
Endocrine Disrupting Chemicals Influence Hub Genes Associated with Aggressive Prostate Cancer



Abstract
:1. Introduction
2. Results
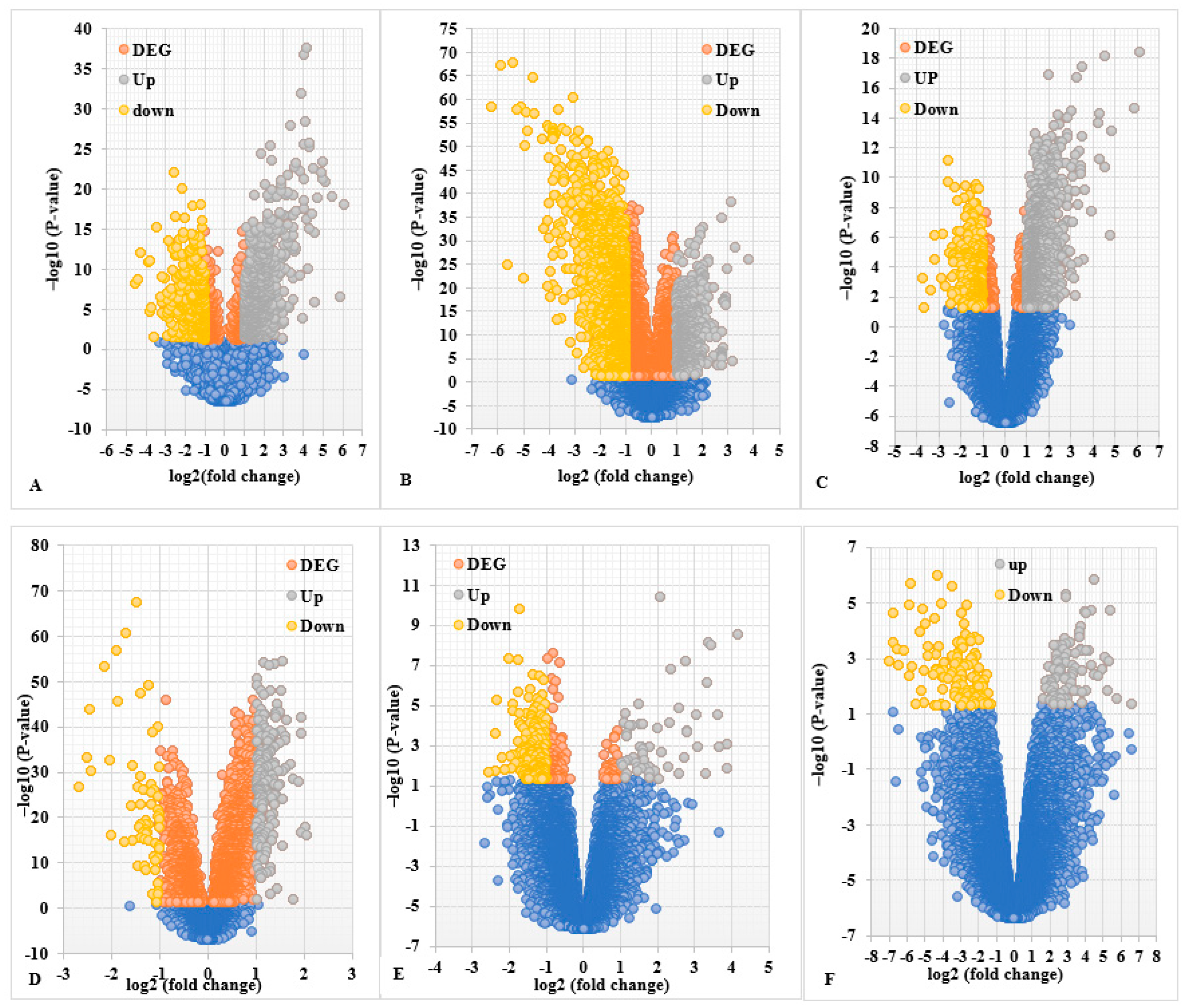
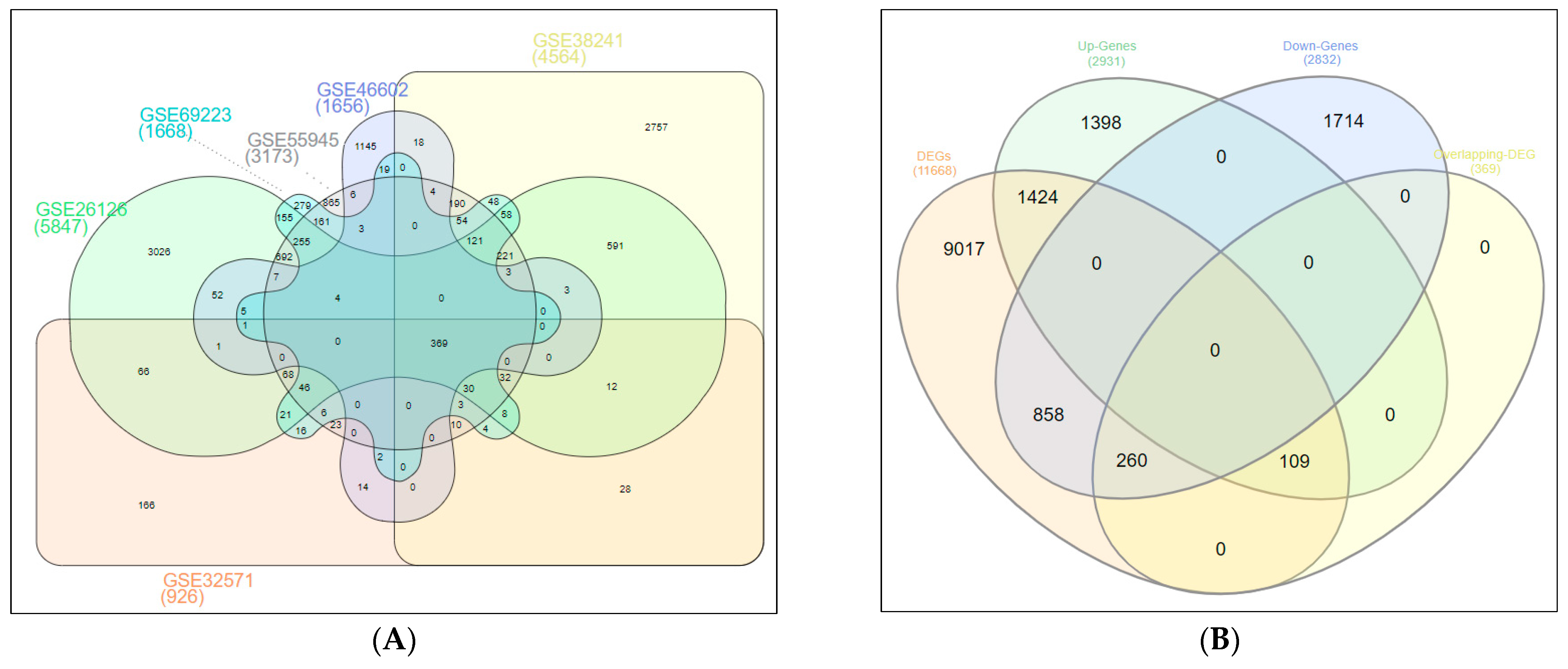
2.1. Screening of DEGs (Up- and Down-Regulated Genes)
2.2. Gene Ontology Enrichment Analysis for DEGs in PCa
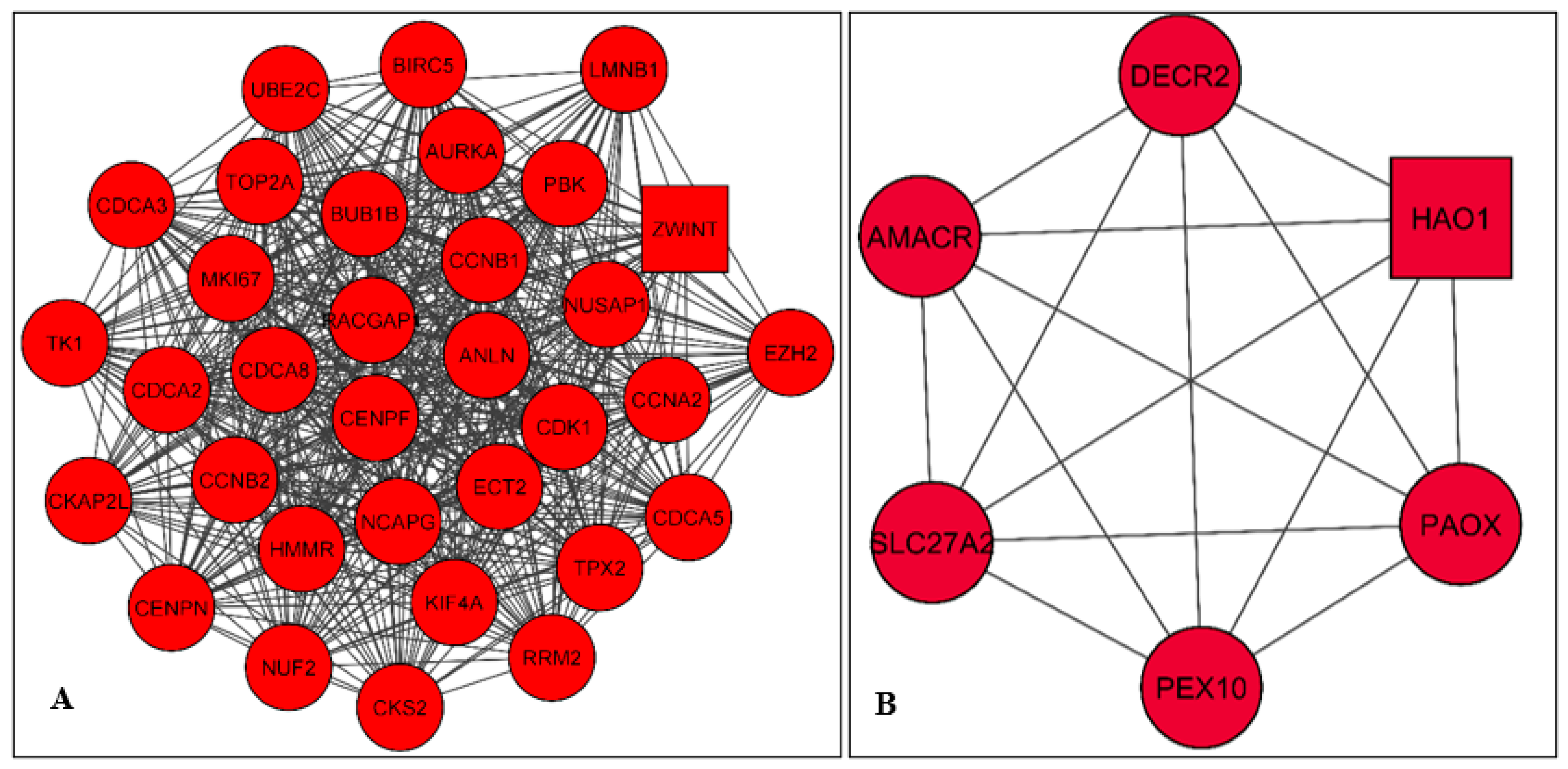
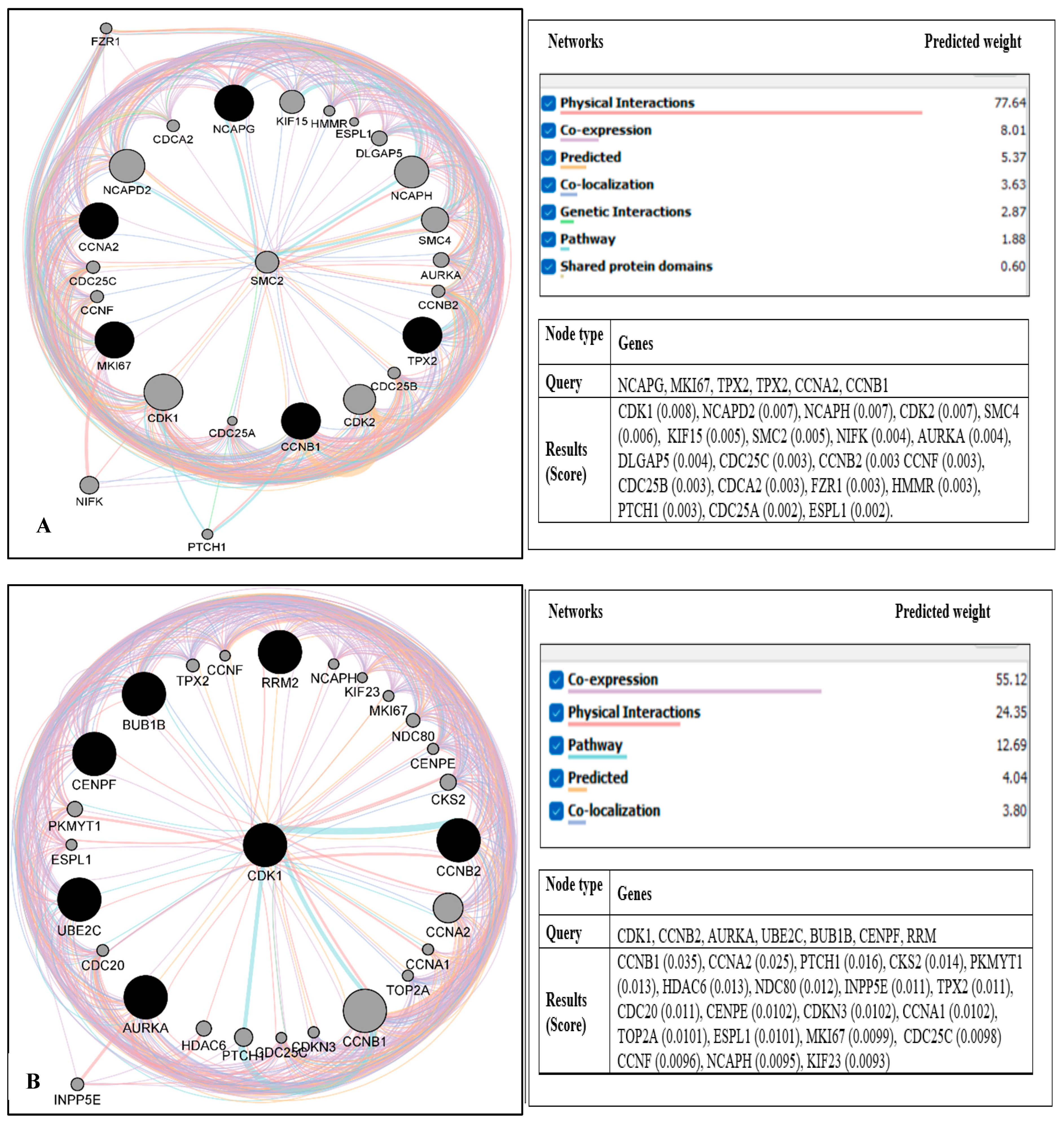
2.3. PPI Network Construction and Module Analysis
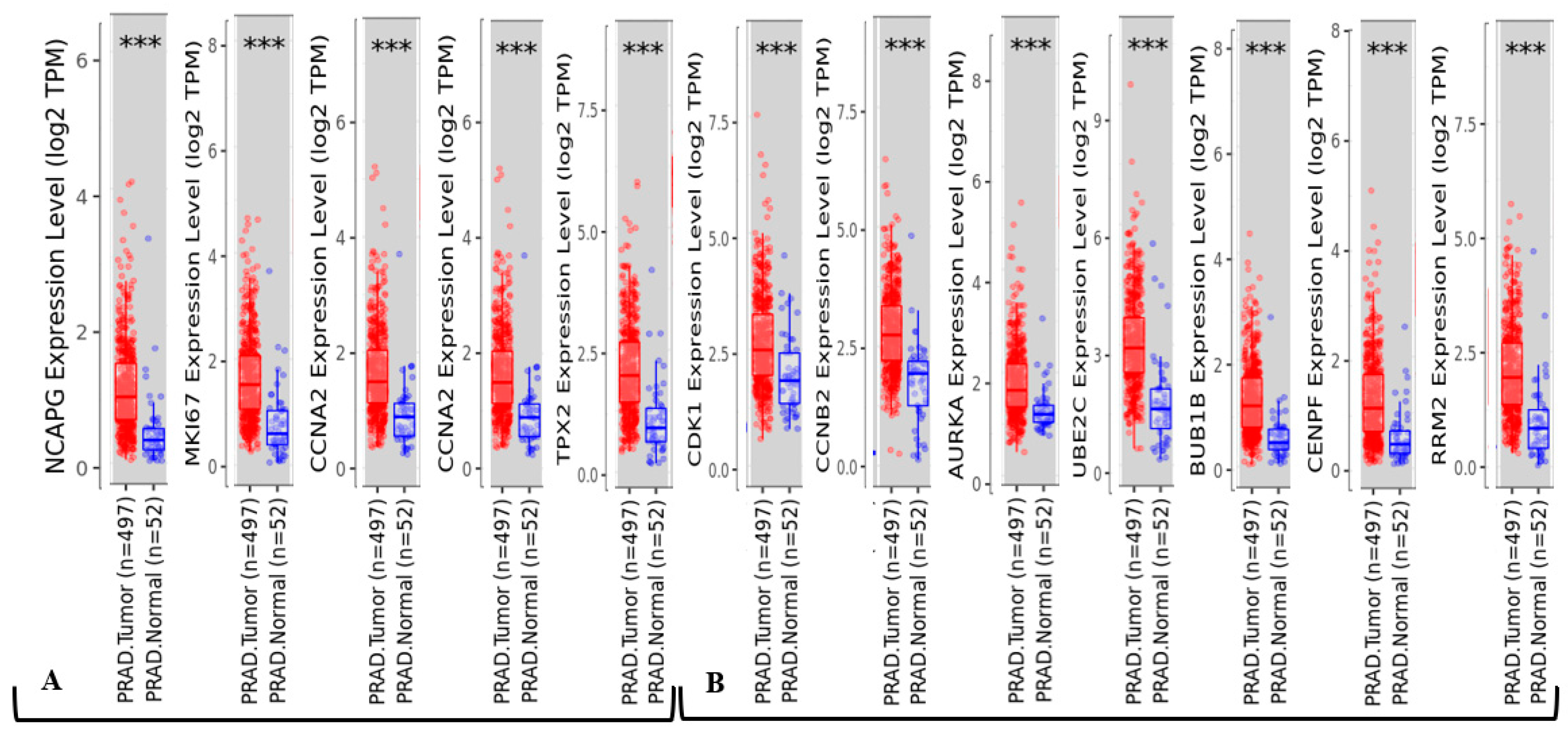
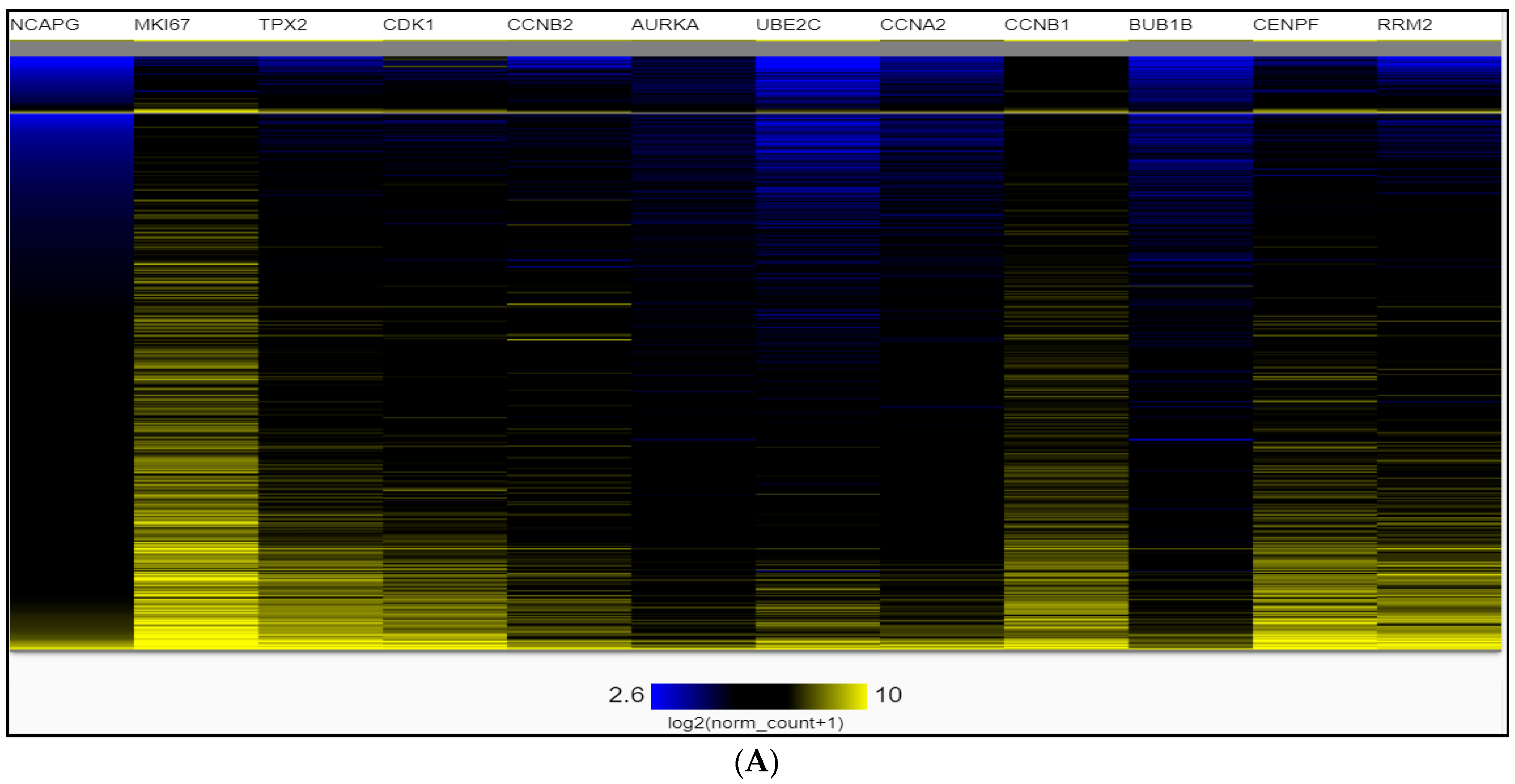
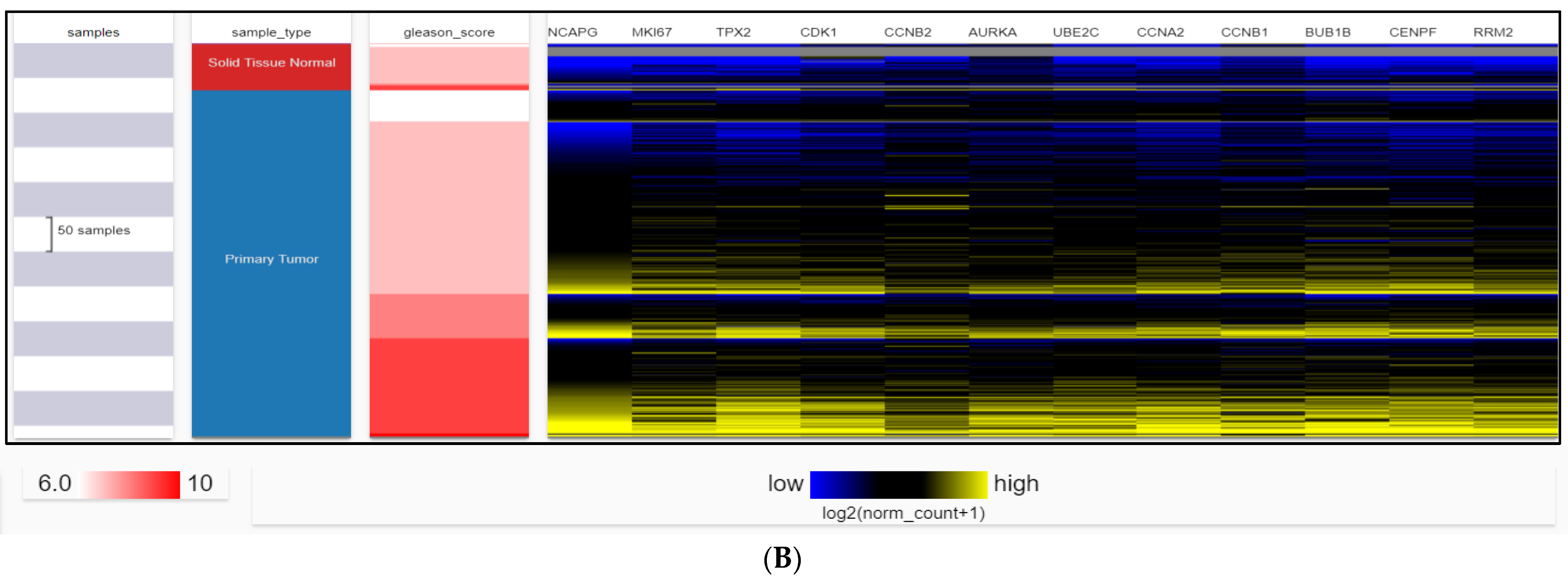
2.4. Dataset Validation for Expression of Hub Genes in PCa Tissues
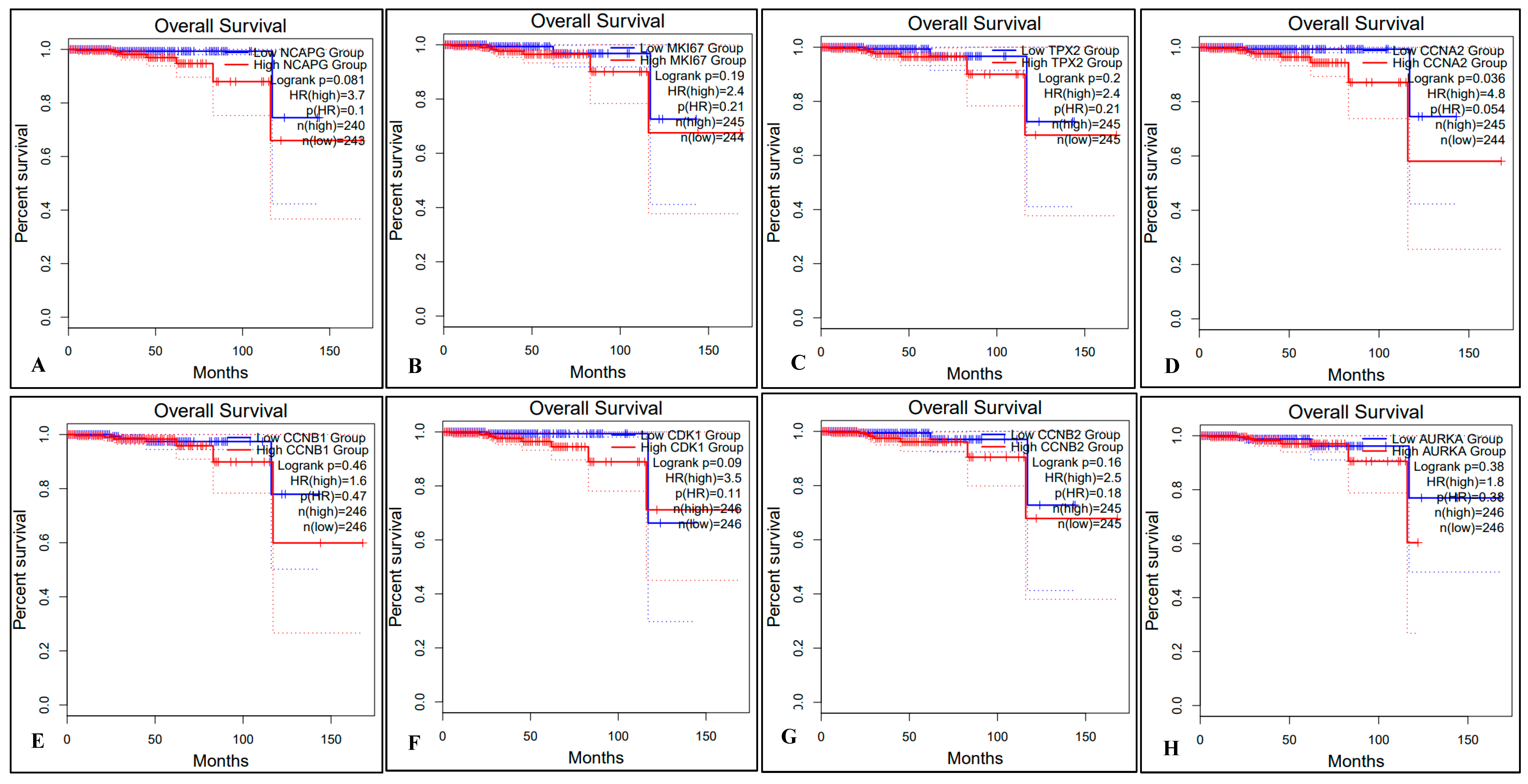
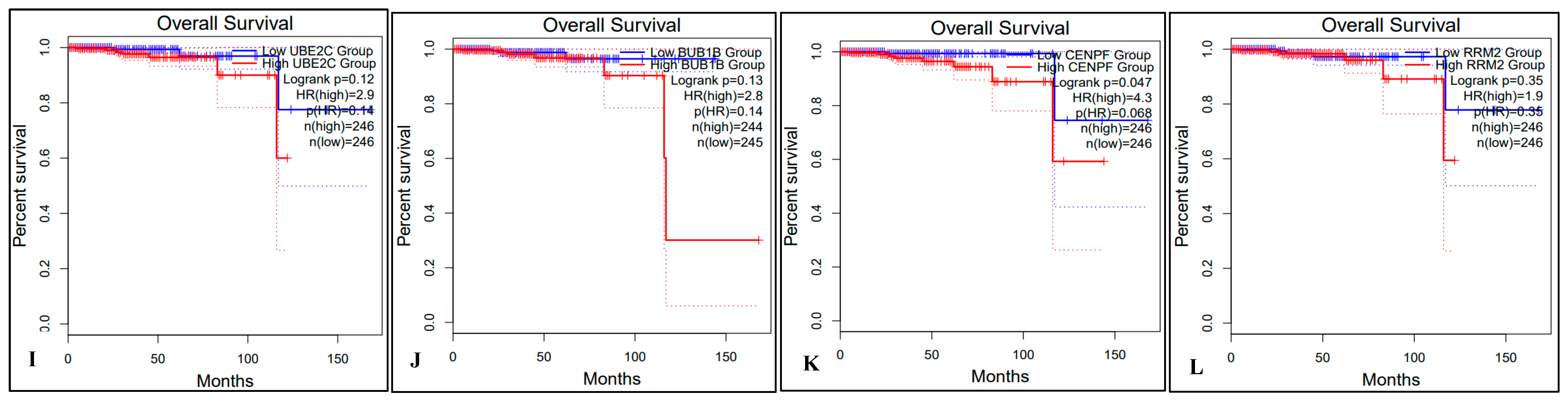
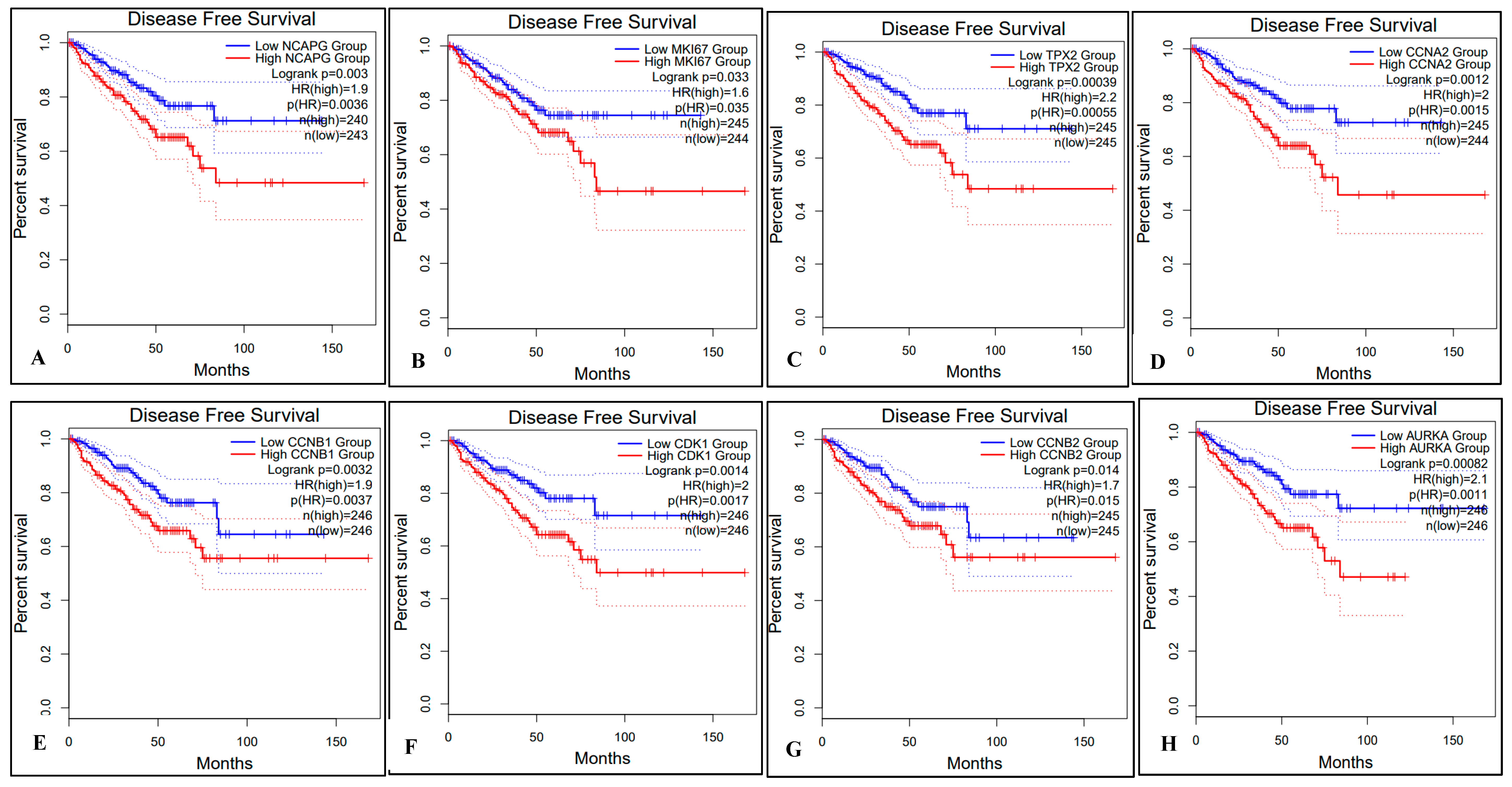
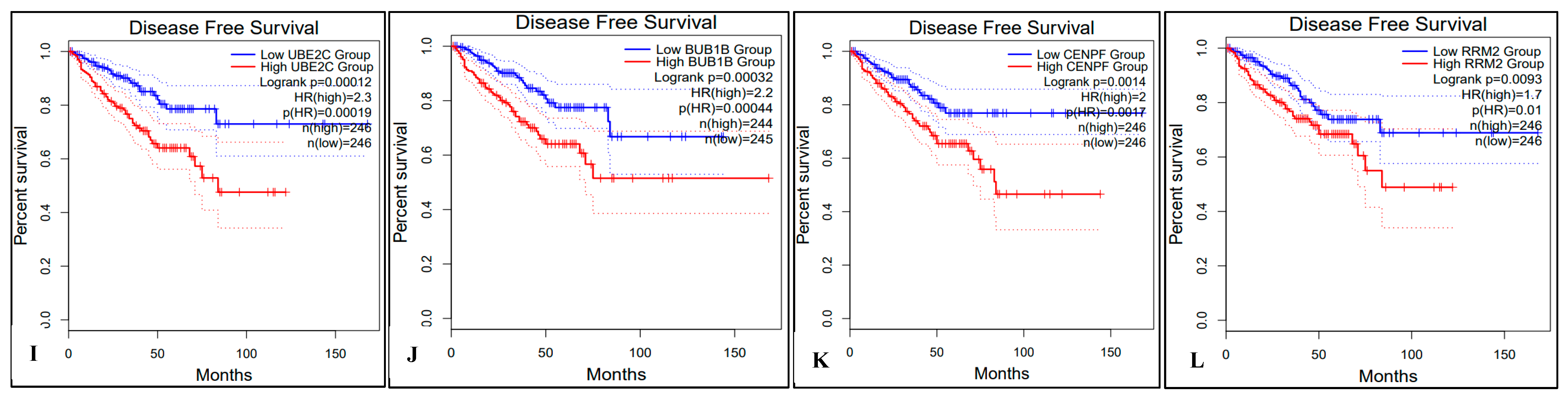
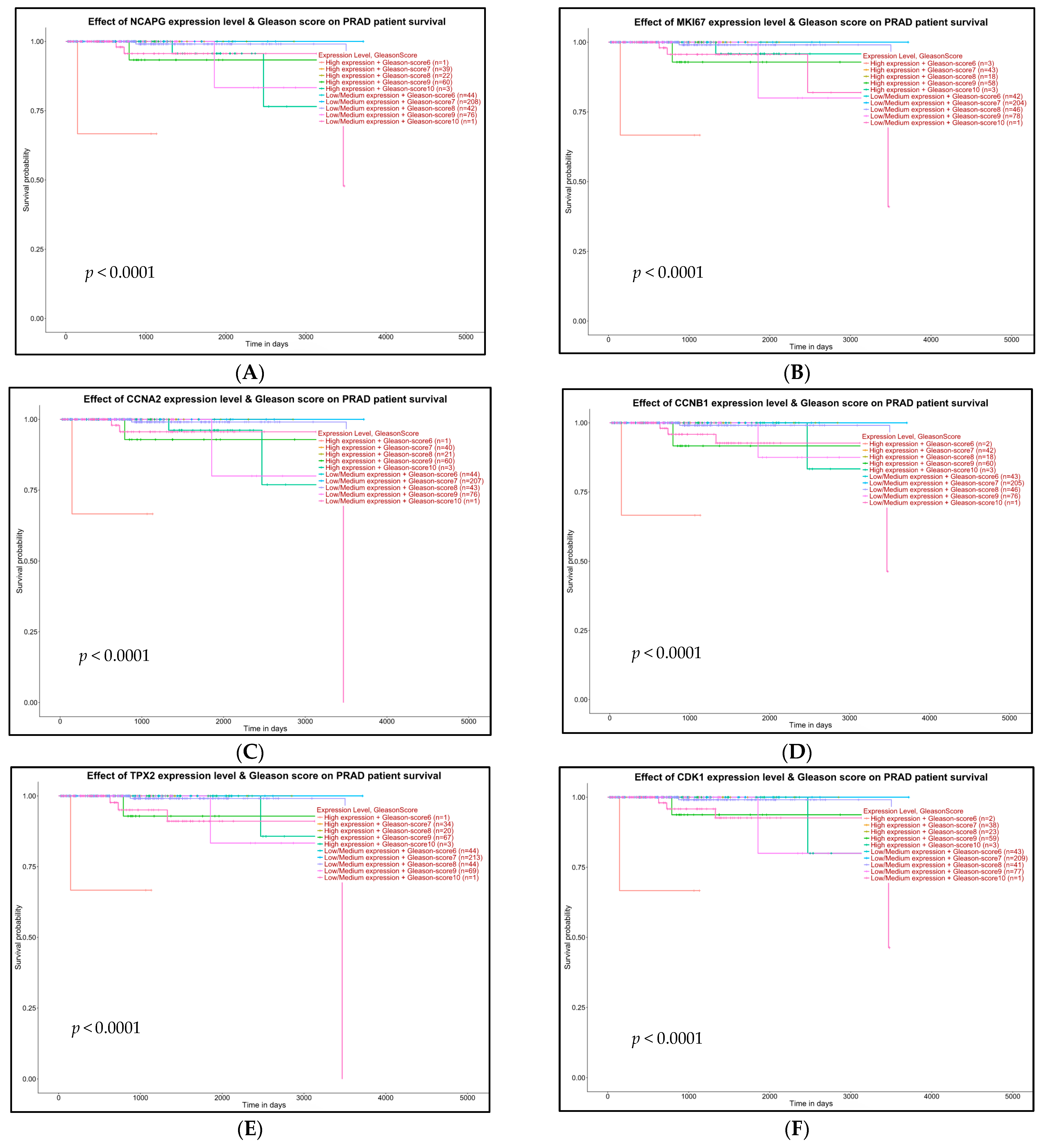
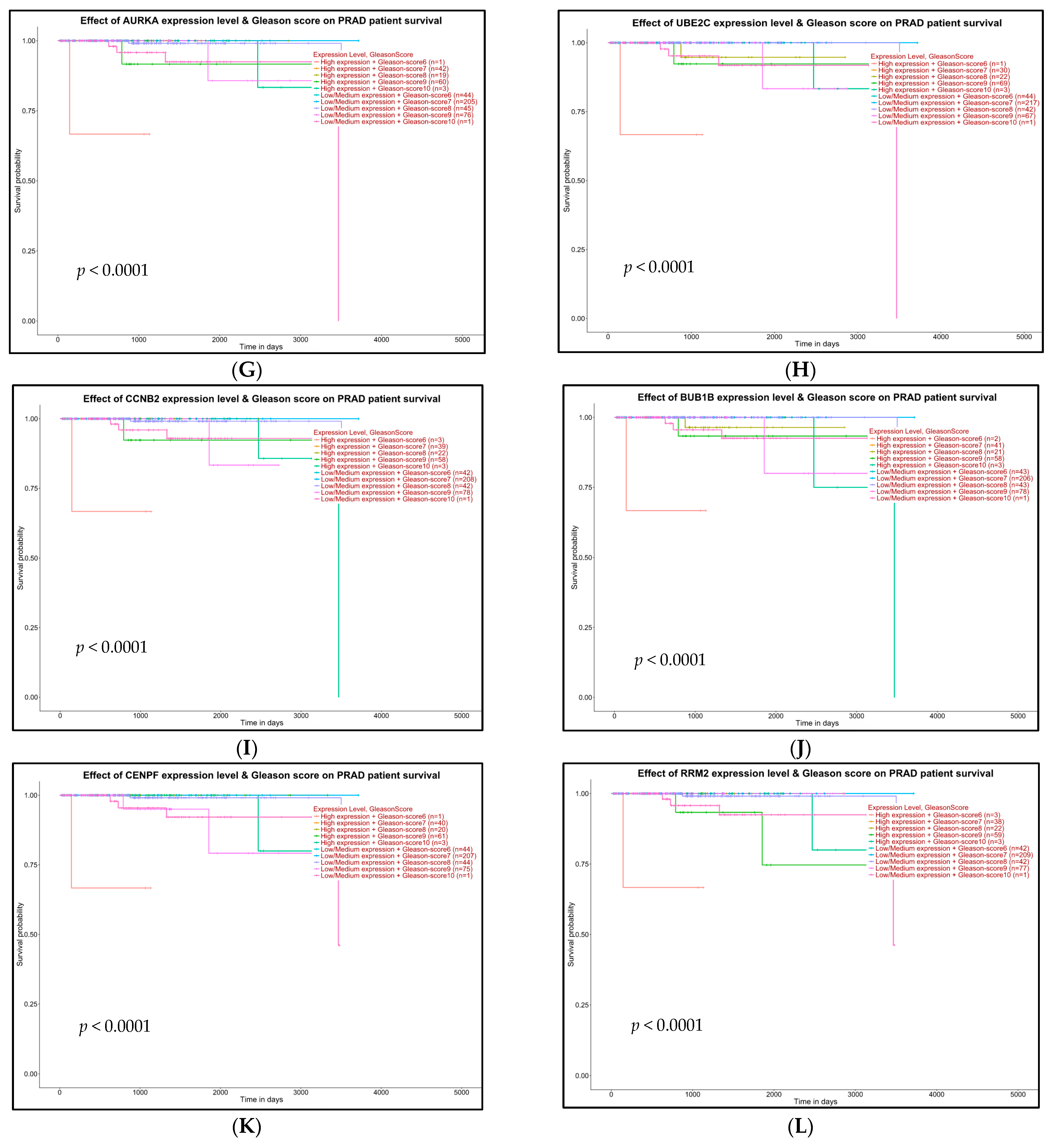
2.5. Survival Analysis of Hub Genes
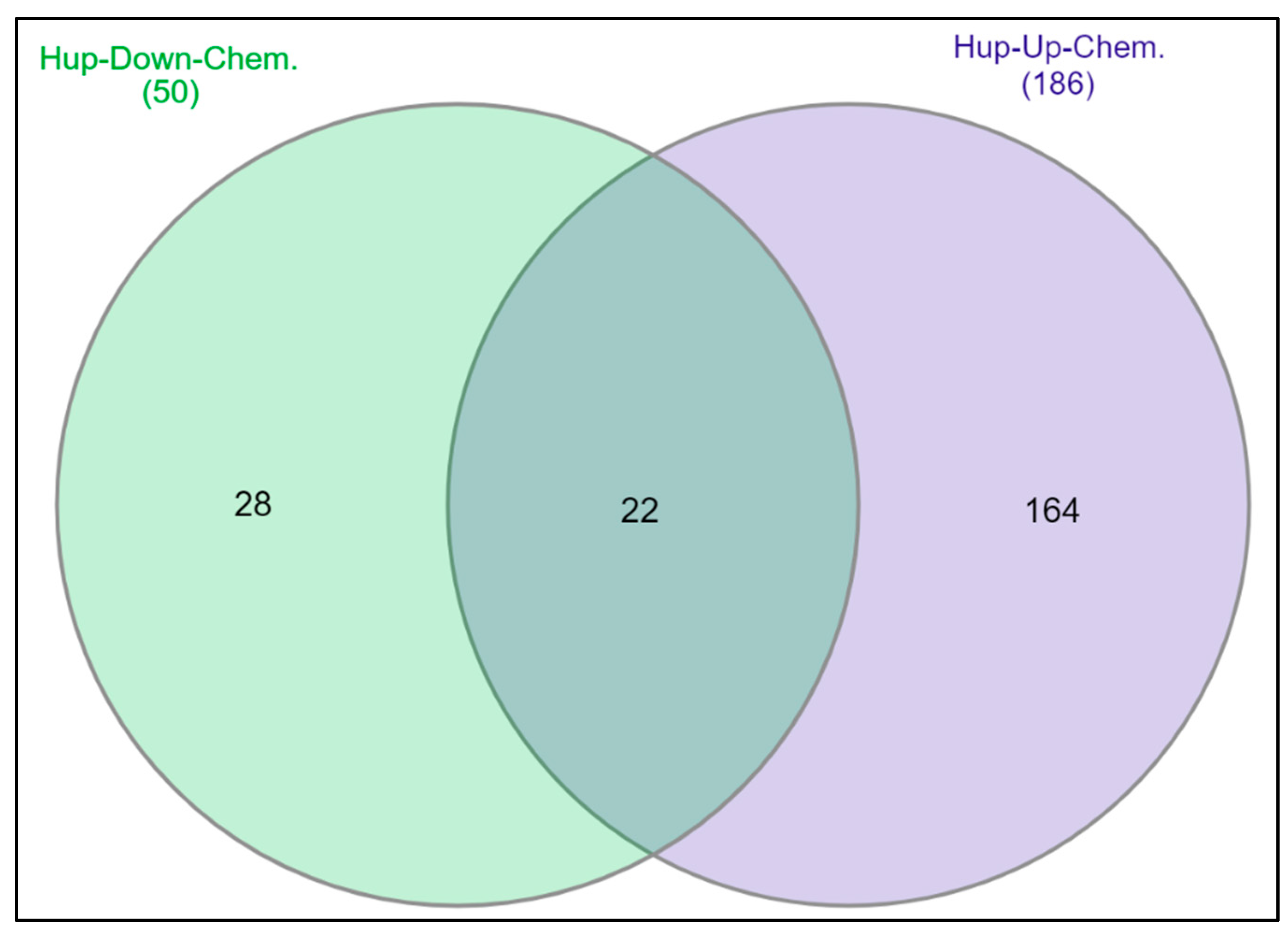
2.6. Chemical-Gene Interaction Analysis for DEGs
3. Discussion
4. Materials and Methods
4.1. Microarray Datasets: Downloaded
4.2. Data Processing: Screening and Identification of DEGs
4.3. DEGs: GO, Biological Functional and Enrichment Analysis
4.4. Protein–Protein Interaction (PPI) Network Construction
4.5. Modules Selection and Clustering Analysis
4.6. External Dataset Validation and Evaluation of the Analysis of Hub Genes
4.7. Survival Analysis with Hub Genes
- High expression and Gleason score 6 (n)
- High expression and Gleason score 7 (n)
- High expression and Gleason score 8 (n)
- High expression and Gleason score 9 (n)
- High expression and Gleason score 10 (n)
- Low/medium expression and Gleason score 6 (n)
- Low/medium expression and Gleason score 7 (n)
- Low/medium expression and Gleason score 8 (n)
- Low/medium expression and Gleason score 9 (n)
- Low/medium expression and Gleason score 10 (n)
4.8. Chemical-Gene Interaction Analysis for DEGs in PCa
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AR | Androgen receptor |
| AURKA | Aurora kinase A |
| BP | Biological process |
| BUB1B | BUB1 mitotic checkpoint serine/threonine kinase B |
| CRPC | Castrate-resistant prostate cancers |
| CC | Cellular component |
| CCNA2 | Cyclin A2 |
| CCNB1 | Cyclin B1 |
| CCNB2 | Cyclin B2 |
| CDK1 | Cyclin dependent kinase 1 |
| COX | Cyclooxygenases |
| CENPF | Centromere protein F |
| CTD | Comparative Toxicology Database |
| DEGs | Differentially expressed genes |
| DNMC | Degree, density of maximum neighborhood component |
| EDCs | Endocrine-disrupting chemicals |
| EPC | Edge percolated component |
| FDR | False discovery rates |
| GEO | Gene expression omnibus |
| GO | Gene ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LOX | Lipoxygenases |
| MCODE | Molecular complex detection |
| MYCMAX | MAX Gene—MYC-associated factor X |
| MKI67 | Marker of proliferation Ki-67 |
| MF | Molecular function |
| NCAPG | Non-SMC condensin I complex subunit G |
| NFY | Nuclear transcription factor Y |
| PCa | Prostate cancer |
| POPs | Persistent organic pollutants |
| PPI | Protein–protein interaction |
| PSA | Prostate-specific antigen |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| RRM2 | Ribonucleotide reductase regulatory subunit M2 |
| STRING | Search tool for the retrieval of interacting genes/proteins |
| TCGA-PRAD | The Cancer Genome Atlas Prostate Adenocarcinoma |
| TF | Transcription factor |
| TPM | Transcript per million |
| TPX2 | Targeting protein for Xenopus kinesin-like protein 2 |
| FGFR2 | Type 2 fibroblast growth factor receptor |
| UBE2C | Ubiquitin-conjugating enzyme E2 C |
| UCSC-TFBS | University of California, Santa Cruz–transcription factor binding sites |
References
- Viale, P.H. The American Cancer Society’s Facts & Figures: 2021 Edition. J. Adv. Pract. Oncol. 2020, 11, 135–136. [Google Scholar] [PubMed]
- Farhood, B.; Mortezaee, K.; Haghi-Aminjan, H.; Khanlarkhani, N.; Salehi, E.; Nashtaei, M.S.; Najafi, M.; Sahebkar, A. A systematic review of radiation-induced testicular toxicities following radiotherapy for prostate cancer. J. Cell. Physiol. 2019, 234, 14828–14837. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Roobol, M.J.; Steyerberg, E.W.; Kranse, R.; Wolters, T.; van den Bergh, R.C.N.; Bangma, C.H.; Schröder, F.H. A Risk-Based Strategy Improves Prostate-Specific Antigen-Driven Detection of Prostate Cancer. Eur. Urol. 2009, 57, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Gandaglia, G.; Albers, P.; Abrahamsson, P.; Briganti, A.; Catto, J.W.F.; Chapple, C.R.; Montorsi, F.; Mottet, N.; Roobol, M.J.; Sønksen, J.; et al. Structured Population-based Prostate-specific Antigen Screening for Prostate Cancer: The European Association of Urology Position in 2019. Eur. Urol. 2019, 76, 142–150. [Google Scholar] [CrossRef]
- Lu, W.; Ding, Z. Identification of key genes in prostate cancer gene expression profile by bioinformatics. Andrologia 2019, 51, e13169. [Google Scholar] [CrossRef] [PubMed]
- Biddle, C.; Brasel, A.; Underwood, W.; Orom, H. Experiences of Uncertainty in Men with an Elevated PSA. Am. J. Men’s Health 2017, 11, 24–34. [Google Scholar] [CrossRef]
- Saltman, A.; Zegar, J.; Haj-Hamed, M.; Verma, S.; Sidana, A. Prostate cancer biomarkers, and multiparametric MRI: Is there a role for both in prostate cancer management? Ther. Adv. Urol. 2021, 13, 1756287221997186. [Google Scholar] [CrossRef]
- Alwadi, D.; Felty, Q.; Roy, D.; Yoo, C.; Deoraj, A. Environmental Phenol and Paraben Exposure Risks and Their Potential Influence on the Gene Expression Involved in the Prognosis of Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 3679. [Google Scholar] [CrossRef]
- Prins, G.S. Endocrine Disruptors and Prostate Cancer Risk. Endocr. Relat. Cancer 2008, 15, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Tarapore, P.; Ying, J.; Ouyang, B.; Burke, B.; Bracken, B.; Ho, S.-M. Exposure to Bisphenol a Correlates with Early-Onset Prostate Cancer and Promotes Centrosome Amplification and Anchorage-Independent Growth in Vitro. PLoS ONE 2014, 9, e90332. [Google Scholar] [CrossRef] [PubMed]
- Golden, R.; Gandy, J.; Vollmer, G. A Review of the Endocrine Activity of Parabens and Implications for Potential Risks to Human Health. Crit. Rev. Toxicol. 2005, 35, 435–458. [Google Scholar] [CrossRef]
- Ho, S.-M.; Rao, R.; To, S.; Schoch, E.; Tarapore, P. Bisphenol A and Its Analogues Disrupt Centrosome Cycle and Microtubule Dynamics in Prostate Cancer. Endocr. Relat. Cancer 2017, 24, 83–96. [Google Scholar] [CrossRef]
- Feng, T.; Wei, D.; Li, Q.; Yang, X.; Han, Y.; Luo, Y.; Jiang, Y. Four Novel Prognostic Genes Related to Prostate Cancer Identified Using Co-Expression Structure Network Analysis. Front. Genet. 2021, 12, 584164. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xu, Y.; Lu, Y.; Ma, X.; Chen, P.; Luo, S.; Jia, Z.; Liu, Y.; Guo, Y. Identifying differentially expressed genes and small molecule drugs for prostate cancer by a bioinformatics strategy. Asian Pac. J. Cancer Prev. APJCP 2013, 14, 5281–5286. [Google Scholar] [CrossRef]
- He, Z.; Duan, X.; Zeng, G. Identification of potential biomarkers and pivotal biological pathways for prostate cancer using bioinformatics analysis methods. PeerJ 2019, 2019, e7872. [Google Scholar] [CrossRef]
- Song, Z.; Chao, F.; Zhuo, Z.; Ma, Z.; Li, W.; Chen, G. Identification of hub genes in prostate cancer using robust rank aggregation and weighted gene co-expression network analysis. Aging 2019, 11, 4736–4756. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.; Yan, K.; Lin, J.; Zheng, Z.; Bi, J. Identification of core genes associated with prostate cancer progression and outcome via bioinformatics analysis in multiple databases. PeerJ 2020, 2020, e8786. [Google Scholar] [CrossRef] [PubMed]
- Lascorz, J.; Hemminki, K.; Försti, A. Systematic enrichment analysis of gene expression profiling studies identifies consensus pathways implicated in colorectal cancer development. J. Carcinog. 2011, 10, 7. [Google Scholar] [CrossRef]
- Tong, Y.; Song, Y.; Deng, S. Combined analysis and validation for DNA methylation and gene expression profiles associated with prostate cancer. Cancer Cell Int. 2019, 19, 50. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Jin, X.; Wang, K. Integrated Bioinformatics Analysis of Potential Biomarkers for Prostate Cancer. Pathol. Oncol. Res. 2017, 25, 455–460. [Google Scholar] [CrossRef]
- Fang, E.; Zhang, X.; Wang, Q.; Wang, D. Identification of prostate cancer hub genes and therapeutic agents using bioinformatics approach. Cancer Biomark. 2017, 20, 553–561. [Google Scholar] [CrossRef]
- Zhao, R.; Wang, Y.; Zhang, M.; Gu, X.; Wang, W.; Tan, J.; Wei, X.; Jin, N. Screening of potential therapy targets for prostate cancer using integrated analysis of two gene expression profiles. Oncol. Lett. 2017, 14, 5361–5369. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; He, H.; Jiang, F.; Militar, J.; Ran, P.; Qin, G.; Cai, C.; Chen, X.; Zhao, J.; Mo, Z.; et al. Analysis of the specific pathways and networks of prostate cancer for gene expression profiles in the Chinese population. Med. Oncol. 2011, 29, 1972–1984. [Google Scholar] [CrossRef]
- Endo, T.; Uzawa, K.; Suzuki, H.; Tanzawa, H.; Ichikawa, T. Characteristic gene expression profiles of benign prostatic hypertrophy and prostate cancer. Int. J. Oncol. 2009, 35, 499–509. [Google Scholar]
- Doultsinos, D.; Mills, I.G. Derivation and application of molecular signatures to prostate cancer: Opportunities and challenges. Cancers 2021, 13, 495. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Huang, M.; Wang, Y. A Transcription Factor-Based Risk Model for Predicting the Prognosis of Prostate Cancer and Potential Therapeutic Drugs. Evidence-based complementary and alternative medicine. Evid.-Based Complement. Altern. Med. 2021, 2021, 6894278. [Google Scholar] [CrossRef] [PubMed]
- Tagai, E.K.; Miller, S.M.; Kutikov, A.; Diefenbach, M.A.; Gor, R.A.; Al-Saleem, T.; Chen, D.Y.; Fleszar, S.; Roy, G. Prostate Cancer Patients’ Understanding of the Gleason Scoring System: Implications for Shared Decision-Making. J. Cancer Educ. 2018, 34, 441–445. [Google Scholar] [CrossRef]
- McDougal, W.; Scott, J.; Kavoussi, R.; Novick, C.; Partin, W.; Craig, P.; Ramchandani, P. Campbell-Walsh Urology 10th Edition Review; Saunders: Philadelphia, PA, USA, 2011. [Google Scholar]
- Davis, A.P.; Wiegers, T.C.; Johnson, R.J.; Lay, J.M.; Lennon-Hopkins, K.; Saraceni-Richards, C.; Sciaky, D.; Murphy, C.G.; Mattingly, C.J. Text mining effectively scores and ranks the literature for improving chemical-gene-disease curation at the comparative toxicogenomics database. PLoS ONE 2013, 8, e58201. [Google Scholar] [CrossRef]
- Fan, S.; Liang, Z.; Gao, Z.; Pan, Z.; Han, S.; Liu, X.; Zhao, C.; Yang, W.; Pan, Z.; Feng, W. Identification of the key genes and pathways in prostate cancer. Oncol. Lett. 2018, 16, 6663–6669. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y.; Absher, D.M.; Gulzar, Z.G.; Young, S.R.; McKenney, J.K.; Peehl, D.M.; Brooks, J.D.; Myers, R.M.; Sherlock, G. DNA methylation profiling reveals novel biomarkers and important roles for DNA methyltransferases in prostate cancer. Genome Res. 2011, 21, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jia, B.; Zhao, X.; Wang, Y.; Ye, W. miR-93-5p may be an important oncogene in prostate cancer by bioinformatics analysis. J. Cell. Biochem. 2019, 120, 10463–10483. [Google Scholar] [CrossRef]
- Hernández, G.; Ramírez, J.L.; Pedroza-Torres, A.; Herrera, L.A.; Jiménez-Ríos, M.A. The secret life of translation initiation in prostate cancer. Front. Genet. 2019, 10, 14. [Google Scholar] [CrossRef] [PubMed]
- Tai, K.-Y.; Shiah, S.-G.; Shieh, Y.-S.; Kao, Y.-R.; Chi, C.-Y.; Huang, E.; Lee, H.-S.; Chang, L.-C.; Yang, P.-C.; Wu, C.-W. DNA methylation and histone modification regulate silencing of epithelial cell adhesion molecule for tumor invasion and progression. Oncogene 2007, 26, 3989–3997. [Google Scholar] [CrossRef]
- Huang, Y.; Hamana, T.; Liu, J.; Wang, C.; An, L.; You, P.; Chang, J.Y.F.; Xu, J.; Jin, C.; Zhang, Z.; et al. Type 2 Fibroblast Growth Factor Receptor Signaling Preserves Stemness and Prevents Differentiation of Prostate Stem Cells from the Basal Compartment. J. Biol. Chem. 2015, 290, 17753–17761. [Google Scholar] [CrossRef] [PubMed]
- Bonkhoff, H. Estrogen receptor signaling in prostate cancer: Implications for carcinogenesis and tumor progression. Prostate 2018, 78, 2–10. [Google Scholar] [CrossRef]
- Levine, M.S.; Holland, A.J. The impact of mitotic errors on cell proliferation and tumorigenesis. Genes Dev. 2018, 32, 620–638. [Google Scholar] [CrossRef]
- Hatch, E.M.; Fischer, A.H.; Deerinck, T.J.; Hetzer, M.W. Catastrophic Nuclear Envelope Collapse in Cancer Cell Micronuclei. Cell 2013, 154, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhao, Z. Peptide Hormones as Tumor Markers in Clinical Practice. Enzymes 2017, 42, 65–79. [Google Scholar] [PubMed]
- Chesnokova, V.; Melmed, S. Peptide Hormone Regulation of DNA Damage Responses. Endocr. Rev. 2020, 41, 519–537. [Google Scholar] [CrossRef]
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol. 2014, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Ayuningtyas, A.; Kenfield, S.A.; Sesso, H.D.; Campos, H.; Ma, J.; Stampfer, M.J.; Chavarro, J.E. Blood fatty acid patterns are associated with prostate cancer risk in a prospective nested case-control study. Cancer Causes Control 2016, 27, 1153–1161. [Google Scholar] [CrossRef]
- Ferro, M.; Terracciano, D.; Buonerba, C.; Lucarelli, G.; Bottero, D.; Perdonà, S.; Autorino, R.; Serino, A.; Cantiello, F.; Damiano, R.; et al. Anonymous New Prostate Cancer Study Results Reported from European Institute of Oncology (The emerging role of obesity, diet and lipid metabolism in prostate cancer). Obes. Fit. Wellness Week 2017, 13, 285–293. [Google Scholar]
- Liu, Y.; Zuckier, L.S.; Ghesani, N.V. Dominant uptake of fatty acid over glucose by prostate cells: A potential new diagnostic and therapeutic approach. Anticancer Res. 2010, 30, 369–374. [Google Scholar] [PubMed]
- O’Reilly, M.W.; House, P.J.; Tomlinson, J.W. Understanding androgen action in adipose tissue. J. Steroid Biochem. Mol. Biol. 2014, 143, 277–284. [Google Scholar] [CrossRef]
- Gào, X.; Schöttker, B. Reduction-oxidation pathways involved in cancer development: A systematic review of literature reviews. Oncotarget 2017, 8, 51888–51906. [Google Scholar] [CrossRef]
- Speed, N.; Blair, I.A. Cyclooxygenase- and lipoxygenase-mediated DNA damage. Cancer Metastasis Rev. 2011, 30, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Sadras, F.; Monteith, G.R.; Roberts-Thomson, S. An emerging role for calcium Signaling in cancer-associated fibroblasts. Int. J. Mol. Sci. 2021, 22, 11366. [Google Scholar] [CrossRef]
- Deep, G.; Schlaepfer, I.R. Aberrant lipid metabolism promotes prostate cancer: Role in cell survival under hypoxia and extracellular vesicles biogenesis. Int. J. Mol. Sci. 2016, 17, 1061. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, R.; Cheng, K.; Ring, B.Z.; Su, L. Roles of Rap1 signaling in tumor cell migration and invasion. Cancer Biol. Med. 2017, 14, 90–99. [Google Scholar] [PubMed] [Green Version]
- Basu, S.K.; Basu, S.; Johnson, P.F. Localized RAS signaling drives cancer. Oncoscience 2019, 6, 298–300. [Google Scholar] [CrossRef]
- Benatti, P.; Chiaramonte, M.L.; Lorenzo, M.; Hartley, J.A.; Hochhauser, D.; Gnesutta, N.; Mantovani, R.; Imbriano, C.; Dolfini, D. NF-Y activates genes of metabolic pathways altered in cancer cells. Oncotarget 2016, 7, 1633–1650. [Google Scholar] [CrossRef] [PubMed]
- Dolfini, D.; Zambelli, F.; Pavesi, G.; Mantovani, R. A perspective of promoter architecture from the CCAAT box. Cell Cycle 2009, 8, 4127–4137. [Google Scholar] [CrossRef]
- Hogarty, M.D.; Norris, M.D.; Davis, K.; Liu, X.; Evageliou, N.F.; Hayes, C.S.; Pawel, B.; Guo, R.; Zhao, H.; Sekyere, E.; et al. ODC1 is a critical determinant of MYCN oncogenesis and a therapeutic target in neuroblastoma. Cancer Res. 2008, 68, 9735–9745. [Google Scholar] [CrossRef]
- Carabet, L.A.; Lallous, N.; Leblanc, E.; Ban, F.; Morin, H.; Lawn, S.; Ghaidi, F.; Lee, J.; Mills, I.G.; Gleave, M.E.; et al. Computer-aided drug discovery of Myc-Max inhibitors as potential therapeutics for prostate cancer. Eur. J. Med. Chem. 2018, 160, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.M.; Bieberich, C.J.; Dang, C.V.; Nelson, W.G.; Yegnasubramanian, S.; De Marzo, A.M. MYC and prostate cancer. Genes Cancer 2010, 1, 617–628. [Google Scholar] [CrossRef]
- Kim, Y.-A.; Cho, D.-Y.; Dao, P.; Przytycka, T.M. Memcover: Integrated Analysis of Mutual Exclusivity and Functional Network Reveals Dysregulated Pathways across Multiple Cancer Types. Bioinformatics 2015, 31, i284–i292. [Google Scholar] [CrossRef]
- Xie, F.; Xi, J.; Duan, Q. Driver Attribute Filling for Genes in Interaction Network via Modularity Subspace-Based Concept Learning from Small Samples. Complexity 2020, 2020, 6643551. [Google Scholar] [CrossRef]
- Arai, T.; Okato, A.; Yamada, Y.; Sugawara, S.; Kurozumi, A.; Kojima, S.; Yamazaki, K.; Naya, Y.; Ichikawa, T.; Seki, N. Regulation of NCAPG by miR-99a-3p (passenger strand) inhibits cancer cell aggressiveness and is involved in CRPC. Cancer Med. 2018, 7, 1988–2002. [Google Scholar] [CrossRef]
- Zhang, Q.; Su, R.; Shan, C.; Gao, C.; Wu, P. Non-SMC Condensin I Complex, Subunit G (NCAPG) is a Novel Mitotic Gene Required for Hepatocellular Cancer Cell Proliferation and Migration. Oncol. Res. 2018, 26, 269–276. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, H.; Zhang, G.; Zhong, A.; Ma, Q.; Kai, J.; Tong, Y.; Xie, S.; Wang, Y.; Zheng, H.; et al. Targeting TPX2 suppresses proliferation and promotes apoptosis via repression of the PI3k/AKT/P21 signaling pathway and activation of p53 pathway in breast cancer. Biochem. Biophys. Res. Commun. 2018, 507, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Hammarsten, P.; Josefsson, A.; Thysell, E.; Lundholm, M.; Hägglöf, C.; Iglesias-Gato, D.; Flores-Morales, A.; Stattin, P.Ä.; Egevad, L.; Granfors, T.; et al. Immunoreactivity for prostate specific antigen and Ki67 differentiates subgroups of prostate cancer related to outcome. Mod. Pathol. 2019, 32, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Huang, R.; Jiang, F.; Chen, D.; Wang, C.; Han, Z.; Liang, Y.; Zhong, W. Overexpression of TPX2 is associated with progression and prognosis of prostate cancer. Oncol. Lett. 2018, 16, 2823–2832. [Google Scholar] [CrossRef]
- Pan, H.; Su, H.; Hsu, C.; Huang, G.; Wu, T.T. Targeted TPX2 increases chromosome missegregation and suppresses tumor cell growth in human prostate cancer. OncoTargets Ther. 2017, 10, 3531–3543. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, B.; Bjartell, A.; Tuomela, J.; Dizeyi, N.; Tinzl, M.; Helczynski, L.; Nilsson, E.; Otterbein, L.E.; Härkönen, P.; Persson, J.L. Multiple Cellular Mechanisms Related to Cyclin A1 in Prostate Cancer Invasion and Metastasis. J. Natl. Cancer Inst. 2008, 100, 1022–1036. [Google Scholar] [CrossRef]
- Gomez, L.A.; de Las Pozas, A.; Reiner, T.; Burnstein, K.; Perez-Stable, C. Increased expression of cyclin B1 sensitizes prostate cancer cells to apoptosis induced by chemotherapy. Mol. Cancer Ther. 2007, 6, 1534–1543. [Google Scholar] [CrossRef]
- Xie, B.; Wang, S.; Jiang, N.; Li, J.J. Cyclin B1/CDK1-regulated mitochondrial bioenergetics in cell cycle progression and tumor resistance. Cancer Lett. 2019, 443, 56–66. [Google Scholar] [CrossRef]
- Liu, K.; Chen, Y.; Feng, P.; Wang, Y.; Sun, M.; Song, T.; Tan, J.; Li, C.; Liu, S.; Kong, Q.; et al. Identification of Pathologic and Prognostic Genes in Prostate Cancer Based on Database Mining. Front. Genet. 2022, 13, 854531. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.; Tang, Q.; Ren, G. Identification of UBE2C as hub gene in driving prostate cancer by integrated bioinformatics analysis. PLoS ONE 2021, 16, e0247827. [Google Scholar] [CrossRef]
- Park, K.; Chen, Z.; MacDonald, T.Y.; Siddiqui, J.; Ye, H.; Erbersdobler, A.; Shevchuk, M.M.; Robinson, B.D.; Sanda, M.G.; Chinnaiyan, A.M.; et al. Prostate cancer with Paneth cell-like neuroendocrine differentiation has recognizable histomorphology and harbors AURKA gene amplification. Hum. Pathol. 2014, 45, 2136–2143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Song, G.; Xiang, J.; Zhang, H.; Zhao, S.; Zhan, Y. AURKA promotes cancer metastasis by regulating epithelial-mesenchymal transition and cancer stem cell properties in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2017, 486, 514–520. [Google Scholar] [CrossRef]
- Fu, X.; Chen, G.; Cai, Z.; Wang, C.; Liu, Z.; Lin, Z.; Wu, Y.; Liang, Y.; Han, Z.; Liu, J.; et al. Overexpression of BUB1B contributes to progression of prostate cancer and predicts poor outcome in patients with prostate cancer. OncoTargets Ther. 2016, 9, 2211–2220. [Google Scholar]
- Shahid, M.; Lee, M.Y.; Piplani, H.; Andres, A.M.; Zhou, B.; Yeon, A.; Kim, M.; Kim, H.L.; Kim, J. Centromere protein F (CENPF), a microtubule binding protein, modulates cancer metabolism by regulating pyruvate kinase M2 phosphorylation signaling. Cell Cycle 2018, 17, 2802–2818. [Google Scholar] [CrossRef]
- Wu, Y.; Sun, J.; Li, A.; Chen, D. The promoted delivery of RRM2 siRNA to vascular smooth muscle cells through liposome-polycation-DNA complex conjugated with cell penetrating peptides. Biomed. Pharmacother. 2018, 103, 982–988. [Google Scholar] [CrossRef]
- Li, J.; Pang, J.; Liu, Y.; Zhang, J.; Zhang, C.; Shen, G.; Song, L. Suppression of RRM2 inhibits cell proliferation, causes cell cycle arrest and promotes the apoptosis of human neuroblastoma cells and in human neuroblastoma RRM2 is suppressed following chemotherapy. Oncol. Rep. 2018, 40, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Pascale, M.; Aversa, C.; Barbazza, R.; Marongiu, B.; Siracusano, S.; Stoffel, F.; Sulfaro, S.; Roggero, E.; Bonin, S.; Stanta, G. The proliferation marker Ki67, but not neuroendocrine expression, is an independent factor in the prediction of prognosis of primary prostate cancer patients. Radiol. Oncol. 2016, 50, 313–320. [Google Scholar] [CrossRef]
- Huang, C.G.; Li, F.X.; Pan, S.; Xu, C.B.; Dai, J.Q.; Zhao, X.H. Identification of genes associated with castration-resistant prostate cancer by gene expression profile analysis. Mol. Med. Rep. 2017, 16, 6803–6813. [Google Scholar] [CrossRef] [PubMed]
- Diamanti-Kandarakis, E.; Bourguignon, J.P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine disrupting chemicals: An endocrine society scientific statement. Endocr. Rev. 2009, 30, 293–342. [Google Scholar] [CrossRef]
- Scott, H.M.; Mason, J.I.; Sharpe, R.M. Steroidogenesis in the fetal testis and its susceptibility to disruption by exogenous compounds. Endocr. Rev. 2009, 30, 883–925. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.; Deoraj, A.; Felty, Q.; Roy, D. Environmental Estrogen-like Endocrine Disrupting Chemicals and Breast Cancer. Mol. Cell. Endocrinol. 2017, 457, 89–102. [Google Scholar] [CrossRef]
- Roy, D.; Morgan, M.; Yoo, C.; Deoraj, A.; Roy, S.; Yadav, V.; Garoub, M.; Assaggaf, H.; Doke, M. Integrated Bioinformatics, environmental epidemiologic and genomic approaches to identify environmental and molecular links between endometriosis and breast cancer. Int. J. Mol. Sci. 2015, 16, 25285–25322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, C.F.; Christoph, G.R.; Julien, E.; Kelley, J.M.; Kronenberg, J.; McCarthy, J.; Reiss, R. Assessing the risks of exposures to multiple chemicals with a common mechanism of toxicity: How to cumulate? Regul. Toxicol. Pharmacol. 2000, 31, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Feron, V.J.; Groten, J.P. Toxicological evaluation of chemical mixtures. Food Chem. Toxicol. 2002, 40, 825–839. [Google Scholar] [CrossRef]
- Lim, J.E.; Park, S.H.; Jee, S.H.; Park, H. Body concentrations of persistent organic pollutants and prostate cancer: A meta-analysis. Environ. Sci. Pollut. Res. Int. 2015, 22, 11275–11284. [Google Scholar] [CrossRef] [PubMed]
- Prezioso, D.; Denis Louis, J.; Klocker, H.; Sciarra, A.; Reis, M.; Naber, K.; Lobe, B.L.; Pacik, D.; Griffiths, K. Estrogens and aspects of prostate disease. Int. J. Urol. 2007, 14, 1–16. [Google Scholar] [CrossRef]
- Ritchie, J.M.; Vial, S.L.; Fuortes, L.J.; Guo, H.; Reedy, V.E.; Smith, E.M. Organochlorines and risk of prostate cancer. J. Occup. Environ. Med. 2003, 45, 692–702. [Google Scholar] [CrossRef]
- Ritchie, J.M.; Vial, S.L.; Fuortes, L.J.; Robertson, L.W.; Guo, H.; Reedy, V.E.; Smith, E.M. Comparison of proposed frameworks for grouping polychlorinated biphenyl congener data applied to a case-control pilot study of prostate cancer. Environ. Res. 2005, 98, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.-E.; Nam, C.; Yang, J.; Rha, K.H.; Lim, K.-M.; Jee, S.H. Serum persistent organic pollutants (POPs) and prostate cancer risk: A case-cohort study. Int. J. Hyg. Environ. Health 2017, 220, 849–856. [Google Scholar] [CrossRef]
- Lewis-Mikhael, A.M.; Olmedo-Requena, R.; Martínez-Ruiz, V.; Bueno-Cavanillas, A.; Jiménez-Moleón, J.J. Organochlorine pesticides and prostate cancer, Is there an association? A meta-analysis of epidemiological evidence. Cancer Causes Control 2015, 26, 1375–1392. [Google Scholar] [CrossRef]
- Mortensen, M.M.; Høyer, S.; Lynnerup, A.; Ørntoft, T.F.; Sørensen, K.D.; Borre, M.; Dyrskjøt, L. Expression profiling of prostate cancer tissue delineates genes associated with recurrence after prostatectomy. Sci. Rep. 2015, 5, 16018. [Google Scholar] [CrossRef] [PubMed]
- Aryee, M.J.; Liu, W.; Engelmann, J.C.; Nuhn, P.; Gurel, M.; Haffner, M.C.; Esopi, D.; Irizarry, R.A.; Getzenberg, R.H.; Nelson, W.G.; et al. DNA methylation alterations exhibit intraindividual stability and interindividual heterogeneity in prostate cancer metastases. Sci. Transl. Med. 2013, 5, 169ra10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meller, S.; Meyer, H.; Bethan, B.; Dietrich, D.; Maldonado, S.G.; Lein, M.; Montani, M.; Reszka, R.; Schatz, P.; Peter, E.; et al. Integration of tissue metabolomics, transcriptomics and immunohistochemistry reveals ERG- and gleason score-specific metabolomic alterations in prostate cancer. Oncotarget 2016, 7, 1421–1438. [Google Scholar] [CrossRef]
- Kuner, R.; Fälth, M.; Pressinotti, N.C.; Brase, J.C.; Puig, S.B.; Metzger, J.; Gade, S.; Schäfer, G.; Bartsch, G.; Steiner, E.; et al. The maternal embryonic leucine zipper kinase (MELK) is upregulated in high-grade prostate cancer. J. Mol. Med. 2012, 91, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Wang, D.; Yang, X.; Zhang, W.; Huang, Q. Cancer Omic Data Based Explainable AI Drug Recommendation Inference: A Traceability Perspective for Explainability. Biomed. Signal Process. Control. 2023, 79, 104144. [Google Scholar] [CrossRef]
- Lee, H.-R.; Jeung, E.-B.; Cho, M.-H.; Kim, T.-H.; Leung, P.C.; Choi, K.-C. Molecular Mechanism(s) of Endocrine-Disrupting Chemicals and Their Potent Oestrogenicity in Diverse Cells and Tissues That Express Oestrogen Receptors. J. Cell. Mol. Med. 2012, 17, 1–11. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007, 8, R183. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Botstein, D.; Cherry, J.M.; Ashburner, M.; Ball, C.A.; Blake, J.A.; Butler, H.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Bandettini, W.P.; Kellman, P.; Mancini, C.; Booker, O.J.; Vasu, S.; Leung, S.W.; Wilson, J.R.; Shanbhag, S.M.; Chen, M.Y.; Arai, A.E. MultiContrast Delayed Enhancement (MCODE) improves detection of subendocardial myocardial infarction by late gadolinium enhancement cardiovascular magnetic resonance: A clinical validation study. J. Cardiovasc. Magn. Reson. 2012, 14, 83. [Google Scholar] [CrossRef]
- Chin, C.; Chen, S.; Wu, H.; Ho, C.; Ko, M.; Lin, C. CytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franz, M.; Rodriguez, H.; Lopes, C.; Zuberi, K.; Montojo, J.; Bader, G.D.; Morris, Q. GeneMANIA update 2018. Nucleic Acids Res. 2018, 46, W60–W64. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Davis, A.P.; King, B.L.; Mockus, S.; Murphy, C.G.; Saraceni-Richards, C.; Rosenstein, M.; Wiegers, T.; Mattingly, C.J. The Comparative Toxicogenomics Database: Update 2011. Nucleic Acids Res. 2011, 39, D1067–D1072. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [PubMed]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. Affy—Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Deoraj, A.; Yoo, C.; Roy, D. Integrated bioinformatics biostatistics and molecular epidemiologic approaches to study how the environment and genes work together to affect the development of complex chronic diseases. In Gene-Environment Interaction Analysis: Methods in Bioinformatics and Computational Biology; Anno, S., Ed.; Pan Stanford Publishing Pte. Ltd.: New York, NY, USA, 2016; pp. 151–191. [Google Scholar]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Term | Count | p-Value | FDR |
|---|---|---|---|---|
| 1 | NFY | 57 | 6.69 × 10−3 | 9.89 × 10−1 |
| 2 | CETS1P54 | 31 | 2.88 × 10−3 | 9.89 × 10−1 |
| 3 | OLF1 | 50 | 3.99 × 10−2 | 9.89 × 10−1 |
| 4 | SRF | 73 | 6.28 × 10−2 | 9.89 × 10−1 |
| 5 | COMP1 | 52 | 8.19 × 10−2 | 9.89 × 10−1 |
| 6 | RP58 | 27 | 6.91 × 10−1 | 1.3 × 10−2 |
| 7 | HMX1 | 21 | 1.23 × 10−1 | 1.99 × 10−2 |
| 8 | NF1 | 30 | 1.32 × 10−1 | 1.99 × 10−2 |
| 9 | PPARA | 23 | 1.68 × 10−1 | 2.25 × 10−2 |
| 10 | GFI1 | 22 | 6.7910−1 | 7.61 × 10−2 |
| # | Term | Count | p-Value | FDR |
|---|---|---|---|---|
| 1 | MYCMAX | 133 | 5.35 × 10−6 | 4.07 × 10−4 |
| 2 | PAX4 | 179 | 7.63 × 10−6 | 4.07 × 10−4 |
| 3 | PAX5 | 138 | 1.26 × 10−5 | 4.07 × 10−4 |
| 4 | USF | 142 | 1.27 × 10−5 | 4.07 × 10−4 |
| 5 | NRSF | 149 | 2.76 × 10−5 | 6.44 × 10−4 |
| 6 | HEN1 | 140 | 3.02 × 10−5 | 6.44 × 10−4 |
| 7 | P300 | 97 | 4.28 × 10−5 | 7.83 × 10−4 |
| 8 | MAZR | 61 | 1.55 × 10−4 | 2.37 × 10−3 |
| 9 | AP4 | 148 | 1.83 × 10−4 | 2.37 × 10−3 |
| 10 | NMYC | 82 | 1.94 × 10−4 | 2.37 × 10−3 |
| Chemical Name | Group | EDCs/Carcinogenic | |
|---|---|---|---|
| 1 | Arsenic | Heavy metals | EDCs |
| 2 | Copper | EDCs | |
| 3 | Cadmium | EDCs | |
| 4 | Zinc | EDCs | |
| 5 | Benzo(a)pyrene | Polycyclic aromatic hydrocarbons (PAH) | EDCs |
| 6 | Benzophenone-3 | Environmental phenols | EDCs |
| 7 | Bisphenol A | EDCs | |
| 8 | Methylparaben | EDCs | |
| 9 | Propylparaben | EDCs | |
| 10 | Sodium arsenate | Inorganic compounds | Carcinogenic |
| 11 | Copper sulfate | No | |
| 12 | Dietary fats | Type of nutrient | No |
| 13 | Diethylstilbestrol | Synthetic (manufactured) form of estrogen | EDCs |
| 14 | Dihydrotestosterone | Steroid hormone | No |
| 15 | Testosterone | No | |
| 16 | Estradiol | Estrogenic steroid | EDCs |
| 17 | Genistein | Polyphenolic isoflavone | EDCs |
| 18 | DDT | Pesticides | EDCs |
| 19 | Heptachlor | EDCs | |
| 20 | Aldrin | EDCs | |
| 21 | Chlordecone | EDCs | |
| 22 | Phthalates | Polyvinyl chloride (PVC)/plasticizers | EDCs |
| GEO Profile | Case | Control | Platform | Annotation Platform | References |
|---|---|---|---|---|---|
| GSE46602 | 36 | 14 | GPL570 | Affymetrix Human Genome U133 Plus 2.0 Array | [93] |
| GSE38241 | 18 | 21 | GPL4133 | Agilent-014850 Whole Human Genome Microarray | [94] |
| GSE69223 | 15 | 15 | GPL570 | Affymetrix Human Genome U133 Plus 2.0 Array | [95] |
| GSE32571 | 95 | 39 | GPL6947 | Illumina HumanHT-12 V3.0 expression BeadChip | [96] |
| GSE55945 | 4 | 4 | GPL570 | Affymetrix Human Genome U133 Plus 2.0 Array | [33] |
| GSE26126 | 95 | 98 | GPL8490 | Illumina HumanMethylation27 BeadChip | [34] |
| Total | 227 | 191 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alwadi, D.; Felty, Q.; Yoo, C.; Roy, D.; Deoraj, A. Endocrine Disrupting Chemicals Influence Hub Genes Associated with Aggressive Prostate Cancer. Int. J. Mol. Sci. 2023, 24, 3191. https://doi.org/10.3390/ijms24043191
Alwadi D, Felty Q, Yoo C, Roy D, Deoraj A. Endocrine Disrupting Chemicals Influence Hub Genes Associated with Aggressive Prostate Cancer. International Journal of Molecular Sciences. 2023; 24(4):3191. https://doi.org/10.3390/ijms24043191
Chicago/Turabian StyleAlwadi, Diaaidden, Quentin Felty, Changwon Yoo, Deodutta Roy, and Alok Deoraj. 2023. "Endocrine Disrupting Chemicals Influence Hub Genes Associated with Aggressive Prostate Cancer" International Journal of Molecular Sciences 24, no. 4: 3191. https://doi.org/10.3390/ijms24043191
APA StyleAlwadi, D., Felty, Q., Yoo, C., Roy, D., & Deoraj, A. (2023). Endocrine Disrupting Chemicals Influence Hub Genes Associated with Aggressive Prostate Cancer. International Journal of Molecular Sciences, 24(4), 3191. https://doi.org/10.3390/ijms24043191