
Ubiquitin-Specific Proteases (USPs) and Metabolic Disorders


Abstract
:
1. Introduction
2. Obesity and Adipogenesis
3. Diabetes Mellitus and Insulin Resistance
3.1. T1DM
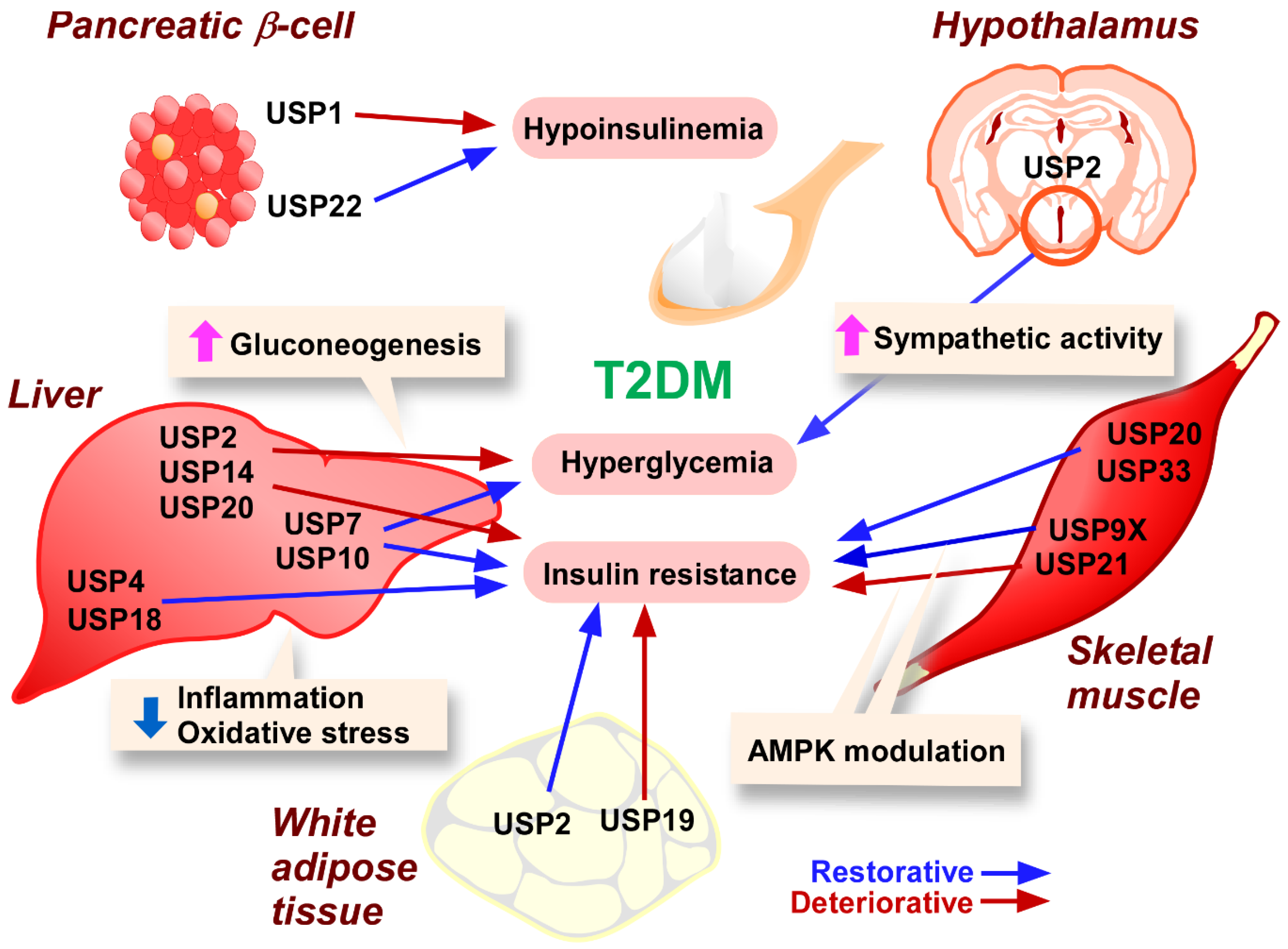
3.2. T2DM
3.2.1. Pancreatic β-Cells
3.2.2. Adipose Tissue
3.2.3. Skeletal Muscle
3.2.4. Liver
3.2.5. Hypothalamus
4. Diabetes-Associated Disorders
4.1. Nephropathy
4.2. Retinopathy
4.3. Neuropathy
4.4. Myopathy
4.5. Cardiomyopathy
4.6. Foot Ulcers
5. NAFLD
6. Atherosclerosis
7. Cushing Disease
8. Perspectives
9. Short Summary and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | Acetyl-CoA carboxylase |
| ACLY | ATP citrate synthase |
| ACTH | Adrenocorticotropic hormone |
| AGE | Advanced glycation end product |
| AMPK | AMP-activated kinase |
| ApoE | Apolipoprotein E |
| ATF4 | Activating transcription factor 4 |
| ATG | Autophagy target gene |
| BAX | Bcl2-associated X protein |
| Bcl-2 | B-cell/CLL lymphoma 2 |
| BMAL-1 | Brain and muscle arnt-like 1 |
| CBP | 3′,5′-cyclic monophosphate-responsive element binding protein |
| C/EBP | CCAAT-enhancer-binding protein |
| CHOP | C/EBP homologous protein |
| CREB | cAMP-responsive element binding protein |
| CRL | Cullin RING ubiquitin ligase |
| CSN | COP9 signalosome |
| DIM | 3′3-Diindolylmethane |
| DNA-PKcs | DNA-dependent protein kinase catalytic subunit |
| DOCK4 | Dedicator of cytokinesis 4 |
| DUB | Deubiquitinating enzyme |
| ECM | Extracellular matrix |
| EDL | Extensor digitorum longus |
| EMT | Epithelial-to-mesenchymal transition |
| ER | Endoplasmic reticulum |
| ERAP1 | Endoplasmic reticulum aminopeptidase 1 |
| ERK | Extracellular signal-regulated kinase |
| FABP4 | Fatty acid binding protein 4 |
| FASN | Fatty acid synthase |
| FoxP3 | Forkhead box P3 |
| FSTL1 | Follistatin-like protein 1 |
| GATA1 | GATA binding protein 1 |
| GLUT | Glucose transporter |
| GM-CSF | Granulocyte macrophage-colony stimulating factor |
| G6PC | Glucose-6-phosphatase catalytic subunit |
| HAUSP | Herpesvirus-associated ubiquitin-specific protease |
| HCC | Hepatocellular carcinoma |
| HDL | High-density lipoprotein |
| HIF1α | Hypoxia-inducible factor 1α |
| HMGCR | HMG-CoA reductase |
| HOMA-IR | Homeostasis model assessment-insulin resistance |
| HOX1 | Heme oxygenase-1 |
| HSD1 | 11β-hydroxysteroid dehydrogenase 1 |
| HUVEC | Human umbilical vein endothelial cell |
| IDOL | Inducible degrader of the LDLR |
| IFN | Interferon |
| IκB-α | Nuclear factor of κ light polypeptide gene enhancer in B-cells inhibitor-α |
| IL | Interleukin |
| IRS1 | Insulin receptor substrate 1 |
| JAK | Janus kinase |
| JAMM | JAB1/MPN/Mov34 metalloproteases |
| JNK | c-Jun N-terminal kinase |
| KO | Knockout |
| KLF4 | Krüppel-like factor 4 |
| LC3B | Microtubule-associated protein light chain 3B-1 |
| LDL | Low-density lipoprotein |
| LDLR | LDL receptor |
| LKB1 | Liver kinase B1 |
| LncRNA | Long non-coding RNA |
| LPS | Lipopolysaccharide |
| MAFLD | Metabolic-dysfunction-associated fatty liver disease |
| MAPK | Mitogen-activated kinase |
| MAT | Methionine adenosyltransferase |
| MCDD | Methionine and choline-deficient diet |
| MCPIP | Monocyte chemotactic protein-induced protein |
| MDA5 | Melanoma differentiation-associated protein 5 |
| Mdm2 | Murine double minute 2 |
| MINDY | Motif interacting with ubiquitin-containing novel DUB |
| miR-320 | Micro RNA-320 |
| MJD | Machado-Joseph disease protein domain protease |
| mTOR | Mammalian target of rapamycin |
| Myh | Myosin heavy chain |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NAFL | Non-alcoholic fatty liver |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NICD1 | Noch1 intracellular domain 1 |
| NLRC5 | NOD-like receptor family cascade recruitment domain family domain containing 5 |
| NQO1 | NAD(P)H: quinone oxidoreductase 1 |
| NF-κB | Nuclear factor of κ light polypeptide gene enhancer in B-cells |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| OTU | Ovarian tumor domain-containing proteases |
| PCK | Phosphoenolpyruvate carboxykinase 1 |
| PD | Primaquine diphosphate |
| PDGF-BB | Platelet-derived growth factor-BB |
| PDK1 | 3-Phosphoinositide-dependent protein kinase 1 |
| PGC | Peroxisome proliferator-activated receptor γ coactivator |
| PI3K | Phosphatidyl inositol 3 kinase |
| PiT1 | Phosphate inorganic transporter 1 |
| PKA | cAMP-dependent kinase |
| POMC | Proopiomelanocortin |
| PPARγ | Peroxisome proliferator-activated receptor γ |
| PRDX3 | Peroxiedoxin-3 |
| PSMD | 26S Proteasome non-ATPase regulatory subunit |
| RAC1 | RAS-related C3 botulinus toxin substrate 1 |
| RAGE | Receptor for AGEs |
| RIPK | Receptor-interacting protein kinase 1 |
| ROS | Reactive oxygen species |
| shRNA | Short hairpin RNA |
| Sirt | Sirtuin |
| SORT | Sterol O-acetyltransferase |
| SPAG5 | Sperm-associated antigen 5 |
| SPAG-AS1 | SPAG-antisense1 |
| SRA-1 | Class A1 scavenger receptor |
| SREBP1c | Sterol regulatory element binding protein 1c |
| STAT | Signal transducer and activator of transcription |
| STZ | Streptozotocin |
| TAK1 | Transforming growth factor-β activated kinase 1 |
| T1DM | Type 1 Diabetes Mellitus |
| T2DM | Type 2 Diabetes Mellitus |
| TEC | Tubular epithelial cell |
| TGF-β | Transforming growth factor-β |
| TNF-α | Tumor necrosis factor-α |
| TRAF6 | Tumor necrosis factor receptor-associated factor 6 |
| Treg | Regulatory T-cell |
| UBA | Ubiquitin-associated domain |
| UCH | Ubiquitin C-terminal hydrolase |
| UIM | Ubiquitin-interacting motif |
| UPS | Ubiquitin–proteasome system |
| USP | Ubiquitin-specific protease |
| VCAM1 | Vascular cell adhesion molecule 1 |
| VEGF | Vascular endothelial cell growth factor |
| VMH | Ventromedial hypothalamus |
| VLDL | Very low-density lipoprotein |
| WT-1 | William tumor-1 |
| ZNF638 | Zinc finger protein 638 |
| ZnF-UBP | Zinc finger ubiquitin-specific protease domain |
| ZUFSP | Zinc finger and UFSP domain protein |
References
- Huang, P.L. A comprehensive definition for metabolic syndrome. Dis. Model. Mech. 2009, 2, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Neuhauser, H.K. The metabolic syndrome. Lancet 2005, 366, 1415–1428. [Google Scholar] [CrossRef]
- Ahmed, M.; Kumari, N.; Mirgani, Z.; Saeed, A.; Ramadan, A.; Ahmed, M.H.; Almobarak, A.O. Metabolic syndrome; Definition, Pathogenesis, Elements, and the Effects of medicinal plants on it’s elements. J. Diabetes Metab. Disord. 2022, 21, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Powell-Wiley, T.M.; Poirier, P.; Burke, L.E.; Després, J.P.; Gordon-Larsen, P.; Lavie, C.J.; Lear, S.A.; Ndumele, C.E.; Neeland, I.J.; Sanders, P.; et al. Obesity and Cardiovascular Disease A Scientific Statement From the American Heart Association. Circulation 2021, 143, E984–E1010. [Google Scholar] [CrossRef] [PubMed]
- Sivan, E. Free Fatty Acids and Insulin Resistance during Pregnancy. J. Clin. Endocrinol. Metab. 1998, 83, 2338–2342. [Google Scholar] [CrossRef]
- Dalen, J.E.; Alpert, J.S.; Goldberg, R.J.; Weinstein, R.S. The epidemic of the 20th century: Coronary heart disease. Am. J. Med. 2014, 127, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Lawes, C.M.; Bennett, D.A.; Barker-Collo, S.L.; Parag, V. Worldwide stroke incidence and early case fatality reported in 56 population-based studies: A systematic review. Lancet Neurol. 2009, 8, 355–369. [Google Scholar] [CrossRef]
- Srinivasan, V.A.R.; Raghavan, V.A.; Parthasarathy, S. Biochemical Basis and Clinical Consequences of Glucolipotoxicity: A Primer. Heart Fail. Clin. 2012, 8, 501–511. [Google Scholar] [CrossRef]
- Delarue, J.; Magnan, C. Free fatty acids and insulin resistance. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 142–148. [Google Scholar] [CrossRef]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [Green Version]
- McCracken, E.; Monaghan, M.; Sreenivasan, S. Pathophysiology of the metabolic syndrome. Clin. Dermatol. 2018, 36, 14–20. [Google Scholar] [CrossRef]
- Myers, M.G.; Leibel, R.L.; Seeley, R.J.; Schwartz, M.W. Obesity and leptin resistance: Distinguishing cause from effect. Trends Endocrinol. Metab. 2010, 21, 643–651. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Kazamel, M.; Stino, A.M.; Smith, A.G. Metabolic syndrome and peripheral neuropathy. Muscle Nerve 2021, 63, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Otsu, K. Inflammation andmetabolic cardiomyopathy. Cardiovasc. Res. 2017, 113, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Palomer, X.; Salvadó, L.; Barroso, E.; Vázquez-Carrera, M. An overview of the crosstalk between inflammatory processes and metabolic dysregulation during diabetic cardiomyopathy. Int. J. Cardiol. 2013, 168, 3160–3172. [Google Scholar] [CrossRef]
- Nosalski, R.; Guzik, T.J. Perivascular adipose tissue inflammation in vascular disease. Br. J. Pharmacol. 2017, 174, 3496–3513. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Invest. 2017, 127, 1–4. [Google Scholar] [CrossRef]
- Cantó, C.; Auwerx, J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Prentice, K.J.; Saksi, J.; Hotamisligil, G.S. Adipokine FABP4 integrates energy stores and counterregulatory metabolic responses. J. Lipid Res. 2019, 60, 734–740. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Ballantyne, C.M. Metabolic Inflammation and Insulin Resistance in Obesity. Circ. Res. 2020, 126, 1549–1564. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, R.A.; Weeks, K.L. Histone deacetylases in cardiovascular and metabolic diseases. J. Mol. Cell. Cardiol. 2019, 130, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Viscarra, J.; Sul, H.S. Epigenetic regulation of hepatic lipogenesis: Role in hepatosteatosis and diabetes. Diabetes 2020, 69, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Shi, Z.; Ji, X.; Zhang, W.; Luan, J.; Zahr, T.; Qiang, L. Adipokines, adiposity, and atherosclerosis. Cell. Mol. Life Sci. 2022, 79, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Priest, C.; Tontonoz, P. Inter-organ cross-talk in metabolic syndrome. Nat. Metab. 2019, 1, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed]
- Hopps, E.; Caimi, G. Protein oxidation in metabolic syndrome. Clin. Investig. Med. 2013, 36, 1–8. [Google Scholar] [CrossRef]
- Ruan, H.B.; Nie, Y.; Yang, X. Regulation of protein degradation by O-GlcNAcylation: Crosstalk with ubiquitination. Mol. Cell. Proteom. 2013, 12, 3489–3497. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Bae, J.S.; Kim, T.H.; Park, J.M.; Ahn, Y.H. Role of transcription factor modifications in the pathogenesis of insulin resistance. Exp. Diabetes Res. 2012, 2012, 1–16. [Google Scholar] [CrossRef]
- Cruz, L.; Soares, P.; Correia, M. Ubiquitin-specific proteases: Players in cancer cellular processes. Pharmaceuticals 2021, 14, 848. [Google Scholar] [CrossRef]
- Sun, J.; Shi, X.; Mamun, M.; Gao, Y. The role of deubiquitinating enzymes in gastric cancer (Review). Oncol. Lett. 2019, 19, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Yan, F.; Ying, M.; Cao, J.; He, Q.; Zhu, H.; Yang, B. Inhibition of Ubiquitin-Specific Proteases as a Novel Anticancer Therapeutic Strategy. Front. Pharmacol. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Liu, Y.; Zhou, H. Advances in the Development Ubiquitin-Specific Peptidase (USP) Inhibitors. Int. J. Mol. Sci. 2021, 22, 4546. [Google Scholar] [CrossRef]
- Hu, B.; Zhang, D.; Zhao, K.; Wang, Y.; Pei, L.; Fu, Q.; Ma, X. Spotlight on USP4: Structure, Function, and Regulation. Front. Cell Dev. Biol. 2021, 9, 1–15. [Google Scholar] [CrossRef]
- Ning, F.; Xin, H.; Liu, J.; Lv, C.; Xu, X.; Wang, M.; Wang, Y.; Zhang, W.; Zhang, X. Structure and function of USP5: Insight into physiological and pathophysiological roles. Pharmacol. Res. 2020, 157, 104557. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Gao, Y.; Zhou, L.; Chen, J.; Xie, Z.; Ye, Z.; Wang, Y. USP30: Structure, Emerging Physiological Role, and Target Inhibition. Front. Pharmacol. 2022, 13, 1–13. [Google Scholar] [CrossRef]
- Kitamura, H.; Hashimoto, M. USP2-related cellular signaling and consequent pathophysiological outcomes. Int. J. Mol. Sci. 2021, 22, 1209. [Google Scholar] [CrossRef] [PubMed]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef] [PubMed]
- Jung, U.J.; Choi, M.S. Obesity and its metabolic complications: The role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 6184–6223. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, F.M.; Smas, C.M.; Sul, H.S. Understanding adipocyte differentiation. Physiol. Rev. 1998, 78, 783–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ron, D.; Habener, J.F. CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev. 1992, 6, 439–453. [Google Scholar] [CrossRef]
- Huang, X.; Ordemann, J.; Müller, J.M.; Dubiel, W. The COP9 signalosome, cullin 3 and Keap1 supercomplex regulates CHOP stability and adipogenesis. Biol. Open 2012, 1, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, N.F.; Tian, W.; Wu, T.; Sun, Z.; Lau, A.; Chapman, E.; Fang, D.; Zhang, D.D. USP15 negatively regulates Nrf2 through Deubiquitination of Keap1. Mol. Cell 2013, 51, 68–79. [Google Scholar] [CrossRef]
- Yu, K.; Mo, D.; Wu, M.; Chen, H.; Chen, L.; Li, M.; Chen, Y. Activating transcription factor 4 regulates adipocyte differentiation via altering the coordinate expression of CCATT/enhancer binding protein β and peroxisome proliferator-activated receptor γ. FEBS J. 2014, 281, 2399–2409. [Google Scholar] [CrossRef]
- Chen, H.; Yuan, R.; Zhang, Y.; Zhang, X.; Chen, L.; Zhou, X.; Yuan, Z.; Nie, Y.; Li, M.; Mo, D.; et al. ATF4 regulates SREBP1c expression to control fatty acids synthesis in 3T3-L1 adipocytes differentiation. Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 1459–1469. [Google Scholar] [CrossRef]
- Yang, H.; Seo, S.G.; Shin, S.H.; Min, S.; Kang, M.J.; Yoo, R.; Kwon, J.Y.; Yue, S.; Kim, K.H.; Cheng, J.X.; et al. 3,3′-Diindolylmethane suppresses high-fat diet-induced obesity through inhibiting adipogenesis of pre-adipocytes by targeting USP2 activity. Mol. Nutr. Food Res. 2017, 61, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Zhao, W.; Gu, W. Suppression of Cancer Cell Growth by Promoting Cyclin D1 Degradation. Mol. Cell 2009, 36, 469–476. [Google Scholar] [CrossRef]
- Graner, E.; Tang, D.; Rossi, S.; Baron, A.; Migita, T.; Weinstein, L.J.; Lechpammer, M.; Huesken, D.; Zimmermann, J.; Signoretti, S.; et al. The isopeptidase USP2a regulates the stability of fatty acid synthase in prostate cancer. Cancer Cell 2004, 5, 253–261. [Google Scholar] [CrossRef]
- Coyne, E.S.; Bédard, N.; Gong, Y.J.; Faraj, M.; Tchernof, A.; Wing, S.S. The deubiquitinating enzyme USP19 modulates adipogenesis and potentiates high-fat-diet-induced obesity and glucose intolerance in mice. Diabetologia 2019, 62, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.Y.; Shi, X.J.; Hu, A.; Wang, J.Q.; Ding, Y.; Jiang, W.; Sun, M.; Zhao, X.; Luo, J.; Qi, W.; et al. Feeding induces cholesterol biosynthesis via the mTORC1–USP20–HMGCR axis. Nature 2020, 588, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Pierce, K.A.; Jedrychowski, M.P.; Garrity, R.; Winther, S.; Vidoni, S.; Yoneshiro, T.; Spinelli, J.B.; Lu, G.Z.; Kazak, L.; et al. Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature 2018, 560, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Lin, L.; Li, Q.; Xue, Y.; Zheng, F.; Wang, G.; Zheng, C.; Du, L.; Hu, M.; Huang, Y.; et al. Scd1 controls de novo beige fat biogenesis through succinate-dependent regulation of mitochondrial complex II. Proc. Natl. Acad. Sci. USA 2020, 117, 2462–2472. [Google Scholar] [CrossRef] [PubMed]
- Bolton, J.; Montastier, E.; Carayol, J.; Bonnel, S.; Mir, L.; Marques, M.A.; Astrup, A.; Saris, W.; Iacovoni, J.; Villa-Vialaneix, N.; et al. Molecular biomarkers for weight control in obese individuals subjected to a multiphase dietary intervention. J. Clin. Endocrinol. Metab. 2017, 102, 2751–2761. [Google Scholar] [CrossRef] [PubMed]
- Petersmann, A.; Müller-Wieland, D.; Müller, U.A.; Landgraf, R.; Nauck, M.; Freckmann, G.; Heinemann, L.; Schleicher, E. Definition, Classification and Diagnosis of Diabetes Mellitus. Exp. Clin. Endocrinol. Diabetes 2019, 127, S1–S7. [Google Scholar] [CrossRef] [PubMed]
- Katsarou, A.; Gudbjörnsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, A. Type 1 diabetes mellitus. Nat. Rev. Dis. Prim. 2017, 3, 1–18. [Google Scholar] [CrossRef]
- Cortez, J.T.; Montauti, E.; Shifrut, E.; Gatchalian, J.; Zhang, Y.; Shaked, O.; Xu, Y.; Roth, T.L.; Simeonov, D.R.; Zhang, Y.; et al. CRISPR screen in regulatory T cells reveals modulators of Foxp3. Nature 2020, 582, 416–420. [Google Scholar] [CrossRef]
- Lee, G.R. The balance of th17 versus treg cells in autoimmunity. Int. J. Mol. Sci. 2018, 19, 730. [Google Scholar] [CrossRef]
- Santin, I.; Moore, F.; Grieco, F.A.; Marchetti, P.; Brancolini, C.; Eizirik, D.L. USP18 is a key regulator of the interferon-driven gene network modulating pancreatic beta cell inflammation and apoptosis. Cell Death Dis. 2012, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martín, C. Pathophysiology of type 2 diabetes mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef] [PubMed]
- Inaishi, J.; Saisho, Y. Beta-cell mass in obesity and type 2 diabetes, and its relation to pancreas fat: A mini-review. Nutrients 2020, 12, 3846. [Google Scholar] [CrossRef]
- Abdul, M.; Khan, B.; Hashim, M.J.; King, J.K.; Govender, R.D.; Mustafa, H.; Kaabi, J. Al Epidemiology of type 2 diabetes—Global burden of disease and forecasted trends. J. Epidemiol. Glob. Health 2020, 10, 107–111. [Google Scholar]
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef]
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.N.; Chen, H.; Li, Y.; Zhou, L.; McGibbon, M.A.; Taylor, S.I.; Quon, M.J. Physiological role of AKT in insulin-stimulated translocation of GLUT4 in transfected rat adipose cells. Mol. Endocrinol. 1997, 11, 1881–1890. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Tang, Z.; Shen, A.; Tao, T.; Wan, C.; Zhu, X.; Huang, J.; Zhang, W.; Xia, N.; Wang, S.; et al. The role of PTP1B O-GlcNAcylation in hepatic insulin resistance. Int. J. Mol. Sci. 2015, 16, 22856–22869. [Google Scholar] [CrossRef]
- Kaneto, H.; Katakami, N.; Matsuhisa, M.; Matsuoka, T.A. Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediators Inflamm. 2010, 2010, 1–11. [Google Scholar] [CrossRef]
- Newsholme, P.; Keane, K.N.; Carlessi, R.; Cruzat, V. Oxidative stress pathways in pancreatic β-cells and insulin-sensitive cells and tissues: Importance to cell metabolism, function, and dysfunction. Am. J. Physiol. Cell Physiol. 2019, 317, C420–C433. [Google Scholar] [CrossRef] [PubMed]
- Elumalai, S.; Karunakaran, U.; Moon, J.S.; Won, K.C. High glucose-induced PRDX3 acetylation contributes to glucotoxicity in pancreatic β-cells: Prevention by Teneligliptin. Free Radic. Biol. Med. 2020, 160, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Gorrepati, K.D.D.; Lupse, B.; Annamalai, K.; Yuan, T.; Maedler, K.; Ardestani, A. Loss of Deubiquitinase USP1 Blocks Pancreatic β-Cell Apoptosis by Inhibiting DNA Damage Response. iScience 2018, 1, 72–86. [Google Scholar] [CrossRef]
- Tobe, K.; Nemoto, S.; Kadowaki, T. Molecular mechanism of insulin resistance. Nippon. Rinsho. Jpn. J. Clin. Med. 2005, 63 (Suppl. S2), 114–130. [Google Scholar]
- Lee, Y.S.; Olefsky, J. Chronic tissue inflammation and metabolic disease. Genes Dev. 2021, 35, 307–328. [Google Scholar] [CrossRef] [PubMed]
- Kahn, C.R.; Wang, G.; Lee, K.Y. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J. Clin. Invest. 2019, 129, 3990–4000. [Google Scholar] [CrossRef]
- Kiran, S.; Kumar, V.; Kumar, S.; Price, R.L.; Singh, U.P. Adipocyte, immune cells, and mirna crosstalk: A novel regulator of metabolic dysfunction and obesity. Cells 2021, 10, 1004. [Google Scholar] [CrossRef] [PubMed]
- Strissel, K.J.; Stancheva, Z.; Miyoshi, H.; Perfield, J.W.; DeFuria, J.; Jick, Z.; Greenberg, A.S.; Obin, M.S. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 2007, 56, 2910–2918. [Google Scholar] [CrossRef] [PubMed]
- Rotter, V.; Nagaev, I.; Smith, U. Interleukin-6 (IL-6) Induces Insulin Resistance in 3T3-L1 Adipocytes and Is, Like IL-8 and Tumor Necrosis Factor-α, Overexpressed in Human Fat Cells from Insulin-resistant Subjects. J. Biol. Chem. 2003, 278, 45777–45784. [Google Scholar] [CrossRef] [PubMed]
- Krogh-Madsen, R.; Plomgaard, P.; Møller, K.; Mittendorfer, B.; Pedersen, B.K. Influence of TNF-α and IL-6 infusions on insulin sensitivity and expression of IL-18 in humans. Am. J. Physiol. Endocrinol. Metab. 2006, 291, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, H.; Ishino, T.; Shimamoto, Y.; Okabe, J.; Miyamoto, T.; Takahashi, E.; Miyoshi, I. Ubiquitin-specific protease 2 modulates the lipopolysaccharide-elicited expression of proinflammatory cytokines in macrophage-like HL-60 cells. Mediators Inflamm. 2017, 2017, 6909415. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, H.; Kimura, S.; Shimamoto, Y.; Okabe, J.; Ito, M.; Miyamoto, T.; Naoe, Y.; Kikuguchi, C.; Meek, B.; Toda, C.; et al. Ubiquitin-specific protease 2ߝ69 in macrophages potentially modulates metainflammation. FASEB J. 2013, 27, 4940–4953. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Kimura, S.; Miyamoto, T.; Fukushima, S.; Amagasa, M.; Shimamoto, Y.; Nishioka, C.; Okamoto, S.; Toda, C.; Washio, K.; et al. Macrophage ubiquitin-specific protease 2 modifies insulin sensitivity in obese mice. Biochem. Biophys. Rep. 2017, 9, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Chadt, A.; Al-Hasani, H. Glucose transporters in adipose tissue, liver, and skeletal muscle in metabolic health and disease. Pflugers Arch. Eur. J. Physiol. 2020, 472, 1273–1298. [Google Scholar] [CrossRef]
- Blaauw, B.; Schiaffino, S.; Reggiani, C. Mechanisms modulating skeletal muscle phenotype. Compr. Physiol. 2013, 3, 1645–1687. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.Y.; Luo, J.; Lei, H.G.; Jiang, Y.Z.; Bai, L.; Li, M.Z.; Tang, G.Q.; Li, X.W.; Zhang, S.H.; Zhu, L. Effects of muscle fiber type on glycolytic potential and meat quality traits in different tibetan pig muscles and their association with glycolysis-related gene expression. Genet. Mol. Res. 2015, 14, 14366–14378. [Google Scholar] [CrossRef]
- Moriggi, M.; Belloli, S.; Barbacini, P.; Murtaj, V.; Torretta, E.; Chaabane, L.; Canu, T.; Penati, S.; Malosio, M.L.; Esposito, A.; et al. Skeletal muscle proteomic profile revealed gender-related metabolic responses in a diet-induced obesity animal model. Int. J. Mol. Sci. 2021, 22, 4680. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Koo, J.H.; Jin, X.; Kim, W.; Park, S.Y.; Park, S.; Rhee, E.P.; Choi, C.S.; Kim, S.G. Ablation of USP21 in skeletal muscle promotes oxidative fibre phenotype, inhibiting obesity and type 2 diabetes. J. Cachexia. Sarcopenia Muscle 2021, 12, 1669–1689. [Google Scholar] [CrossRef]
- Madeo, F.; Carmona-Gutierrez, D.; Hofer, S.J.; Kroemer, G. Caloric Restriction Mimetics against Age-Associated Disease: Targets, Mechanisms, and Therapeutic Potential. Cell Metab. 2019, 29, 592–610. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Bang, E.; Jung, H.J.; Noh, S.G.; Yu, B.P.; Choi, Y.J.; Chung, H.Y. Anti-aging effects of calorie restriction (CR) and CR mimetics based on the senoinflammation concept. Nutrients 2020, 12, 422. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, R.Y.; Song, J.; Guan, Y.F.; Xu, T.Y.; Du, H.; Viollet, B.; Miao, C.Y. Loss of AMP-activated protein kinase-α2 impairs the insulin-sensitizing effect of calorie restriction in skeletal muscle. Diabetes 2012, 61, 1051–1061. [Google Scholar] [CrossRef]
- Gancheva, S.; Jelenik, T.; Álvarez-Hernández, E.; Roden, M. Interorgan metabolic crosstalk in human insulin resistance. Physiol. Rev. 2018, 98, 1371–1415. [Google Scholar] [CrossRef]
- Sell, H.; Eckel, J.; Dietze-Schroeder, D. Pathways leading to muscle insulin resistance—The muscle-fat connection. Arch. Physiol. Biochem. 2006, 112, 105–113. [Google Scholar] [CrossRef]
- Lan, F.; Misu, H.; Chikamoto, K.; Takayama, H.; Kikuchi, A.; Mohri, K.; Takata, N.; Hayashi, H.; Matsuzawa-Nagata, N.; Takeshita, Y.; et al. LECT2 functions as a hepatokine that links obesity to skeletal muscle insulin resistance. Diabetes 2014, 63, 1649–1664. [Google Scholar] [CrossRef]
- Plomgaard, P.; Nielsen, A.R.; Fischer, C.P.; Mortensen, O.H.; Broholm, C.; Penkowa, M.; Krogh-Madsen, R.; Erikstrup, C.; Lindegaard, B.; Petersen, A.M.W.; et al. Associations between insulin resistance and TNF-α in plasma, skeletal muscle and adipose tissue in humans with and without type 2 diabetes. Diabetologia 2007, 50, 2562–2571. [Google Scholar] [CrossRef]
- Hadcock, J.R.; Maibon, C.C. Regulation of receptor expression by agonists: Transcriptional and post-transcriptional controls. Trends Neurosci. 1991, 14, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Brennesvik, E.O.; Ktori, C.; Ruzzin, J.; Jebens, E.; Shepherd, P.R.; Jensen, J. Adrenaline potentiates insulin-stimulated PKB activation via cAMP and Epac: Implications for cross talk between insulin and adrenaline. Cell. Signal. 2005, 17, 1551–1559. [Google Scholar] [CrossRef]
- Berthouze, M.; Venkataramanan, V.; Li, Y.; Shenoy, S.K. The deubiquitinases USP33 and USP20 coordinate Β2 adrenergic receptor recycling and resensitization. EMBO J. 2009, 28, 1684–1696. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Jiang, H.; Yin, H.; Wang, F.; Hu, R.; Hu, X.; Peng, B.; Shu, Y.; Li, Z.; Chen, S.; et al. Hepatokine ERAP1 Disturbs Skeletal Muscle Insulin Sensitivity Via Inhibiting USP33-Mediated ADRB2 Deubiquitination. Diabetes 2022, 71, 921–933. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira Dos Santos, A.R.; Zanuso, B.D.O.; Miola, V.F.B.; Barbalho, S.M.; Santos Bueno, P.C.; Flato, U.A.P.; Detregiachi, C.R.P.; Buchaim, D.V.; Buchaim, R.L.; Tofano, R.J.; et al. Adipokines, myokines, and hepatokines: Crosstalk and metabolic repercussions. Int. J. Mol. Sci. 2021, 22, 2639. [Google Scholar] [CrossRef]
- Rui, L. Energy Metabolism in the Liver. In Comprehensive Physiology; Wiley & Sons: Hoboken, NJ, USA, 2014; Volume 176, pp. 177–197. [Google Scholar]
- Di Dalmazi, G.; Pagotto, U.; Pasquali, R.; Vicennati, V. Glucocorticoids and type 2 diabetes: From physiology to pathology. J. Nutr. Metab. 2012, 2012, 1–9. [Google Scholar] [CrossRef]
- Forand, A.; Koumakis, E.; Rousseau, A.; Sassier, Y.; Journe, C.; Merlin, J.F.; Leroy, C.; Boitez, V.; Codogno, P.; Friedlander, G.; et al. Disruption of the Phosphate Transporter Pit1 in Hepatocytes Improves Glucose Metabolism and Insulin Signaling by Modulating the USP7/IRS1 Interaction. Cell Rep. 2016, 16, 2736–2748. [Google Scholar] [CrossRef]
- Lee, K.W.; Cho, J.G.; Kim, C.M.; Kang, A.Y.; Kim, M.; Ahn, B.Y.; Chung, S.S.; Lim, K.H.; Baek, K.H.; Sung, J.H.; et al. Herpesvirus-associated ubiquitin-specific protease (HAUSP) modulates peroxisome proliferator-activated receptor γ (PPARγ) stability through its deubiquitinating activity. J. Biol. Chem. 2013, 288, 32886–32896. [Google Scholar] [CrossRef]
- Hall, J.A.; Tabata, M.; Rodgers, J.T.; Puigserver, P. USP7 attenuates hepatic gluconeogenesis through modulation of FoxO1 gene promoter occupancy. Mol. Endocrinol. 2014, 28, 912–924. [Google Scholar] [CrossRef]
- Molusky, M.M.; Li, S.; Ma, D.; Yu, L.; Lin, J.D. Ubiquitin-specific protease 2 regulates hepatic gluconeogenesis and diurnal glucose metabolism through 11β-hydroxysteroid dehydrogenase 1. Diabetes 2012, 61, 1025–1035. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, Z.; Hu, Y.; Lu, Y.; Li, D.; Liu, J.; Liao, S.; Hu, M.; Wang, Y.; Zhang, D.; et al. Sustained ER stress promotes hyperglycemia by increasing glucagon action through the deubiquitinating enzyme USP14. Proc. Natl. Acad. Sci. USA 2019, 116, 21732–21738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, C.B.; Takahashi, J.S.; Bass, J. The Meter of Metabolism. Cell 2008, 134, 728–742. [Google Scholar] [CrossRef]
- Kida, K.; Nishio, T.; Yokozawa, T.; Nagai, K.; Matsuda, H.; Nakagawa, H. The circadian change of gluconeogenesis in the liver: In vivo in fed rats. J. Biochem. 1980, 88, 1009–1013. [Google Scholar] [CrossRef] [PubMed]
- Molusky, M.M.; Ma, D.; Buelow, K.; Yin, L.; Lin, J.D. Peroxisomal Localization and Circadian Regulation of Ubiquitin-Specific Protease 2. PLoS ONE 2012, 7, 1–10. [Google Scholar] [CrossRef]
- Chapman, K.; Holmes, M.; Seckl, J. 11Β-Hydroxysteroid Dehydrogenases Intracellular Gate-Keepers of Tissue Glucocorticoid Action. Physiol. Rev. 2013, 93, 1139–1206. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Jiang, S.; Li, M.; Xiong, X.; Zhu, M.; Li, D.; Zhao, L.; Qian, L.; Zhai, L.; Li, J.; et al. Proteome-wide analysis of USP14 substrates revealed its role in hepatosteatosis via stabilization of FASN. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, F.; Gao, L.; Xu, L.; Tong, R.; Lin, N.; Su, Y.; Yan, Y.; Gao, Y.; He, J.; et al. Ubiquitin-Specific Protease 4 Is an Endogenous Negative Regulator of Metabolic Dysfunctions in Nonalcoholic Fatty Liver Disease in Mice. Hepatology 2018, 68, 897–917. [Google Scholar] [CrossRef] [PubMed]
- Tanti, J.F.; Jager, J. Cellular mechanisms of insulin resistance: Role of stress-regulated serine kinases and insulin receptor substrates (IRS) serine phosphorylation. Curr. Opin. Pharmacol. 2009, 9, 753–762. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Zhao, L.P.; Shen, L.J.; Wang, S.; Zhang, K.; Qi, Y.; Zheng, J.; Zhang, X.J.; Zhu, X.Y.; Bao, R.; et al. USP18 protects against hepatic steatosis and insulin resistance through its deubiquitinating activity. Hepatology 2017, 66, 1866–1884. [Google Scholar] [CrossRef]
- Zhang, Y.; Wan, J.; Xu, Z.; Hua, T.; Sun, Q. Exercise ameliorates insulin resistance via regulating TGFβ-activated kinase 1 (TAK1)-mediated insulin signaling in liver of high-fat diet-induced obese rats. J. Cell. Physiol. 2019, 234, 7467–7474. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Mo, K.; Wang, G.; Chen, W.; Zhang, W.; Guo, Y.; Sun, Z. Intervention of Gastrodin in Type 2 Diabetes Mellitus and Its Mechanism. Front. Pharmacol. 2021, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Qin, C.; Zhu, L.; Fang, C.; Zhang, Y.; Zhang, H.; Pei, F.; Tian, S.; Zhu, X.Y.; Gong, J.; et al. Ubiquitin-Specific Peptidase 10 (USP10) Inhibits Hepatic Steatosis, Insulin Resistance, and Inflammation Through Sirt6. Hepatology 2018, 68, 1786–1803. [Google Scholar] [CrossRef]
- Ignacio-Souza, L.M.; Bombassaro, B.; Pascoal, L.B.; Portovedo, M.A.; Razolli, D.S.; Coope, A.; Victorio, S.C.; De Moura, R.F.; Nascimento, L.F.; Arruda, A.P.; et al. Defective regulation of the ubiquitin/proteasome system in the hypothalamus of obese male mice. Endocrinology 2014, 155, 2831–2844. [Google Scholar] [CrossRef] [PubMed]
- Mastaitis, J.W.; Wurmbach, E.; Cheng, H.; Sealfon, S.C.; Mobbs, C.V. Acute induction of gene expression in brain and liver by insulin-induced hypoglycemia. Diabetes 2005, 54, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Fujimoto, M.; Konno, K.; Lee, M.L.; Yamada, Y.; Yamashita, K.; Toda, C.; Tomura, M.; Watanabe, M.; Inanami, O.; et al. Ubiquitin-Specific Protease 2 in the Ventromedial Hypothalamus Modifies Blood Glucose Levels by Controlling Sympathetic Nervous Activation. J. Neurosci. 2022, 42, 4607–4618. [Google Scholar] [CrossRef] [PubMed]
- Stirban, A.; Gawlowski, T.; Roden, M. Vascular effects of advanced glycation endproducts: Clinical effects and molecular mechanisms. Mol. Metab. 2014, 3, 94–108. [Google Scholar] [CrossRef]
- Tomlinson, D.R.; Gardiner, N.J. Glucose neurotoxicity. Nat. Rev. Neurosci. 2008, 9, 36–45. [Google Scholar] [CrossRef]
- Weiswasser, J.M.; Arora, S.; Shuman, C.; Kellicut, D.; Sidawy, A.N. Diabetic neuropathy. Nat. Rev. Dis. Prim. 2019, 5, 42. [Google Scholar] [CrossRef]
- Eftekhari, A.; Vahed, S.Z.; Kavetskyy, T.; Rameshrad, M.; Jafari, S.; Chodari, L.; Hosseiniyan, S.M.; Derakhshankhah, H.; Ahmadian, E.; Ardalan, M. Cell junction proteins: Crossing the glomerular filtration barrier in diabetic nephropathy. Int. J. Biol. Macromol. 2020, 148, 475–482. [Google Scholar] [CrossRef]
- Wolf, G.; Ziyadeh, F.N. Cellular and molecular mechanisms of proteinuria in diabetic nephropathy. Nephron—Physiol. 2007, 106, p26–p31. [Google Scholar] [CrossRef]
- Zhou, G.; Li, C.; Cai, L. Advanced glycation end-products induce connective tissue growth factor-mediated renal fibrosis predominantly through transforming growth factor β-independent pathway. Am. J. Pathol. 2004, 165, 2033–2043. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, Y.S.; Sun, L.; Xie, P.; Liu, F.Y.; Chen, S. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 395–423. [Google Scholar] [CrossRef] [PubMed]
- Selby, N.M.; Taal, M.W. An updated overview of diabetic nephropathy: Diagnosis, prognosis, treatment goals and latest guidelines. Diabetes, Obes. Metab. 2020, 22, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Ritz, E.; Zeng, X.X.; Rychlík, I. Clinical manifestation and natural history of diabetic nephropathy. Contrib. Nephrol. 2011, 170, 19–27. [Google Scholar] [CrossRef]
- Masakane, I.; Nakai, S.; Ogata, S.; Kimata, N.; Hanafusa, N.; Hamano, T.; Wakai, K.; Wada, A.; Nitta, K. An Overview of Regular Dialysis Treatment in Japan (As of 31 December 2013). Ther. Apher. Dial. 2015, 19, 540–574. [Google Scholar] [CrossRef] [PubMed]
- Shepard, B.D. Sex differences in diabetes and kidney disease: Mechanisms and consequences. Am. J. Physiol. Ren. Physiol. 2019, 317, F456–F462. [Google Scholar] [CrossRef] [PubMed]
- Mankhey, R.W.; Bhatti, F.; Maric, C. 17β-Estradiol replacement improves renal function and pathology associated with diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2005, 288, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Diamond-Stanic, M.K.; Romero-Aleshire, M.J.; Hoyer, P.B.; Greer, K.; Hoying, J.B.; Brooks, H.L. Midkine, a heparin-binding protein, is increased in the diabetic mouse kidney postmenopause. Am. J. Physiol. Ren. Physiol. 2011, 300, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Takata, T.; Nakazawa, Y.; Nakamura, Y.; Guo, X.; Yamada, S.; Ishigaki, Y.; Takeuchi, M.; Miyazawa, K. Potential of an interorgan network mediated by toxic advanced glycation end-products in a rat model. Nutrients 2021, 13, 80. [Google Scholar] [CrossRef]
- Goru, S.K.; Kadakol, A.; Pandey, A.; Malek, V.; Sharma, N.; Gaikwad, A.B. Histone H2AK119 and H2BK120 monoubiquitination modulate SET7/9 and SUV39H1 in type 1 diabetes-induced renal fibrosis. Biochem. J. 2016, 473, 3937–3949. [Google Scholar] [CrossRef]
- Atanassov, B.S.; Koutelou, E.; Dent, S.Y. The role of deubiquitinating enzymes in chromatin regulation. FEBS Lett. 2011, 585, 2016–2023. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Huang, J.; Xie, X.; Wang, S.; Chen, C.; Shen, X.; Liu, P.; Huang, H. Sirt1 resists advanced glycation end products-induced expressions of fibronectin and TGF-β1 by activating the Nrf2/ARE pathway in glomerular mesangial cells. Free Radic. Biol. Med. 2013, 65, 528–540. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.P.; Chen, C.; Hao, J.; Huang, J.Y.; Liu, P.Q.; Huang, H.Q. AGEs-rage system down-regulates sirt1 through the ubiquitin-proteasome pathway to promote fn and tgf-ß1 expression in male rat glomerular mesangial cells. Endocrinology 2015, 156, 268–279. [Google Scholar] [CrossRef]
- Yu, W.C.; Huang, R.Y.; Chou, T.C. Oligo-fucoidan improves diabetes-induced renal fibrosis via activation of sirt-1, glp-1r, and nrf2/ho-1: An in vitro and in vivo study. Nutrients 2020, 12, 3068. [Google Scholar] [CrossRef] [PubMed]
- Mao, R.; Shen, J.; Hu, X. BMSCs-derived exosomal microRNA-let-7a plays a protective role in diabetic nephropathy via inhibition of USP22 expression. Life Sci. 2021, 268, 118937. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.X.; Wang, Q.J.; Li, H.; Huang, Q. Silencing of USP22 suppresses high glucose-induced apoptosis, ROS production and inflammation in podocytes. Mol. Biosyst. 2016, 12, 1445–1456. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.H.; Xiao, H.M.; Zhang, M.; Lin, Z.Y.; Yang, Y.; Chen, R.; Liu, P.Q.; Huang, K.P.; Huang, H.Q. USP9X deubiquitinates connexin43 to prevent high glucose-induced epithelial-to-mesenchymal transition in NRK-52E cells. Biochem. Pharmacol. 2021, 188, 114562. [Google Scholar] [CrossRef]
- Carew, R.M.; Wang, B.; Kantharidis, P. The role of EMT in renal fibrosis. Cell Tissue Res. 2012, 347, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Zhao, X. USP9X prevents AGEs-induced upregulation of FN and TGF-β1 through activating Nrf2-ARE pathway in rat glomerular mesangial cells. Exp. Cell Res. 2020, 393, 112100. [Google Scholar] [CrossRef]
- Xu, X.; Xiao, L.; Xiao, P.; Yang, S.; Chen, G.; Liu, F.; Kanwar, Y.S.; Sun, L. A Glimpse of Matrix Metalloproteinases in Diabetic Nephropathy. Curr. Med. Chem. 2014, 21, 3244–3260. [Google Scholar] [CrossRef]
- Shelton, L.M.; Park, B.K.; Copple, I.M. Role of Nrf2 in protection against acute kidney injury. Kidney Int. 2013, 84, 1090–1095. [Google Scholar] [CrossRef]
- Zhu, S.; Hou, S.; Lu, Y.; Sheng, W.; Cui, Z.; Dong, T.; Feng, H.; Wan, Q. USP36-Mediated Deubiquitination of DOCK4 Contributes to the Diabetic Renal Tubular Epithelial Cell Injury via Wnt/β-Catenin Signaling Pathway. Front. Cell Dev. Biol. 2021, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Sun, L.; Xiao, L.; Han, Y.; Fu, X.; Xiong, X.; Xu, X.; Liu, Y.; Yang, S.; Liu, F.; et al. Insights into the Mechanisms Involved in the Expression and Regulation of Extracellular Matrix Proteins in Diabetic Nephropathy. Curr. Med. Chem. 2015, 22, 2858–2870. [Google Scholar] [CrossRef]
- Barutta, F.; Bellini, S.; Gruden, G. Mechanisms of podocyte injury and implications for diabetic nephropathy. Clin. Sci. 2022, 136, 493–520. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Deng, Y.; Wang, Y.; Sun, X.; Chen, S.; Fu, G. SPAG5-AS1 inhibited autophagy and aggravated apoptosis of podocytes via SPAG5/AKT/mTOR pathway. Cell Prolif. 2020, 53, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Banu, K.; Ni, Z.; Leventhal, J.S.; Menon, M.C. Podocyte autophagy in homeostasis and disease. J. Clin. Med. 2021, 10, 1184. [Google Scholar] [CrossRef]
- Xu, D.; Shan, B.; Sun, H.; Xiao, J.; Zhu, K.; Xie, X.; Li, X.; Liang, W.; Lu, X.; Qian, L.; et al. USP14 regulates autophagy by suppressing K63 ubiquitination of Beclin. Genes Dev. 2016, 30, 1718–1730. [Google Scholar] [CrossRef]
- Xu, E.; Yin, C.; Yi, X.; Liu, Y. Inhibition of USP15 ameliorates high-glucose-induced oxidative stress and inflammatory injury in podocytes through regulation of the Keap1/Nrf2 signaling. Environ. Toxicol. 2022, 37, 765–775. [Google Scholar] [CrossRef]
- Vom Hagen, F.; Feng, Y.; Hillenbrand, A.; Hoffmann, S.; Shani, M.; Deutsch, U.; Hammes, H.P. Early loss of arteriolar smooth muscle cells: More than just a pericyte loss in diabetic retinopathy. Exp. Clin. Endocrinol. Diabetes 2005, 113, 573–576. [Google Scholar] [CrossRef]
- Flaxman, S.R.; Bourne, R.R.A.; Resnikoff, S.; Ackland, P.; Braithwaite, T.; Cicinelli, M.V.; Das, A.; Jonas, J.B.; Keeffe, J.; Kempen, J.; et al. Global causes of blindness and distance vision impairment 1990–2020: A systematic review and meta-analysis. Lancet Glob. Heal. 2017, 5, e1221–e1234. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Su, X.; Wu, Q.; Shan, H.; Lv, L.; Yu, T.; Zhao, X.; Sun, J.; Yang, R.; Zhang, L.; et al. LncRNA 2810403D21Rik/Mirf promotes ischemic myocardial injury by regulating autophagy through targeting Mir26a. Autophagy 2020, 16, 1077–1091. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mishra, M.; Kumar, B. Diabetic retinopathy and transcriptional regulation of a small molecular weight G-Protein, Rac1. Exp. Eye Res. 2016, 147, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, Y.; Jin, W.; Xing, Y.; Yang, A. Catechin weakens diabetic retinopathy by inhibiting the expression of nf-κb signaling pathway-mediated inflammatory factors. Ann. Clin. Lab. Sci. 2018, 48, 594–600. [Google Scholar]
- Li, J.; Chen, Y.; Zhang, X.; Ye, S.; Yi, J.; Chen, Q.; Liu, Q. Inhibition of acetylcholinesterase attenuated retinal inflammation via suppressing NF-κB activation. Exp. Eye Res. 2020, 195, 108003. [Google Scholar] [CrossRef] [PubMed]
- Terasaki, H.; Kase, S.; Shirasawa, M.; Otsuka, H.; Hisatomi, T.; Sonoda, S.; Ishida, S.; Ishibashi, T.; Sakamoto, T. TNF-α Decreases VEGF Secretion in Highly Polarized RPE Cells but Increases It in Non-Polarized RPE Cells Related to Crosstalk between JNK and NF-κB Pathways. PLoS ONE 2013, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, K.; Naumann, M. CSN-associated USP48 confers stability to nuclear NF-κB/RelA by trimming K48-linked Ub-chains. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 453–469. [Google Scholar] [CrossRef]
- Mirra, S.; Sánchez-Bellver, L.; Casale, C.; Pescatore, A.; Marfany, G. Ubiquitin Specific Protease USP48 Destabilizes NF-κB/p65 in Retinal Pigment Epithelium Cells. Int. J. Mol. Sci. 2022, 23, 9682. [Google Scholar] [CrossRef]
- Noh, M.; Zhang, H.; Kim, H.; Park, S.; Kim, Y.M.; Kwon, Y.G. Primaquine Diphosphate, a Known Antimalarial Drug, Blocks Vascular Leakage Acting Through Junction Stabilization. Front. Pharmacol. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Wang, K.; Wang, Y.; Hu, Z.; Zhang, L.; Li, G.; Dang, L.; Tan, Y.; Cao, X.; Shi, F.; Zhang, S.; et al. Bone-targeted lncRNA OGRU alleviates unloading-induced bone loss via miR-320-3p/Hoxa10 axis. Cell Death Dis. 2020, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Zheng, Y.; Sun, Y.; Lai, M.; Qiu, J.; Gui, F.; Zeng, Q.; Liu, F. Suppressing long noncoding RNA OGRU ameliorates diabetic retinopathy by inhibition of oxidative stress and inflammation via miR-320/USP14 axis. Free Radic. Biol. Med. 2021, 169, 361–381. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Kanda, A.; Liu, Y.; Noda, K.; Murata, M.; Ishida, S. Involvement of müller glial autoinduction of TGF-β in diabetic fibrovascular proliferation via glial-mesenchymal transition. Investig. Ophthalmol. Vis. Sci. 2020, 61, 29. [Google Scholar] [CrossRef]
- Andersen, H. Motor Neuropathy, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 126. [Google Scholar]
- Dauch, J.R.; Yanik, B.M.; Hsieh, W.; Oh, S.S.; Cheng, H.T. Neuron-astrocyte signaling network in spinal cord dorsal horn mediates painful neuropathy of type 2 diabetes. Glia 2012, 60, 1301–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Caballero, A.; Gadotti, V.M.; Stemkowski, P.; Weiss, N.; Souza, I.A.; Hodgkinson, V.; Bladen, C.; Chen, L.; Hamid, J.; Pizzoccaro, A.; et al. The Deubiquitinating Enzyme USP5 Modulates Neuropathic and Inflammatory Pain by Enhancing Cav3.2 Channel Activity. Neuron 2014, 83, 1144–1158. [Google Scholar] [CrossRef] [PubMed]
- Gadotti, V.M.; Caballero, A.G.; Berger, N.D.; Gladding, C.M.; Chen, L.; Pfeifer, T.A.; Zamponi, G.W. Small Organic Molecule Disruptors of Cav3.2—USP5 Interactions Reverse Inflammatory and Neuropathic Pain. Mol. Pain 2015, 11, 1–13. [Google Scholar] [CrossRef]
- Leenders, M.; Verdijk, L.B.; Van Der Hoeven, L.; Adam, J.J.; Van Kranenburg, J.; Nilwik, R.; Van Loon, L.J.C. Patients with type 2 diabetes show a greater decline in muscle mass, muscle strength, and functional capacity with aging. J. Am. Med. Dir. Assoc. 2013, 14, 585–592. [Google Scholar] [CrossRef]
- Severinsen, M.C.K.; Pedersen, B.K. Muscle–Organ Crosstalk: The Emerging Roles of Myokines. Endocr. Rev. 2020, 41, 594–609. [Google Scholar] [CrossRef] [PubMed]
- Sieber, C.C. Malnutrition and sarcopenia. Aging Clin. Exp. Res. 2019, 31, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.J.; Hastings, M.K. The application of exercise training for diabetic peripheral neuropathy. J. Clin. Med. 2021, 10, 793–798. [Google Scholar] [CrossRef]
- Izzo, A.; Massimino, E.; Riccardi, G.; Della Pepa, G. A Narrative Review on Sarcopenia in Type 2 Diabetes Mellitus: Prevalence and Associated Factors. Nutrients 2021, 13, 183. [Google Scholar] [CrossRef]
- Giha, H.A.; Sater, M.S.; Alamin, O.A.O. Diabetes mellitus tendino-myopathy: Epidemiology, clinical features, diagnosis and management of an overlooked diabetic complication. Acta Diabetol. 2022, 59, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Combaret, L.; Adegoke, O.A.J.; Bedard, N.; Baracos, V.; Attaix, D.; Wing, S.S. USP19 is a ubiquitin-specific protease regulated in rat skeletal muscle during catabolic states. Am. J. Physiol. Endocrinol. Metab. 2005, 288, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Henning, R.J. Type-2 diabetes mellitus and cardiovascular disease. Future Cardiol. 2018, 14, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Ma, J.; Liu, Y.; Shao, Y.; Xiong, X.; Duan, W.; Gao, E.; Yang, Q.; Chen, S.; Yang, J.; et al. FSTL1-USP10-Notch1 Signaling Axis Protects Against Cardiac Dysfunction Through Inhibition of Myocardial Fibrosis in Diabetic Mice. Front. Cell Dev. Biol. 2021, 9, 1–14. [Google Scholar] [CrossRef]
- Lim, R.; Sugino, T.; Nolte, H.; Andrade, J.; Zimmermann, B.; Shi, C.; Doddaballapur, A.; Ong, Y.T.; Wilhelm, K.; Fasse, J.W.D.; et al. Deubiquitinase USP10 regulates notch signaling in the endothelium. Science 2019, 364, 188–193. [Google Scholar] [CrossRef]
- Bandyk, D.F. The diabetic foot: Pathophysiology, evaluation, and treatment. Semin. Vasc. Surg. 2018, 31, 43–48. [Google Scholar] [CrossRef]
- Li, X.; Wang, T.; Tao, Y.; Wang, X.; Li, L.; Liu, J. Inhibition of USP7 suppresses advanced glycation end-induced cell cycle arrest and senescence of human umbilical vein endothelial cells through ubiquitination of p53. Acta Biochim. Biophys. Sin. 2022, 54, 311–320. [Google Scholar] [CrossRef]
- Francque, S.M.; Marchesini, G.; Kautz, A.; Walmsley, M.; Dorner, R.; Lazarus, J.V.; Zelber-Sagi, S.; Hallsworth, K.; Busetto, L.; Frühbeck, G.; et al. Non-alcoholic fatty liver disease: A patient guideline. JHEP Rep. 2021, 3, 100322. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Sakurai, Y.; Kubota, N.; Yamauchi, T.; Kadowaki, T. Role of insulin resistance in mafld. Int. J. Mol. Sci. 2021, 22, 4156. [Google Scholar] [CrossRef]
- Ballestri, S.; Zona, S.; Targher, G.; Romagnoli, D.; Baldelli, E.; Nascimbeni, F.; Roverato, A.; Guaraldi, G.; Lonardo, A. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2016, 31, 936–944. [Google Scholar] [CrossRef]
- Ni, W.; Lin, S.; Bian, S.; Zheng, W.; Qu, L.; Fan, Y.; Lu, C.; Xiao, M.; Zhou, P. USP7 mediates pathological hepatic de novo lipogenesis through promoting stabilization and transcription of ZNF638. Cell Death Dis. 2020, 11, 843. [Google Scholar] [CrossRef]
- Martínez-López, N.; Varela-Rey, M.; Fernández-Ramos, D.; Woodhoo, A.; Vázquez-Chantada, M.; Embade, N.; Espinosa-Hevia, L.; Bustamante, F.J.; Parada, L.A.; Rodriguez, M.S.; et al. Activation of LKB1-Akt pathway independent of phosphoinositide 3-kinase plays a critical role in the proliferation of hepatocellular carcinoma from nonalcoholic steatohepatitis. Hepatology 2010, 52, 1621–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.C.; Alvarez, L.; Huang, Z.Z.; Chen, L.; An, W.; Corrales, F.J.; Avila, M.A.; Kanel, G.; Mato, J.M. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proc. Natl. Acad. Sci. USA 2001, 98, 5560–5565. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Chantar, M.L.; Corrales, F.J.; Martínez-Cruz, L.A.; García-Trevijano, E.R.; Huang, Z.Z.; Chen, L.; Kanel, G.; Avila, M.A.; Mato, J.M.; Lu, S.C. Spontaneous oxidative stress and liver tumors in mice lacking methionine adenosyltransferase 1A. FASEB J. 2002, 16, 1292–1294. [Google Scholar] [CrossRef] [PubMed]
- Meruvu, S.; Hugendubler, L.; Mueller, E. Regulation of adipocyte differentiation by the zinc finger protein ZNF638. J. Biol. Chem. 2011, 286, 26516–26523. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.; Zhou, X.; Xu, J.; Gao, L. Rodent Models of Nonalcoholic Fatty Liver Disease. Digestion 2020, 101, 522–535. [Google Scholar] [CrossRef]
- Jacobs, A.; Warda, A.S.; Verbeek, J.; Cassiman, D.; Spincemaille, P. An Overview of Mouse Models of Nonalcoholic Steatohepatitis: From Past to Present. Curr. Protoc. Mouse Biol. 2016, 6, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Xin, S.L.; Yu, Y.Y. Ubiquitin-specific peptidase 10 ameliorates hepatic steatosis in nonalcoholic steatohepatitis model by restoring autophagic activity. Dig. Liver Dis. 2022, 54, 1021–1029. [Google Scholar] [CrossRef]
- Chen, D.; Ning, Z.; Chen, H.; Lu, C.; Liu, X.; Xia, T.; Qi, H.; Wang, W.; Ling, T.; Guo, X.; et al. An integrative pan-cancer analysis of biological and clinical impacts underlying ubiquitin-specific-processing proteases. Oncogene 2020, 39, 587–602. [Google Scholar] [CrossRef] [PubMed]
- Ning, Z.; Guo, X.; Liu, X.; Lu, C.; Wang, A.; Wang, X.; Wang, W.; Chen, H.; Qin, W.; Liu, X.; et al. USP22 regulates lipidome accumulation by stabilizing PPARγ in hepatocellular carcinoma. Nat. Commun. 2022, 13, 2187. [Google Scholar] [CrossRef]
- Gao, Y.; Lin, L.P.; Zhu, C.H.; Chen, Y.; Hou, Y.T.; Ding, J. Growth arrest induced by C75, a fatty acid synthase inhibitor, was partially modulated by p38 MAPK but not by p53 in human hepatocellular carcinoma. Cancer Biol. Ther. 2006, 5, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Yang, Y.M.; Han, C.Y.; Koo, J.H.; Oh, H.; Kim, S.S.; You, B.H.; Choi, Y.H.; Park, T.S.; Lee, C.H.; et al. Gα12 ablation exacerbates liver steatosis and obesity by suppressing USP22/SIRT1-regulated mitochondrial respiration. J. Clin. Invest. 2018, 128, 5587–5602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.F.; Infante, J.R. Molecular pathways: Fatty acid synthase. Clin. Cancer Res. 2015, 21, 5434–5438. [Google Scholar] [CrossRef] [PubMed]
- Seeßle, J.; Liebisch, G.; Schmitz, G.; Stremmel, W.; Chamulitrat, W. Palmitate activation by fatty acid transport protein 4 as a model system for hepatocellular apoptosis and steatosis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2015, 1851, 549–565. [Google Scholar] [CrossRef]
- Baldini, S.F.; Wavelet, C.; Hainault, I.; Guinez, C.; Lefebvre, T. The Nutrient-Dependent O-GlcNAc Modification Controls the Expression of Liver Fatty Acid Synthase. J. Mol. Biol. 2016, 428, 3295–3304. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S.A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011, 140, 1071–1083. [Google Scholar] [CrossRef]
- Lu, X.J.; Shi, Y.; Chen, J.L.; Ma, S. Krüppel-like factors in hepatocellular carcinoma. Tumor Biol. 2015, 36, 533–541. [Google Scholar] [CrossRef]
- Yang, H.; Park, D.; Ryu, J.; Park, T. USP11 degrades KLF4 via its deubiquitinase activity in liver diseases. J. Cell. Mol. Med. 2021, 25, 6976–6987. [Google Scholar] [CrossRef]
- Yue, S.; Li, J.; Lee, S.-Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl Ester Accumulation Induced by PTEN Loss and PI3K/AKT Activation Underlies Human Prostate Cancer Aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef]
- Geng, F.; Cheng, X.; Wu, X.; Yoo, J.Y.; Cheng, C.; Guo, J.Y.; Mo, X.; Ru, P.; Hurwitz, B.; Kim, S.H.; et al. Inhibition of SOAT1 suppresses glioblastoma growth via blocking SREBP-1-mediated lipogenesis. Clin. Cancer Res. 2016, 22, 5337–5348. [Google Scholar] [CrossRef]
- Jiang, Y.; Sun, A.; Zhao, Y.; Ying, W.; Sun, H.; Yang, X.; Xing, B.; Sun, W.; Ren, L.; Hu, B.; et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature 2019, 567, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Gu, L.; Lin, X.; Zhou, X.; Lu, B.; Liu, C.; Li, Y.; Prochownik, E.V.; Karin, M.; Wang, F.; et al. P53 deficiency affects cholesterol esterification to exacerbate hepatocarcinogenesis. Hepatology 2022, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T. Pathophysiology of diabetic dyslipidemia. J. Atheroscler. Thromb. 2018, 25, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Rong, J.X.; Shapiro, M.; Trogan, E.; Fisher, E.A. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc. Natl. Acad. Sci. USA 2003, 100, 13531–13536. [Google Scholar] [CrossRef] [PubMed]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.C.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef]
- Kim, K.; Ivanov, S.; Williams, J.W. Monocyte Recruitment, Specification, and Function in Atherosclerosis. Cells 2020, 10, 15. [Google Scholar] [CrossRef]
- Bäck, M.; Yurdagul, A.; Tabas, I.; Öörni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 176, 139–148. [Google Scholar] [CrossRef]
- Bos, D.; Arshi, B.; Van Den Bouwhuijsen, Q.J.A.; Ikram, M.K.; Selwaness, M.; Vernooij, M.W.; Kavousi, M.; Van Der Lugt, A. Atherosclerotic Carotid Plaque Composition and Incident Stroke and Coronary Events. J. Am. Coll. Cardiol. 2021, 77, 1426–1435. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.K.; Sorrentino, V.; Trezza, R.A.; Heride, C.; Urbe, S.; Distel, B.; Zelcer, N. The deubiquitylase USP2 regulates the ldlr pathway by counteracting the E3-Ubiquitin Ligase IDOL. Circ. Res. 2016, 118, 410–419. [Google Scholar] [CrossRef]
- Zelcer, N.; Hong, C.; Boyadjian, R.; Tontonoz, P. LXR Regulates Cholesterol Uptake Through Idol-Dependent Ubiquitination of the LDL Receptor. Science 2009, 325, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. TLR signaling pathways. Semin. Immunol. 2004, 16, 3–9. [Google Scholar] [CrossRef]
- Jean-Charles, P.Y.; Zhang, L.; Wu, J.H.; Han, S.O.; Brian, L.; Freedman, N.J.; Shenoy, S.K. Ubiquitin-specific protease 20 regulates the reciprocal functions of β-Arrestin2 in toll-like receptor 4-promoted nuclear factor kB (NFkB) activation. J. Biol. Chem. 2016, 291, 7450–7464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jean-Charles, P.Y.; Wu, J.H.; Zhang, L.; Kaur, S.; Nepliouev, I.; Stiber, J.A.; Brian, L.; Qi, R.; Wertman, V.; Shenoy, S.K.; et al. USP20 (ubiquitin-specific protease 20) inhibits TNF (tumor necrosis factor)-triggered smooth muscle cell inflammation and attenuates atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2295–2305. [Google Scholar] [CrossRef]
- Liu, H.; Li, X.; Yan, G.; Lun, R. Knockdown of USP14 inhibits PDGF-BB-induced vascular smooth muscle cell dedifferentiation: Via inhibiting mTOR/P70S6K signaling pathway. RSC Adv. 2019, 9, 36649–36657. [Google Scholar] [CrossRef]
- Shi, K.; Zhang, J.Z.; Zhao, R.L.; Yang, L.; Guo, D. PSMD7 downregulation induces apoptosis and suppresses tumorigenesis of esophageal squamous cell carcinoma via the mTOR/p70S6K pathway. FEBS Open Bio. 2018, 8, 533–543. [Google Scholar] [CrossRef]
- Kingsley, K.; Huff, J.L.; Rust, W.L.; Carroll, K.; Martinez, A.M.; Fitchmun, M.; Plopper, G.E. ERK1/2 mediates PDGF-BB stimulated vascular smooth muscle cell proliferation and migration on laminin-5. Biochem. Biophys. Res. Commun. 2002, 293, 1000–1006. [Google Scholar] [CrossRef]
- Frismantiene, A.; Philippova, M.; Erne, P.; Resink, T.J. Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell. Signal. 2018, 52, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Qiu, J.; Wu, J.; Zhang, L.; Wei, F.; Lu, L.; Wang, C.; Zeng, Z.; Liang, S.; Zheng, J. USP14-mediated NLRC5 upregulation inhibits endothelial cell activation and inflammation in atherosclerosis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2022, 159258. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Cai, C.; Sun, T.; Wang, Q.; Xie, W.; Wang, R.; Cui, J. Reversible ubiquitination shapes NLRC5 function and modulates NF-κB activation switch. J. Cell Biol. 2015, 211, 1025–1040. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tang, X.; Yao, L.; Wang, Y.; Chen, Z.; Li, M.; Wu, N.; Wu, D.; Dai, X.; Jiang, H.; et al. Disruption of USP9X in macrophages promotes foam cell formation and atherosclerosis. J. Clin. Invest. 2022, 132. [Google Scholar] [CrossRef] [PubMed]
- Akoumianakis, I.; Sanna, F.; Margaritis, M.; Badi, I.; Akawi, N.; Herdman, L.; Coutinho, P.; Fagan, H.; Antonopoulos, A.S.; Oikonomou, E.K.; et al. Adipose tissue-derived WNT5A regulates vascular redox signaling in obesity via USP17/RAC1-mediated activation of NADPH oxidases. Sci. Transl. Med. 2019, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Shibli-Rahhal, A.; Van Beek, M.; Schlechte, J.A. Cushing’s syndrome. Clin. Dermatol. 2006, 24, 260–265. [Google Scholar] [CrossRef]
- Perez-Rivas, L.G.; Theodoropoulou, M.; Ferraù, F.; Nusser, C.; Kawaguchi, K.; Stratakis, C.A.; Rueda Faucz, F.; Wildemberg, L.E.; Assié, G.; Beschorner, R.; et al. The gene of the ubiquitin-specific protease 8 is frequently mutated in adenomas causing cushing’s disease. J. Clin. Endocrinol. Metab. 2015, 100, E997–E1004. [Google Scholar] [CrossRef]
- Simon, J.; Theodoropoulou, M. Genetics of Cushing’s disease. J. Neuroendocrinol. 2022, 34, 1–8. [Google Scholar] [CrossRef]
- Sbiera, S.; Deutschbein, T.; Weigand, I.; Reincke, M.; Fassnacht, M.; Allolio, B. The New Molecular Landscape of Cushing’s Disease. Trends Endocrinol. Metab. 2015, 26, 573–583. [Google Scholar] [CrossRef]
- Reincke, M.; Sbiera, S.; Hayakawa, A.; Theodoropoulou, M.; Osswald, A.; Beuschlein, F.; Meitinger, T.; Mizuno-Yamasaki, E.; Kawaguchi, K.; Saeki, Y.; et al. Mutations in the deubiquitinase gene usp8 cause cushing’s disease. Nat. Genet. 2015, 47, 31–38. [Google Scholar] [CrossRef]
- Ma, Z.Y.; Song, Z.J.; Chen, J.H.; Wang, Y.F.; Li, S.Q.; Zhou, L.F.; Mao, Y.; Li, Y.M.; Hu, R.G.; Zhang, Z.Y.; et al. Recurrent gain-of-function USP8 mutations in Cushing’s disease. Cell Res. 2015, 25, 306–317. [Google Scholar] [CrossRef]
- Araki, T.; Tone, Y.; Yamamoto, M.; Kameda, H.; Ben-Shlomo, A.; Yamada, S.; Takeshita, A.; Yamamoto, M.; Kawakami, Y.; Tone, M.; et al. Two Distinctive POMC Promoters Modify Gene Expression in Cushing Disease. J. Clin. Endocrinol. Metab. 2021, 106, E3346–E3363. [Google Scholar] [CrossRef]
- Weigand, I.; Knobloch, L.; Flitsch, J.; Saeger, W.; Monoranu, C.M.; Höfner, K.; Herterich, S.; Rotermund, R.; Ronchi, C.L.; Buchfelder, M.; et al. Impact of USP8 Gene Mutations on Protein Deregulation in Cushing Disease. J. Clin. Endocrinol. Metab. 2019, 104, 2535–2546. [Google Scholar] [CrossRef] [PubMed]
- Treppiedi, D.; Di Muro, G.; Marra, G.; Barbieri, A.M.; Mangili, F.; Catalano, R.; Serban, A.; Ferrante, E.; Locatelli, M.; Lania, A.G.; et al. USP8 inhibitor RA-9 reduces ACTH release and cell growth in tumor corticotrophs. Endocr. Relat. Cancer 2021, 28, 573–582. [Google Scholar] [CrossRef]
- Sbiera, S.; Tryfonidou, M.A.; Weigand, I.; Grinwis, G.C.M.; Broeckx, B.; Herterich, S.; Allolio, B.; Deutschbein, T.; Fassnacht, M.; Meij, B.P. Lack of ubiquitin specific protease 8 (USP8) mutations in canine corticotroph pituitary adenomas. PLoS ONE 2016, 11, 1–14. [Google Scholar] [CrossRef]
- Barber, T.M.; Adams, E.; Ansorge, O.; Byrne, J.V.; Karavitaki, N.; Wass, J.A.H. Nelson’s syndrome. Eur. J. Endocrinol. 2010, 163, 495–507. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Rivas, L.G.; Theodoropoulou, M.; Puar, T.H.; Fazel, J.; Stieg, M.R.; Ferraù, F.; Assié, G.; Gadelha, M.R.; Deutschbein, T.; Fragoso, M.C.; et al. Somatic USP8 mutations are frequent events in corticotroph tumor progression causing Nelson’s tumor. Eur. J. Endocrinol. 2018, 178, 57–63. [Google Scholar] [CrossRef]
- Sbiera, S.; Perez-Rivas, L.G.; Taranets, L.; Weigand, I.; Flitsch, J.; Graf, E.; Monoranu, C.M.; Saeger, W.; Hagel, C.; Honegger, J.; et al. Driver mutations in USP8 wild-type Cushing’s disease. Neuro. Oncol. 2019, 21, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Xiong, J.; Zhan, J.; Yuan, F.; Tang, M.; Zhang, C.; Cao, Z.; Chen, Y.; Lu, X.; Li, Y.; et al. Ubiquitin-specific peptidase 7 (USP7)-mediated deubiquitination of the histone deacetylase SIRT7 regulates gluconeogenesis. J. Biol. Chem. 2017, 292, 13296–13311. [Google Scholar] [CrossRef] [PubMed]
- Kessler, B.M. Selective and reversible inhibitors of ubiquitin-specific protease 7: A patent evaluation (WO2013030218). Expert Opin. Ther. Pat. 2014, 24, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Kitamura, H.; Kikuguchi, C.; Hase, K.; Ohno, H.; Ohara, O. SP600125 Inhibits Cap-dependent Translation Independently of the c-Jun N-terminal Kinase Pathway. Cell Struct. Funct. 2011, 36, 27–33. [Google Scholar] [CrossRef]
- Bain, J.; McLauchlan, H.; Elliott, M.; Cohen, P. The specificities of protein kinase inhibitors: An update. Biochem. J. 2003, 371, 199–204. [Google Scholar] [CrossRef]
- De Poot, S.A.H.; Tian, G.; Finley, D. Meddling with Fate: The Proteasomal Deubiquitinating Enzymes. J. Mol. Biol. 2017, 429, 3525–3545. [Google Scholar] [CrossRef] [PubMed]
- Sakata, E.; Stengel, F.; Fukunaga, K.; Zhou, M.; Saeki, Y.; Förster, F.; Baumeister, W.; Tanaka, K.; Robinson, C.V. The Catalytic Activity of Ubp6 Enhances Maturation of the Proteasomal Regulatory Particle. Mol. Cell 2011, 42, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Koulich, E.; Li, X.; DeMartino, G.N. Relative Structural and Functional Roles of Multiple Deubiquitylating Proteins Associated with Mammalian 26S Proteasome. Mol. Biol. Cell 2008, 19, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ning, S.; Yu, B.; Wang, Y. USP14: Structure, Function, and Target Inhibition. Front. Pharmacol. 2022, 12, 1–23. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. MTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Bedard, N.; Yang, Y.; Gregory, M.; Cyr, D.G.; Suzuki, J.; Yu, X.; Chian, R.-C.; Hermo, L.; O’Flaherty, C.; Smith, C.E.; et al. Mice lacking the USP2 deubiquitinating enzyme have severe male subfertility associated with defects in fertilization and sperm motility. Biol. Reprod. 2011, 85, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Kimura, S.; Kanno, C.; Yanagawa, Y.; Watanabe, T.; Okabe, J.; Takahashi, E.; Nagano, M.; Kitamura, H. Macrophage ubiquitin-specific protease 2 contributes to motility, hyperactivation, capacitation, and in vitro fertilization activity of mouse sperm. Cell. Mol. Life Sci. 2021, 78, 2929–2948. [Google Scholar] [CrossRef]
- Hashimoto, M.; Saito, N.; Ohta, H.; Yamamoto, K.; Tashiro, A.; Nakazawa, K.; Inanami, O.; Kitamura, H. Inhibition of ubiquitin-specific protease 2 causes accumulation of reactive oxygen species, mitochondria dysfunction, and intracellular ATP decrement in C2C12 myoblasts. Physiol. Rep. 2019, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Prasun, P. Mitochondrial dysfunction in metabolic syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165838. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | USPs | Expressing Cells/Tissues | Restorative/Deteriorative | Direct Targets | References |
|---|---|---|---|---|---|
| Obesity | USP2 | Adipocyte | Deteriorative | Cyclin D | [46] |
| USP7 | Adipocyte | Deteriorative | SREBP1c | [45] | |
| USP15 | Adipocyte | Deteriorative | Keap1 | [42] | |
| USP19 | Adipocyte | Deteriorative | ? | [49] | |
| USP20 | Hepatocyte | Deteriorative | HMGCR | [50] | |
| USP53 | Adipose tissue | Restorative | ? | [53] | |
| T1DM | USP18 | β-cell | Restorative | ? | [58] |
| USP22 | Treg | Restorative | FoxP3 | [56] | |
| T2DM | USP1 | β-cell | Deteriorative | ? | [69] |
| USP22 | β-cell | Restorative | Sirt-1 | [68] | |
| USP2 | Adipose tissue macrophage | Restorative | ? | [78,79] | |
| USP19 | Adipocyte | Deteriorative | ? | [49] | |
| USP9X | Myocyte | Restorative | AMPKα2 | [87] | |
| USP20 | Myocyte | Restorative | β2 adrenoreceptor | [94] | |
| USP21 | Myocyte | Deteriorative | DNA-PKcs, ACLY | [84] | |
| USP33 | Myocyte | Restorative | β2 adrenoreceptor | [94,95] | |
| USP2 | Hepatocyte | Deteriorative | C/EBPα | [102] | |
| USP4 | Hepatocyte | Restorative | TAK1 | [109] | |
| USP7 | Hepatocyte | Restorative | IRS1 | [99] | |
| Restorative | PPARγ | [100] | |||
| Restorative | FoxO1 | [101] | |||
| USP10 | Hepatocyte | Restorative | Sirt-6 | [114] | |
| USP14 | Hepatocyte | Deteriorative | CBP | [103] | |
| Deteriorative | FASN | [108] | |||
| USP18 | Hepatocyte | Restorative | TAK1 | [111] | |
| USP20 | Hepatocyte | Deteriorative | HMGCR | [50] | |
| USP2 | Hypothalamus | Restorative | ? | [117] | |
| Diabetic nephropathy | USP2 | Kidney | ? | ? | [130] |
| USP7 | Kidney | ? | H2A, H2B? | [132] | |
| USP9X | Renal epithelial cell | Restorative | Connexin43 | [139] | |
| Mesangial cell | Restorative | Nrf2 | [141] | ||
| USP14 | Podocyte | Deteriorative | SPAG5-AS | [147] | |
| USP15 | Podocyte | Deteriorative | Keap1 | [150] | |
| USP16 | Kidney | ? | H2A, H2B? | [132] | |
| USP21 | Kidney | ? | H2A, H2B? | [132] | |
| USP22 | Kidney | ? | H2A? H2B | [132] | |
| Mesangial cell | Restorative | Sirt-1 | [135] | ||
| Renal epithelial cell | Restorative | Sirt-1 | [136] | ||
| Kidney | Deteriorative | ? | [137] | ||
| Podocyte | Deteriorative | ? | [138] | ||
| USP36 | Renal epithelial cell | Deteriorative | DOCK4 | [144] | |
| Diabetic retinopathy | USP1 | Vascular endothelial cells | Deteriorative | ? | [160] |
| USP14 | Müller cells | Deteriorative | TGF-β receptor, IκBα, Nrf2 | [162] | |
| USP48 | Pigment epithelial cell | Restorative | NFκBp65 | [159] | |
| Diabetic neuropathy | USP5 | Spinal dorsal horn | Deteriorative | Cav3.2 | [167] |
| Diabetic myopathy | USP19 | Myocyte | ? | ? | [174] |
| USP21 | Myocyte | Deteriorative | DNA-PKcs, ACLY | [84] | |
| Diabetic cardiomyopathy | USP10 | Cardiomyocyte | Restorative | NICD1 | [176] |
| Diabetic foot ulcers | USP7 | Vascular endothelial cells | Deteriorative | p53 | [179] |
| NAFLD | USP2 | Hepatocytes | Deteriorative | FASN | [198,199] |
| USP4 | Hepatocytes | Restorative | TAK1 | [109] | |
| USP7 | Hepatocyte | Restorative | IRS1 | [99] | |
| Deteriorative | Mdm2 | [185] | |||
| Deteriorative | PPARγ | [100] | |||
| Deteriorative | ZNF638, | [184] | |||
| CREB, | |||||
| SREBP1c | |||||
| USP10 | Hepatocyte | Restorative | Sirt-6 | [114] | |
| Restorative | ? | [191] | |||
| USP11 | Hepatocyte | Deteriorative | KLF4 | [201] | |
| USP14 | Hepatocyte | Deteriorative | FASN | [108] | |
| USP18 | Hepatocyte | Restorative | TAK1 | [111] | |
| USP19 | Hepatocyte | Deteriorative | SOAT1 | [204] | |
| USP20 | Hepatocyte | Deteriorative | HMGCR | [50] | |
| USP22 | Hepatocyte | Restorative | Sirt-1 | [195] | |
| Hepatocyte | Deteriorative | PPARγ | [193] | ||
| Atherosclerosis | USP2 | Hepatocyte | Restorative | IDOL | [212] |
| USP9X | Macrophage | Restorative | SR-A1 | [223] | |
| USP14 | Vascular endothelial cell | Restorative | NLRC5 | [221] | |
| Vascular smooth muscle cell | Deteriorative | PMSD7 | [217] | ||
| USP17 | Vascular smooth muscle cell | Deteriorative | ? | [224] | |
| USP20 | Vascular smooth muscle cell | Restorative | TRAF6 | [215] | |
| Restorative | RIPK1 | [216] | |||
| Liver | Deteriorative? | HMGCR | [50] | ||
| Cushing disease | USP8 | Corticotroph adenoma | Deteriorative | ? | [228,229,230,231,232,233,234,235,236,237,238] |
| USP48 | Corticotroph adenoma | Deteriorative | ? | [237] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitamura, H. Ubiquitin-Specific Proteases (USPs) and Metabolic Disorders. Int. J. Mol. Sci. 2023, 24, 3219. https://doi.org/10.3390/ijms24043219
Kitamura H. Ubiquitin-Specific Proteases (USPs) and Metabolic Disorders. International Journal of Molecular Sciences. 2023; 24(4):3219. https://doi.org/10.3390/ijms24043219
Chicago/Turabian StyleKitamura, Hiroshi. 2023. "Ubiquitin-Specific Proteases (USPs) and Metabolic Disorders" International Journal of Molecular Sciences 24, no. 4: 3219. https://doi.org/10.3390/ijms24043219
APA StyleKitamura, H. (2023). Ubiquitin-Specific Proteases (USPs) and Metabolic Disorders. International Journal of Molecular Sciences, 24(4), 3219. https://doi.org/10.3390/ijms24043219