive Analysis of Tumor Microenvironment Based on the Cancer Genome Atlas (TCGA) Database. Front. Med. 2020, 7, 64. [Google Scholar] [CrossRef]
- Miller, D.T.; Lee, K.; Gordon, A.S.; Amendola, L.M.; Adelman, K.; Bale, S.J.; Chung, W.K.; Gollob, M.H.; Harrison, S.M.; Herman, G.E.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2021 update: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Anesthesia Analg. 2021, 23, 1391–1398. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Anesthesia Analg. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Cristofoli, F.; Sorrentino, E.; Guerri, G.; Miotto, R.; Romanelli, R.; Zulian, A.; Cecchin, S.; Paolacci, S.; Miertus, J.; Bertelli, M.; et al. Variant Selection and Interpretation: An Example of Modified VarSome Classifier of ACMG Guidelines in the Diagnostic Setting. Genes 2021, 12, 1885. [Google Scholar] [CrossRef]
- Rheinbay, E.; Nielsen, M.M.; Abascal, F.; Wala, J.A.; Shapira, O.; Tiao, G.; Hornshøj, H.; Hess, J.M.; Juul, R.I.; Lin, Z.; et al. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature 2020, 578, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J.; Szabo, C. Poly(ADP-ribose) polymerase inhibition: Past, present and future. Nat. Rev. Drug Discov. 2020, 19, 711–736. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Lheureux, S.; Moore, K.N. PARP Inhibitors for Ovarian Cancer: Current Indications, Future Combinations, and Novel Assets in Development to Target DNA Damage Repair. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, e116–e131. [Google Scholar] [CrossRef] [PubMed]
- Nizialek, E.; Antonarakis, E.S. PARP Inhibitors in Metastatic Prostate Cancer: Evidence to Date. Cancer Manag. Res. 2020, 12, 8105–8114. [Google Scholar] [CrossRef] [PubMed]
- Teyssonneau, D.; Margot, H.; Cabart, M.; Anonnay, M.; Sargos, P.; Vuong, N.-S.; Soubeyran, I.; Sevenet, N.; Roubaud, G. Prostate cancer and PARP inhibitors: Progress and challenges. J. Hematol. Oncol. 2021, 14, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wei, M.; Xu, J.; Hua, J.; Liang, C.; Meng, Q.; Zhang, Y.; Liu, J.; Zhang, B.; Yu, X.; et al. PARP inhibitors in pancreatic cancer: Molecular mechanisms and clinical applications. Mol. Cancer 2020, 19, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.; Chung, S.Y.; Parakrama, R.; Fayyaz, F.; Jose, J.; Saif, M.W. The role of PARP inhibitors in BRCA mutated pancreatic cancer. Ther. Adv. Gastroenterol. 2021, 14. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Balasubramaniam, S.; Zhang, H.; Berman, T.; Narayan, P.; Suzman, D.; Bloomquist, E.; Tang, S.; Gong, Y.; Sridhara, R.; et al. FDA Approval Summary: Olaparib Monotherapy or in Combination with Bevacizumab for the Maintenance Treatment of Patients with Advanced Ovarian Cancer. Oncol. 2020, 26, e164–e172. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Murthy, P.; Muggia, F. PARP inhibitors: Clinical development, emerging differences, and the current therapeutic issues. Cancer Drug Resist 2019, 2, 665–679. [Google Scholar] [CrossRef]
- Yi, M.; Dong, B.; Qin, S.; Chu, Q.; Wu, K.; Luo, S. Advances and perspectives of PARP inhibitors. Exp. Hematol. Oncol. 2019, 8, 1–12. [Google Scholar] [CrossRef]
- Kim, D.-S.; Camacho, C.V.; Kraus, W.L. Alternate therapeutic pathways for PARP inhibitors and potential mechanisms of resistance. Exp. Mol. Med. 2021, 53, 42–51. [Google Scholar] [CrossRef]
- Dziadkowiec, K.N.; Gąsiorowska, E.; Nowak-Markwitz, E.; Jankowska, A. PARP inhibitors: Review of mechanisms of action and BRCA1/2 mutation targeting. Menopausal Rev. 2016, 15, 215–219. [Google Scholar] [CrossRef]
- Lee, J.-M.; Ledermann, J.A.; Kohn, E.C. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann. Oncol. 2013, 25, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Toh, M.; Ngeow, J. Homologous Recombination Deficiency: Cancer Predispositions and Treatment Implications. Oncol. 2021, 26, e1526–e1537. [Google Scholar] [CrossRef] [PubMed]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef] [PubMed]
- PRIMA Trial Reports Benefit with Niraparib Across Ovarian Cancer Subsets—The ASCO Post. Available online: https://ascopost.com/issues/september-10-2020-supplement-gynecologic-cancers-almanac/prima-trial-reports-benefit-with-niraparib-across-ovarian-cancer-subsets/ (accessed on 10 December 2022).
- MyChoice CDx|Myriad Genetics. Myriad Oncology. Available online: https://myriad.com/oncology/mychoice-cdx/ (accessed on 10 December 2022).
- Schrijver, I.; Wiita, A.P. Clinical application of high throughput molecular screening techniques for pharmacogenomics. Pharmacogenomics Pers. Med. 2011, 4, 109–121. [Google Scholar] [CrossRef] [Green Version]
- Rezayi, S.; Kalhori, S.R.N.; Saeedi, S. Effectiveness of Artificial Intelligence for Personalized Medicine in Neoplasms: A Systematic Review. BioMed Res. Int. 2022, 2022, 1–34. [Google Scholar] [CrossRef]
- Belizário, J.E.; Sangiuliano, B.A.; Perez-Sosa, M.; Neyra, J.M.; Moreira, D.F. Using Pharmacogenomic Databases for Discovering Patient-Target Genes and Small Molecule Candidates to Cancer Therapy. Front. Pharmacol. 2016, 7, 312. [Google Scholar] [CrossRef]
- Aine, M.; Boyaci, C.; Hartman, J.; Häkkinen, J.; Mitra, S.; Campos, A.B.; Nimeus, E.; Ehinger, A.; Vallon-Christersson, J.; Borg, Å.; et al. Molecular analyses of triple-negative breast cancer in the young and elderly. Breast Cancer Res. 2021, 23, 1–19. [Google Scholar] [CrossRef]
- Pavlovica, K.; Irmejs, A.; Noukas, M.; Palover, M.; Kals, M.; Tonisson, N.; Metspalu, A.; Gronwald, J.; Lubinski, J.; Murmane, D.; et al. Spectrum and frequency of CHEK2 variants in breast cancer affected and general population in the Baltic states region, initial results and literature review. Eur. J. Med Genet. 2022, 65, 104477. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.; Raisen, C.L.; Ba, X.; Sadgrove, N.J.; Padilla-González, G.F.; Simmonds, M.S.J.; Loncaric, I.; Kerschner, H.; Apfalter, P.; Hartl, R.; et al. Emergence of methicillin resistance predates the clinical use of antibiotics. Nature 2022, 602, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Coll, F.; Harrison, E.M.; Toleman, M.S.; Reuter, S.; Raven, K.E.; Blane, B.; Palmer, B.; Kappeler, A.R.M.; Brown, N.M.; Török, M.E.; et al. Longitudinal genomic surveillance of MRSA in the UK reveals transmission patterns in hospitals and the community. Sci. Transl. Med. 2017, 9, eaak9745. [Google Scholar] [CrossRef] [PubMed]
- Blane, B.; E Raven, K.; Leek, D.; Brown, N.; Parkhill, J.; Peacock, S.J. Rapid sequencing of MRSA direct from clinical plates in a routine microbiology laboratory. J. Antimicrob. Chemother. 2019, 74, 2153–2156. [Google Scholar] [CrossRef]
- Eppinger, M.; Pearson, T.; Koenig, S.S.K.; Pearson, O.; Hicks, N.; Agrawal, S.; Sanjar, F.; Galens, K.; Daugherty, S.; Crabtree, J.; et al. Genomic Epidemiology of the Haitian Cholera Outbreak: A Single Introduction Followed by Rapid, Extensive, and Continued Spread Characterized the Onset of the Epidemic. Mbio 2014, 5, e01721-14. [Google Scholar] [CrossRef]
- Orata, F.; Keim, P.S.; Boucher, Y. The 2010 Cholera Outbreak in Haiti: How Science Solved a Controversy. PLoS Pathog. 2014, 10, e1003967. [Google Scholar] [CrossRef]
- Nordgren, J.; Sharma, S.; Kambhampati, A.; Lopman, B.; Svensson, L. Innate Resistance and Susceptibility to Norovirus Infection. PLoS Pathog. 2016, 12, e1005385. [Google Scholar] [CrossRef]
- Nordgren, J.; Svensson, L. Genetic Susceptibility to Human Norovirus Infection: An Update. Viruses 2019, 11, 226. [Google Scholar] [CrossRef] [PubMed]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in COVID. Nature 2021, 591, 92–98. [Google Scholar] [CrossRef]
- Andreakos, E.; Abel, L.; Vinh, D.C.; Kaja, E.; Drolet, B.A.; Zhang, Q.; O’Farrelly, C.; Novelli, G.; Rodríguez-Gallego, C.; Haerynck, F.; et al. A global effort to dissect the human genetic basis of resistance to SARS-CoV-2 infection. Nat. Immunol. 2021, 23, 159–164. [Google Scholar] [CrossRef]
- Liu, Y.-T. Infectious Disease Genomics. In Genetics and Evolution of Infectious Diseases; Elsevier: Amsterdam, The Netherlands, 2017; pp. 211–225. [Google Scholar] [CrossRef]
- Inzaule, S.C.; Tessema, S.K.; Kebede, Y.; Ouma, A.E.O.; Nkengasong, J.N. Genomic-informed pathogen surveillance in Africa: Opportunities and challenges. Lancet Infect. Dis. 2021, 21, e281–e289. [Google Scholar] [CrossRef]
- Simpson, B.N.; Sang, M.E.M.; Puello, Y.C.; Brockmans, E.J.D.; Soto, M.F.D.; Defilló, S.M.R.; Cruz, K.M.T.; Pérez, J.O.S.; Husami, A.; E Day, M.; et al. The 2019–2020 Dengue Fever Epidemic: Genomic Markers Indicating Severity in Dominican Republic Children. J. Pediatr. Infect. Dis. Soc. 2022, piac136. [Google Scholar] [CrossRef]
- Schulz, A.; Sadeghi, B.; Stoek, F.; King, J.; Fischer, K.; Pohlmann, A.; Eiden, M.; Groschup, M.H. Whole-Genome Sequencing of Six Neglected Arboviruses Circulating in Africa Using Sequence-Independent Single Primer Amplification (SISPA) and MinION Nanopore Technologies. Pathogens 2022, 11, 1502. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Kim, J.-M.; Maeda, T.; Goto, M.; Tsuyuki, Y.; Shibata, S.; Shizuno, K.; Okuzumi, K.; Kim, J.-S.; Takahashi, T. Virulence-associated Genome Sequences of Pasteurella canis and Unique Toxin Gene Prevalence of P. canis and Pasteurella multocida Isolated from Humans and Companion Animals. Ann. Lab. Med. 2022, 43, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Al-Trad, E.I.; Hamzah, A.M.C.; Puah, S.M.; Chua, K.H.; Kwong, S.M.; Yeo, C.C.; Chew, C.H. Comparative Genomic Analysis of a Multidrug-Resistant Staphylococcus hominis ShoR14 Clinical Isolate from Terengganu, Malaysia, Led to the Discovery of Novel Mobile Genetic Elements. Pathogens 2022, 11, 1406. [Google Scholar] [CrossRef]
- Souche, E.; Beltran, S.; Brosens, E.; Belmont, J.W.; Fossum, M.; Riess, O.; Gilissen, C.; Ardeshirdavani, A.; Houge, G.; van Gijn, M.; et al. Recommendations for whole genome sequencing in diagnostics for rare diseases. Eur. J. Hum. Genet. 2022, 30, 1017–1021. [Google Scholar] [CrossRef]
- Smedley, D.; Smith, K.R.; Martin, A.; Thomas, E.A.; McDonagh, E.M.; Cipriani, V.; Ellingford, J.M.; Arno, G.; Tucci, A.; Vandrovcova, J.; et al. 100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care — Preliminary Report. N. Engl. J. Med. 2021, 385, 1868–1880. [Google Scholar] [CrossRef]
- Bamshad, M.J.; Shendure, J.A.; Valle, D.; Hamosh, A.; Lupski, J.R.; Gibbs, R.A.; Boerwinkle, E.; Lifton, R.P.; Gerstein, M.; Gunel, M.; et al. The Centers for Mendelian Genomics: A new large-scale initiative to identify the genes underlying rare Mendelian conditions. Am. J. Med Genet. Part A 2012, 158A, 1523–1525. [Google Scholar] [CrossRef]
- Faraji, S.; Patrinos, D.; Hagan, J.; Knoppers, B.M. A centralized rare disease database and whole-genome sequencing as a standard of care: Two essential implementations for the future of health. Facets 2021, 6, 1831–1834. [Google Scholar] [CrossRef]
- Zurek, B.; Ellwanger, K.; Vissers, L.E.L.M.; Schüle, R.; Synofzik, M.; Töpf, A.; de Voer, R.M.; Laurie, S.; Matalonga, L.; Gilissen, C.; et al. Solve-RD: Systematic pan-European data sharing and collaborative analysis to solve rare diseases. Eur. J. Hum. Genet. 2021, 29, 1325–1331. [Google Scholar] [CrossRef]
- Coelho, A.V.C.; Mascaro-Cordeiro, B.; Lucon, D.R.; Nóbrega, M.S.; Reis, R.D.S.; de Alexandre, R.B.; Moura, L.M.S.; de Oliveira, G.S.; Guedes, R.L.M.; Caraciolo, M.P.; et al. The Brazilian Rare Genomes Project: Validation of Whole Genome Sequencing for Rare Diseases Diagnosis. Front. Mol. Biosci. 2022, 9, 821582. [Google Scholar] [CrossRef]
- Takahashi, Y.; Date, H.; Oi, H.; Adachi, T.; Imanishi, N.; Kimura, E.; Takizawa, H.; Kosugi, S.; Matsumoto, N.; Kosaki, K.; et al. Six years’ accomplishment of the Initiative on Rare and Undiagnosed Diseases: Nationwide project in Japan to discover causes, mechanisms, and cures. J. Hum. Genet. 2022, 67, 505–513. [Google Scholar] [CrossRef]
- Roberts, M.; Driggs, D.; Thorpe, M.; Gilbey, J.; Yeung, M.; Ursprung, S.; Aviles-Rivero, A.I.; Etmann, C.; McCague, C.; Beer, L.; et al. Common pitfalls and recommendations for using machine learning to detect and prognosticate for COVID-19 using chest radiographs and CT scans. Nat. Mach. Intell. 2021, 3, 199–217. [Google Scholar] [CrossRef]
- Green, R.C.; Goddard, K.A.; Jarvik, G.P.; Amendola, L.M.; Appelbaum, P.S.; Berg, J.S.; Bernhardt, B.A.; Biesecker, L.G.; Biswas, S.; Blout, C.L.; et al. Clinical Sequencing Exploratory Research Consortium: Accelerating Evidence-Based Practice of Genomic Medicine. Am. J. Hum. Genet. 2016, 98, 1051–1066. [Google Scholar] [CrossRef]
- Stranneheim, H.; Lagerstedt-Robinson, K.; Magnusson, M.; Kvarnung, M.; Nilsson, D.; Lesko, N.; Engvall, M.; Anderlid, B.-M.; Arnell, H.; Johansson, C.B.; et al. Integration of whole genome sequencing into a healthcare setting: High diagnostic rates across multiple clinical entities in 3219 rare disease patients. Genome Med. 2021, 13, 1–15. [Google Scholar] [CrossRef]
- Gilly, A.; Suveges, D.; Kuchenbaecker, K.; Pollard, M.; Southam, L.; Hatzikotoulas, K.; Farmaki, A.-E.; Bjornland, T.; Waples, R.; Appel, E.V.R.; et al. Cohort-wide deep whole genome sequencing and the allelic architecture of complex traits. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Boomsma, D.I.; Wijmenga, C.; Slagboom, E.P.; Swertz, M.A.; Karssen, L.C.; Abdellaoui, A.; Ye, K.; Guryev, V.; Vermaat, M.; van Dijk, F.; et al. The Genome of the Netherlands: Design, and project goals. Eur. J. Hum. Genet. 2013, 22, 221–227. [Google Scholar] [CrossRef]
- Telenti, A.; Pierce, L.C.T.; Biggs, W.H.; di Iulio, J.; Wong, E.H.M.; Fabani, M.M.; Kirkness, E.F.; Moustafa, A.; Shah, N.; Xie, C.; et al. Deep sequencing of 10,000 human genomes. Proc. Natl. Acad. Sci. USA 2016, 113, 11901–11906. [Google Scholar] [CrossRef]
- Besenbacher, S.; Liu, S.; Izarzugaza, J.M.G.; Grove, J.; Belling, K.; Bork-Jensen, J.; Huang, S.; Als, T.D.; Li, S.; Yadav, R.; et al. Novel variation and de novo mutation rates in population-wide de novo assembled Danish trios. Nat. Commun. 2015, 6, 5969. [Google Scholar] [CrossRef]
- Gorzynski, J.E.; Goenka, S.D.; Shafin, K.; Jensen, T.D.; Fisk, D.G.; Grove, M.E.; Spiteri, E.; Pesout, T.; Monlong, J.; Baid, G.; et al. Ultrarapid Nanopore Genome Sequencing in a Critical Care Setting. N. Engl. J. Med. 2022, 386, 700–702. [Google Scholar] [CrossRef]
- Winkler, J.K.; Fink, C.; Toberer, F.; Enk, A.; Deinlein, T.; Hofmann-Wellenhof, R.; Thomas, L.; Lallas, A.; Blum, A.; Stolz, W.; et al. Association Between Surgical Skin Markings in Dermoscopic Images and Diagnostic Performance of a Deep Learning Convolutional Neural Network for Melanoma Recognition. JAMA Dermatol. 2019, 155, 1135–1141. [Google Scholar] [CrossRef]
- Ross CIBM’s Watson Supercomputer Recommended “Unsafe Incorrect” Cancer Treatments Internal Documents Show, S.T.A.T. Available online: https://www.statnews.com/2018/07/25/ibm-watson-recommended-unsafe-incorrect-treatments/ (accessed on 12 December 2022).
- Wong, A.; Otles, E.; Donnelly, J.P.; Krumm, A.; McCullough, J.; DeTroyer-Cooley, O.; Pestrue, J.; Phillips, M.; Konye, J.; Penoza, C.; et al. External Validation of a Widely Implemented Proprietary Sepsis Prediction Model in Hospitalized Patients. JAMA Intern. Med. 2021, 181, 1065–1070. [Google Scholar] [CrossRef]
- Bernstam, E.V.; Shireman, P.K.; Meric-Bernstam, F.; Zozus, M.N.; Jiang, X.; Brimhall, B.B.; Windham, A.K.; Schmidt, S.; Visweswaran, S.; Ye, Y.; et al. Artificial intelligence in clinical and translational science: Successes, challenges and opportunities. Clin. Transl. Sci. 2021, 15, 309–321. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Król, Z.J.; Dobosz, P.; Ślubowska, A.; Mroczek, M. WGS Data Collections: How Do Genomic Databases Transform Medicine? Int. J. Mol. Sci. 2023, 24, 3031. https://doi.org/10.3390/ijms24033031
Król ZJ, Dobosz P, Ślubowska A, Mroczek M. WGS Data Collections: How Do Genomic Databases Transform Medicine? International Journal of Molecular Sciences. 2023; 24(3):3031. https://doi.org/10.3390/ijms24033031
Chicago/Turabian StyleKról, Zbigniew J., Paula Dobosz, Antonina Ślubowska, and Magdalena Mroczek. 2023. "WGS Data Collections: How Do Genomic Databases Transform Medicine?" International Journal of Molecular Sciences 24, no. 3: 3031. https://doi.org/10.3390/ijms24033031
APA StyleKról, Z. J., Dobosz, P., Ślubowska, A., & Mroczek, M. (2023). WGS Data Collections: How Do Genomic Databases Transform Medicine? International Journal of Molecular Sciences, 24(3), 3031. https://doi.org/10.3390/ijms24033031