
Lipids as Targets for Renal Cell Carcinoma Therapy



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
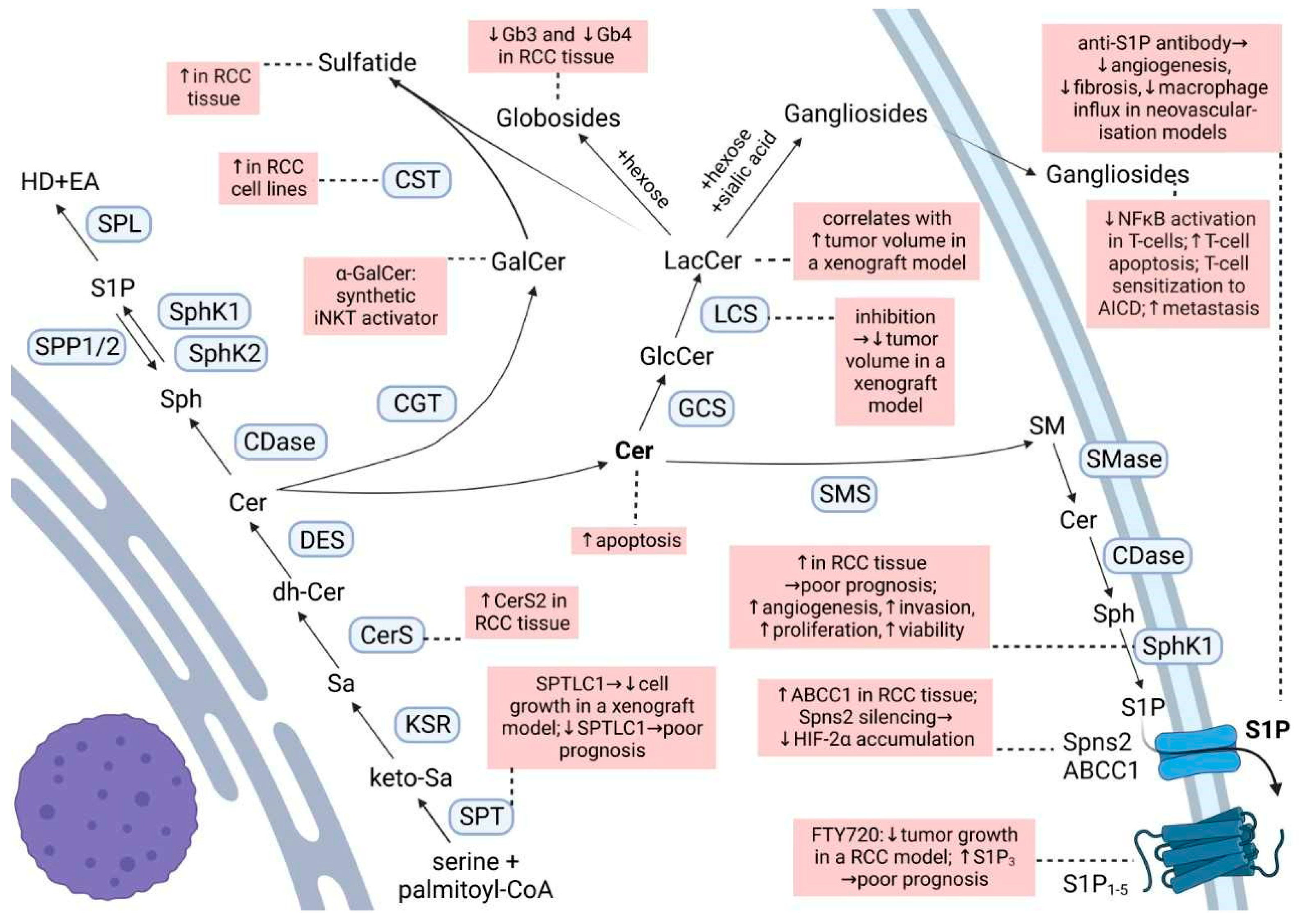
2. Sphingolipids in Renal Cancer
3. Glycosphingolipids in Renal Cancer
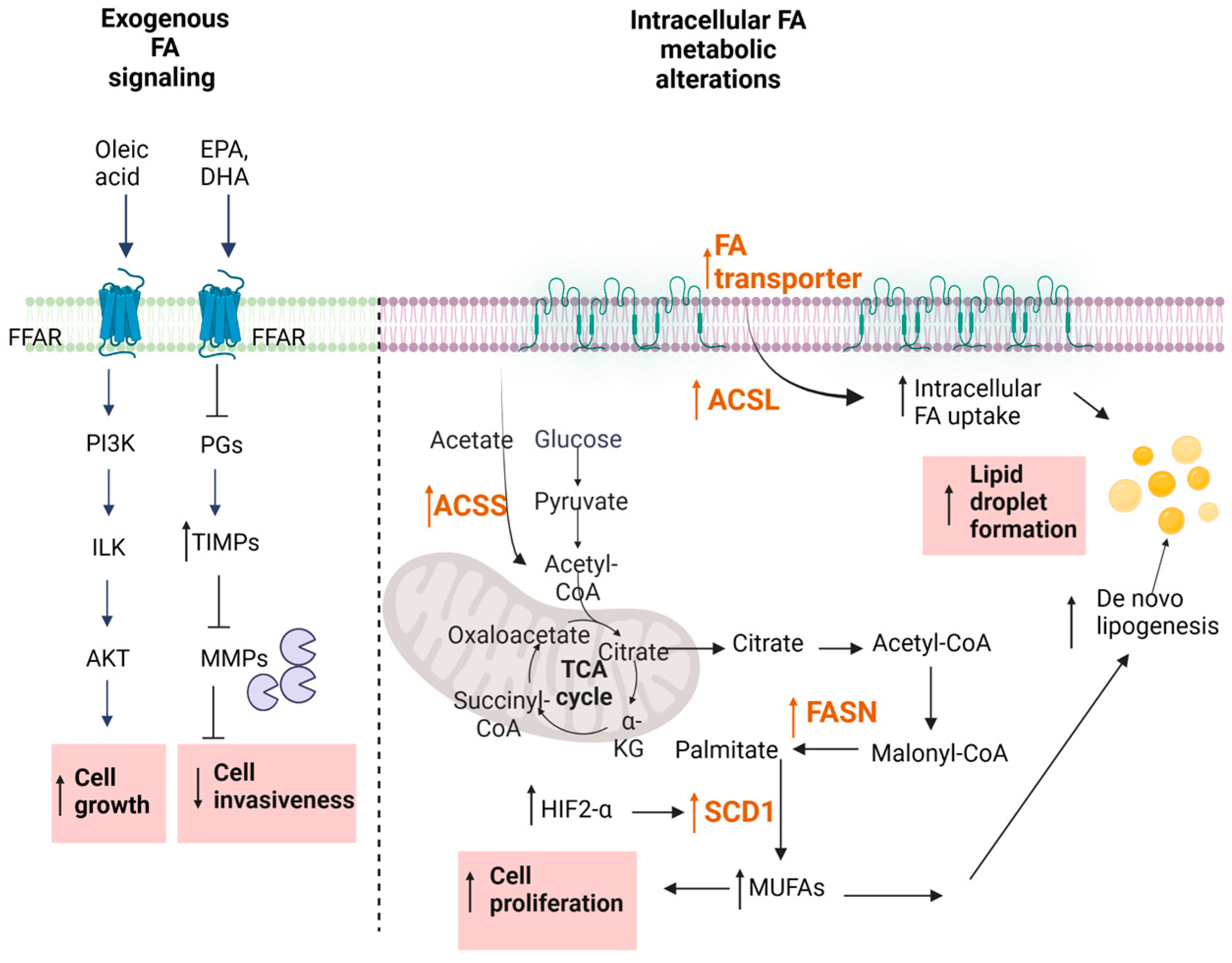
4. Free Fatty Acids in Renal Cancer
4.1. Exogenous Uptake of Fatty Acids
4.2. Regulation of Fatty Acid Signaling in Cancer
4.3. Altered De Novo FA Synthesis
5. Eicosanoids in Renal Cancer
5.1. Prostaglandins
5.2. Thromboxane
5.3. Leukotrienes
5.4. HETE and EETs
6. Cannabinoids in Renal Cancer
7. Cholesterol in Renal Cancer
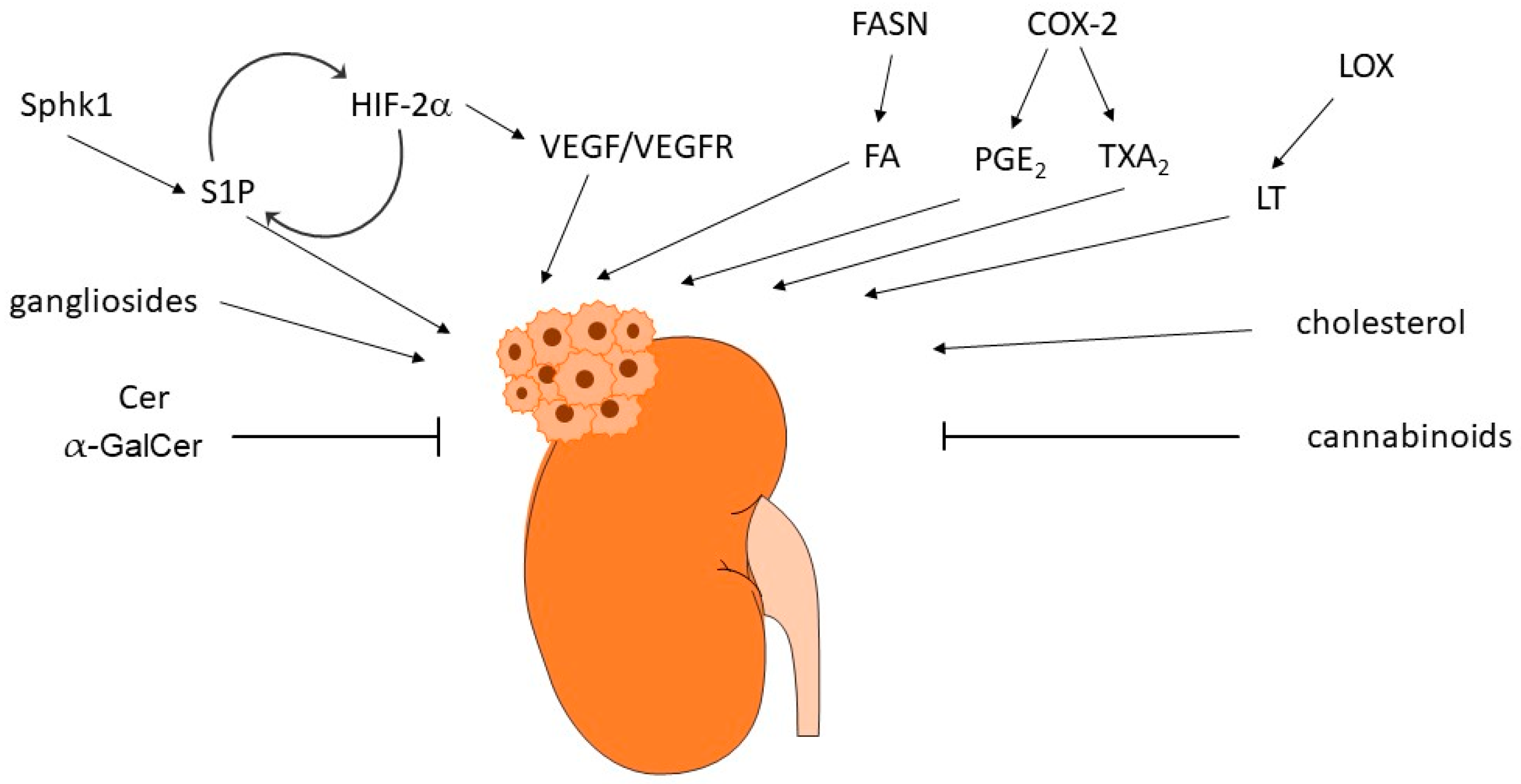
8. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Huang, G.; Wei, W.; Liu, J. Molecular Imaging of Renal Cell Carcinoma in Precision Medicine. Mol. Pharm. 2022, 19, 3457–3470. [Google Scholar] [CrossRef]
- Chow, W.H.; Dong, L.M.; Devesa, S.S. Epidemiology and risk factors for kidney cancer. Nat. Rev. Urol. 2010, 7, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2016, 70, 93–105. [Google Scholar] [CrossRef]
- Capitanio, U.; Bensalah, K.; Bex, A.; Boorjian, S.A.; Bray, F.; Coleman, J.; Gore, J.L.; Sun, M.; Wood, C.; Russo, P. Epidemiology of Renal Cell Carcinoma. Eur. Urol. 2019, 75, 74–84. [Google Scholar] [CrossRef]
- Drabkin, H.A.; Gemmill, R.M. Cholesterol and the development of clear-cell renal carcinoma. Curr. Opin. Pharmacol. 2012, 12, 742–750. [Google Scholar] [CrossRef]
- di Meo, N.A.; Lasorsa, F.; Rutigliano, M.; Loizzo, D.; Ferro, M.; Stella, A.; Bizzoca, C.; Vincenti, L.; Pandolfo, S.D.; Autorino, R.; et al. Renal Cell Carcinoma as a Metabolic Disease: An Update on Main Pathways, Potential Biomarkers, and Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 14360. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, G.; Loizzo, D.; Franzin, R.; Battaglia, S.; Ferro, M.; Cantiello, F.; Castellano, G.; Bettocchi, C.; Ditonno, P.; Battaglia, M. Metabolomic insights into pathophysiological mechanisms and biomarker discovery in clear cell renal cell carcinoma. Expert. Rev. Mol. Diagn. 2019, 19, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260, 1317–1320. [Google Scholar] [CrossRef]
- Gnarra, J.R.; Tory, K.; Weng, Y.; Schmidt, L.; Wei, M.H.; Li, H.; Latif, F.; Liu, S.; Chen, F.; Duh, F.M.; et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat. Genet. 1994, 7, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013, 27, 41–53. [Google Scholar] [CrossRef]
- Wicks, E.E.; Semenza, G.L. Hypoxia-inducible factors: Cancer progression and clinical translation. J. Clin. Investig. 2022, 132, e159839. [Google Scholar] [CrossRef]
- Patel, S.A.; Nilsson, M.B.; Le, X.; Cascone, T.; Jain, R.K.; Heymach, J.V. Molecular Mechanisms and Future Implications of VEGF/VEGFR in Cancer Therapy. Clin. Cancer Res. 2023, 29, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Kaelin, W.G., Jr. Targeting the HIF2-VEGF axis in renal cell carcinoma. Nat. Med. 2020, 26, 1519–1530. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar]
- Wallace, E.M.; Rizzi, J.P.; Han, G.; Wehn, P.M.; Cao, Z.; Du, X.; Cheng, T.; Czerwinski, R.M.; Dixon, D.D.; Goggin, B.S.; et al. A Small-Molecule Antagonist of HIF2alpha Is Efficacious in Preclinical Models of Renal Cell Carcinoma. Cancer Res. 2016, 76, 5491–5500. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 1999, 15, 551–578. [Google Scholar] [CrossRef]
- Huwiler, A.; Kolter, T.; Pfeilschifter, J.; Sandhoff, K. Physiology and pathophysiology of sphingolipid metabolism and signaling. Biochim. Biophys. Acta 2000, 1485, 63–99. [Google Scholar] [CrossRef]
- Huwiler, A.; Pfeilschifter, J. Sphingolipid signaling in renal fibrosis. Matrix Biol. 2018, 68–69, 230–247. [Google Scholar] [CrossRef]
- Henry, B.; Moller, C.; Dimanche-Boitrel, M.T.; Gulbins, E.; Becker, K.A. Targeting the ceramide system in cancer. Cancer Lett. 2013, 332, 286–294. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kim, E.A.; Sohn, U.D.; Yim, C.B.; Im, C. Cytotoxic Activity and Structure Activity Relationship of Ceramide Analogues in Caki-2 and HL-60 Cells. Korean J. Physiol. Pharmacol. 2010, 14, 441–447. [Google Scholar] [CrossRef] [Green Version]
- Stepanovska, B.; Huwiler, A. Targeting the S1P receptor signaling pathways as a promising approach for treatment of autoimmune and inflammatory diseases. Pharmacol. Res. 2020, 154, 104170. [Google Scholar] [CrossRef]
- Takuwa, Y.; Du, W.; Qi, X.; Okamoto, Y.; Takuwa, N.; Yoshioka, K. Roles of sphingosine-1-phosphate signaling in angiogenesis. World J. Biol. Chem. 2010, 1, 298. [Google Scholar] [CrossRef]
- Hoefflin, R.; Harlander, S.; Abhari, B.A.; Peighambari, A.; Adlesic, M.; Seidel, P.; Zodel, K.; Haug, S.; Göcmen, B.; Li, Y. Therapeutic Effects of Inhibition of Sphingosine-1-Phosphate Signaling in HIF-2α Inhibitor-Resistant Clear Cell Renal Cell Carcinoma. Cancers 2021, 13, 4801. [Google Scholar] [CrossRef]
- Ader, I.; Brizuela, L.; Bouquerel, P.; Malavaud, B.; Cuvillier, O. Sphingosine kinase 1: A new modulator of hypoxia inducible factor 1α during hypoxia in human cancer cells. Cancer Res. 2008, 68, 8635–8642. [Google Scholar] [CrossRef] [PubMed]
- Bouquerel, P.; Gstalder, C.; Müller, D.; Laurent, J.; Brizuela, L.; Sabbadini, R.; Malavaud, B.; Pyronnet, S.; Martineau, Y.; Ader, I. Essential role for SphK1/S1P signaling to regulate hypoxia-inducible factor 2α expression and activity in cancer. Oncogenesis 2016, 5, e209. [Google Scholar] [CrossRef]
- Anelli, V.; Gault, C.R.; Cheng, A.B.; Obeid, L.M. Sphingosine kinase 1 is up-regulated during hypoxia in U87MG glioma cells: Role of hypoxia-inducible factors 1 and 2. J. Biol. Chem. 2008, 283, 3365–3375. [Google Scholar] [CrossRef] [PubMed]
- Salama, M.F.; Carroll, B.; Adada, M.; Pulkoski-Gross, M.; Hannun, Y.A.; Obeid, L.M. A novel role of sphingosine kinase-1 in the invasion and angiogenesis of VHL mutant clear cell renal cell carcinoma. FASEB J. 2015, 29, 2803. [Google Scholar] [CrossRef]
- Młynarczyk, G.; Mikłosz, A.; Suchański, J.; Reza, S.; Romanowicz, L.; Sobolewski, K.; Chabowski, A.; Baranowski, M. Grade-dependent changes in sphingolipid metabolism in clear cell renal cell carcinoma. J. Cell. Biochem. 2022, 123, 819–829. [Google Scholar] [CrossRef]
- Xu, Y.; Dong, B.; Wang, J.; Zhang, J.; Xue, W.; Huang, Y. Sphingosine kinase 1 overexpression contributes to sunitinib resistance in clear cell renal cell carcinoma. Oncoimmunology 2018, 7, e1502130. [Google Scholar] [CrossRef] [PubMed]
- Cuvillier, O.; Ader, I.; Bouquerel, P.; Brizuela, L.; Gstalder, C.; Malavaud, B. Hypoxia, therapeutic resistance, and sphingosine 1-phosphate. Adv. Cancer Res. 2013, 117, 117–141. [Google Scholar]
- Zhang, L.; Wang, X.; Bullock, A.J.; Callea, M.; Shah, H.; Song, J.; Moreno, K.; Visentin, B.; Deutschman, D.; Alsop, D.C. Anti-S1P Antibody as a Novel Therapeutic Strategy for VEGFR TKI-Resistant Renal CancerS1P Inhibition as a New Treatment for RCC. Clin. Cancer Res. 2015, 21, 1925–1934. [Google Scholar] [CrossRef] [Green Version]
- Huwiler, A.; Bourquin, F.; Kotelevets, N.; Pastukhov, O.; Capitani, G.; Grütter, M.G.; Zangemeister-Wittke, U. A prokaryotic S1P lyase degrades extracellular S1P in vitro and in vivo: Implication for treating hyperproliferative disorders. PLoS ONE 2011, 6, e22436. [Google Scholar] [CrossRef]
- O’Brien, N.; Jones, S.T.; Williams, D.G.; Cunningham, H.B.; Moreno, K.; Visentin, B.; Gentile, A.; Vekich, J.; Shestowsky, W.; Hiraiwa, M. Production and characterization of monoclonal anti-sphingosine-1-phosphate antibodies 1. J. Lipid Res. 2009, 50, 2245–2257. [Google Scholar] [CrossRef] [PubMed]
- Caballero, S.; Swaney, J.; Moreno, K.; Afzal, A.; Kielczewski, J.; Stoller, G.; Cavalli, A.; Garland, W.; Hansen, G.; Sabbadini, R. Anti-sphingosine-1-phosphate monoclonal antibodies inhibit angiogenesis and sub-retinal fibrosis in a murine model of laser-induced choroidal neovascularization. Exp. Eye Res. 2009, 88, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Shen, J.; Dong, A.; Rashid, A.; Stoller, G.; Campochiaro, P.A. Blockade of sphingosine-1-phosphate reduces macrophage influx and retinal and choroidal neovascularization. J. Cell. Physiol. 2009, 218, 192–198. [Google Scholar] [CrossRef]
- Pal, S.K.; Drabkin, H.A.; Reeves, J.A.; Hainsworth, J.D.; Hazel, S.E.; Paggiarino, D.A.; Wojciak, J.; Woodnutt, G.; Bhatt, R.S. A phase 2 study of the sphingosine-1-phosphate antibody sonepcizumab in patients with metastatic renal cell carcinoma. Cancer 2017, 123, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Huwiler, A.; Zangemeister-Wittke, U. The sphingosine 1-phosphate receptor modulator fingolimod as a therapeutic agent: Recent findings and new perspectives. Pharmacol. Ther. 2018, 185, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Fischl, A.S.; Wang, X.; Falcon, B.L.; Almonte-Baldonado, R.; Bodenmiller, D.; Evans, G.; Stewart, J.; Wilson, T.; Hipskind, P.; Manro, J. Inhibition of Sphingosine Phosphate Receptor 1 Signaling Enhances the Efficacy of VEGF Receptor InhibitionS1P1 Inhibition Improves VEGFR-Targeted Therapy. Mol. Cancer Ther. 2019, 18, 856–867. [Google Scholar] [CrossRef]
- Ying, Y.; Ma, X.; Fang, J.; Chen, S.; Wang, W.; Li, J.; Xie, H.; Wu, J.; Xie, B.; Liu, B. EGR2-mediated regulation of m6A reader IGF2BP proteins drive RCC tumorigenesis and metastasis via enhancing S1PR3 mRNA stabilization. Cell Death Dis. 2021, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Bao, G.; Pei, J.; Cao, Y.; Zhang, C.; Zhao, P.; Zhang, Y.; Damirin, A. NF-κB and EGFR participate in S1PR3-mediated human renal cell carcinomas progression. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2022, 1868, 166401. [Google Scholar] [CrossRef]
- Glueck, M.; Koch, A.; Brunkhorst, R.; Ferreiros Bouzas, N.; Trautmann, S.; Schaefer, L.; Pfeilschifter, W.; Pfeilschifter, J.; Vutukuri, R. The atypical sphingosine 1-phosphate variant, d16: 1 S1P, mediates CTGF induction via S1P2 activation in renal cell carcinoma. FEBS J. 2022, 289, 5670–5681. [Google Scholar] [CrossRef]
- Hanada, K. Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2003, 1632, 16–30. [Google Scholar] [CrossRef]
- Zhu, W.K.; Xu, W.H.; Wang, J.; Huang, Y.Q.; Abudurexiti, M.; Qu, Y.Y.; Zhu, Y.P.; Zhang, H.L.; Ye, D.W. Decreased SPTLC1 expression predicts worse outcomes in ccRCC patients. J. Cell. Biochem. 2020, 121, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Kong, Z.; Guo, X.; Zhao, Z.; Wu, W.; Luo, L.; Zhu, Z.; Yin, S.; Cai, C.; Wu, W.; Wang, D. SPTLC1 inhibits cell growth via modulating Akt/FOXO1 pathway in renal cell carcinoma cells. Biochem. Biophys. Res. Commun. 2019, 520, 1–7. [Google Scholar] [CrossRef]
- Wattenberg, B.W. The long and the short of ceramides. J. Biol. Chem. 2018, 293, 9922–9923. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-K.; Maaß, M.; Quach, A.; Poscic, N.; Prangley, H.; Pallott, E.-C.; Kim, J.L.; Pierce, J.S.; Ogretmen, B.; Futerman, A.H. Dependence of ABCB1 transporter expression and function on distinct sphingolipids generated by ceramide synthases-2 and-6 in chemoresistant renal cancer. J. Biol. Chem. 2022, 298, 101492. [Google Scholar] [CrossRef]
- Schwaab, T.; Ernstoff, M.S. Therapeutic vaccines in renal cell carcinoma. Therapy 2011, 4, 369. [Google Scholar] [CrossRef]
- Companioni, O.; Mir, C.; Garcia-Mayea, Y.; LLeonart, M.E. Targeting Sphingolipids for Cancer Therapy. Front. Oncol. 2021, 11, 745092. [Google Scholar] [CrossRef]
- Lehmann, N.; Paret, C.; El Malki, K.; Russo, A.; Neu, M.A.; Wingerter, A.; Seidmann, L.; Foersch, S.; Ziegler, N.; Roth, L. Tumor Lipids of Pediatric Papillary Renal Cell Carcinoma Stimulate Unconventional T Cells. Front. Immunol. 2020, 11, 1819. [Google Scholar] [CrossRef]
- Vyth-Dreese, F.A.; Sein, J.; van de Kasteele, W.; Dellemijn, T.A.; van den Bogaard, C.; Nooijen, W.J.; de Gast, G.C.; Haanen, J.B.; Bex, A. Lack of anti-tumour reactivity despite enhanced numbers of circulating natural killer T cells in two patients with metastatic renal cell carcinoma. Clin. Exp. Immunol. 2010, 162, 447–459. [Google Scholar] [CrossRef]
- Takahashi, T.; Suzuki, T. Role of sulfatide in normal and pathological cells and tissues. J. Lipid Res. 2012, 53, 1437–1450. [Google Scholar] [CrossRef] [Green Version]
- Sakakibara, N. Glycolipid alterations in human kidney carcinoma. [Hokkaido Igaku Zasshi] Hokkaido J. Med. Sci. 1989, 64, 75–82. [Google Scholar]
- Sakakibara, N.; Gasa, S.; Kamio, K.; Makita, A.; Koyanagi, T. Association of elevated sulfatides and sulfotransferase activities with human renal cell carcinoma. Cancer Res. 1989, 49, 335–339. [Google Scholar]
- Jirasko, R.; Idkowiak, J.; Wolrab, D.; Kvasnicka, A.; Friedecky, D.; Polanski, K.; Studentova, H.; Student, V.; Melichar, B.; Holcapek, M. Altered Plasma, Urine, and Tissue Profiles of Sulfatides and Sphingomyelins in Patients with Renal Cell Carcinoma. Cancers 2022, 14, 4622. [Google Scholar] [CrossRef] [PubMed]
- Porubsky, S.; Nientiedt, M.; Kriegmair, M.C.; Siemoneit, J.-H.H.; Sandhoff, R.; Jennemann, R.; Borgmann, H.; Gaiser, T.; Weis, C.-A.; Erben, P. The prognostic value of galactosylceramide-sulfotransferase (Gal3ST1) in human renal cell carcinoma. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef]
- Jirásko, R.; Holčapek, M.; Khalikova, M.; Vrána, D.; Študent, V.; Prouzová, Z.; Melichar, B. MALDI orbitrap mass spectrometry profiling of dysregulated sulfoglycosphingolipids in renal cell carcinoma tissues. J. Am. Soc. Mass Spectrom. 2017, 28, 1562–1574. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Y.; Patwardhan, G.A.; Xie, P.; Gu, X.; Giuliano, A.E.; Cabot, M.C. Glucosylceramide synthase, a factor in modulating drug resistance, is overexpressed in metastatic breast carcinoma. Int. J. Oncol. 2011, 39, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Y.; Gupta, V.; Patwardhan, G.A.; Bhinge, K.; Zhao, Y.; Bao, J.; Mehendale, H.; Cabot, M.C.; Li, Y.-T.; Jazwinski, S.M. Glucosylceramide synthase upregulates MDR1 expression in the regulation of cancer drug resistance through cSrc and β-catenin signaling. Mol. Cancer 2010, 9, 1–15. [Google Scholar] [CrossRef]
- Jones, J.; Otu, H.; Spentzos, D.; Kolia, S.; Inan, M.; Beecken, W.D.; Fellbaum, C.; Gu, X.; Joseph, M.; Pantuck, A.J. Gene signatures of progression and metastasis in renal cell cancer. Clin. Cancer Res. 2005, 11, 5730–5739. [Google Scholar] [CrossRef]
- Liu, Y.-Y.; Han, T.Y.; Yu, J.Y.; Bitterman, A.; Le, A.; Giuliano, A.E.; Cabot, M.C. Oligonucleotides blocking glucosylceramide synthase expression selectively reverse drug resistance in cancer cells. J. Lipid Res. 2004, 45, 933–940. [Google Scholar] [CrossRef]
- Chatterjee, S.; Alsaeedi, N.; Hou, J.; Bandaru, V.V.R.; Wu, L.; Halushka, M.K.; Pili, R.; Ndikuyeze, G.; Haughey, N.J. Use of a glycolipid inhibitor to ameliorate renal cancer in a mouse model. PLoS ONE 2013, 8, e63726. [Google Scholar] [CrossRef] [PubMed]
- Uzzo, R.G.; Rayman, P.; Kolenko, V.; Clark, P.E.; Cathcart, M.K.; Bloom, T.; Novick, A.C.; Bukowski, R.M.; Hamilton, T.; Finke, J.H. Renal cell carcinoma–derived gangliosides suppress nuclear factor-κB activation in T cells. J. Clin. Investig. 1999, 104, 769–776. [Google Scholar] [CrossRef]
- Finke, J.H.; Rayman, P.; George, R.; Tannenbaum, C.S.; Kolenko, V.; Uzzo, R.; Novick, A.C.; Bukowski, R.M. Tumor-induced sensitivity to apoptosis in T cells from patients with renal cell carcinoma: Role of nuclear factor-κB suppression. Clin. Cancer Res. 2001, 7, 940s–946s. [Google Scholar]
- Kudo, D.; Rayman, P.; Horton, C.; Cathcart, M.K.; Bukowski, R.M.; Thornton, M.; Tannenbaum, C.; Finke, J.H. Gangliosides expressed by the renal cell carcinoma cell line SK-RC-45 are involved in tumor-induced apoptosis of T cells. Cancer Res. 2003, 63, 1676–1683. [Google Scholar]
- Biswas, S.; Biswas, K.; Richmond, A.; Ko, J.; Ghosh, S.; Simmons, M.; Rayman, P.; Rini, B.; Gill, I.; Tannenbaum, C.S. Elevated levels of select gangliosides in T cells from renal cell carcinoma patients is associated with T cell dysfunction. J. Immunol. 2009, 183, 5050–5058. [Google Scholar] [CrossRef] [Green Version]
- Biswas, K.; Richmond, A.; Rayman, P.; Biswas, S.; Thornton, M.; Sa, G.; Das, T.; Zhang, R.; Chahlavi, A.; Tannenbaum, C.S. GM2 expression in renal cell carcinoma: Potential role in tumor-induced T-cell dysfunction. Cancer Res. 2006, 66, 6816–6825. [Google Scholar] [CrossRef]
- Hoon, D.S.; Okun, E.; Neuwirth, H.; Morton, D.L.; Irie, R.F. Aberrant expression of gangliosides in human renal cell carcinomas. J. Urol. 1993, 150, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Handa, K.; Withers, D.A.; Satoh, M.; Hakomori, S.-i. Binding specificity of siglec7 to disialogangliosides of renal cell carcinoma: Possible role of disialogangliosides in tumor progression. FEBS Lett. 2001, 504, 82–86. [Google Scholar] [CrossRef]
- Wu, D.-Y.; Adak, A.K.; Kuo, Y.-T.; Shen, Y.-J.; Li, P.-J.; Hwu, J.R.; Lin, C.-C. A Modular Chemoenzymatic Synthesis of Disialosyl Globopentaosylceramide (DSGb5Cer) Glycan. J. Org. Chem. 2020, 85, 15920–15935. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Ito, A.; Kakoi, N.; Shimada, S.; Itoh, J.; Mitsuzuka, K.; Arai, Y. Ganglioside, disialosyl globopentaosylceramide (DSGb5), enhances the migration of renal cell carcinoma cells. Tohoku J. Exp. Med. 2015, 236, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Itoh, J.; Ito, A.; Shimada, S.; Kawasaki, Y.; Kakoi, N.; Saito, H.; Mitsuzuka, K.; Watanabe, M.; Satoh, M.; Saito, S. Clinicopathological significance of ganglioside DSGb5 expression in renal cell carcinoma. Glycoconj. J. 2017, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, R.; Saito, S.; Bilim, V.; Hara, N.; Itoi, T.; Yamana, K.; Nishiyama, T.; Arai, Y.; Takahashi, K.; Tomita, Y. High incidence of GalNAc disialosyl lactotetraosylceramide in metastatic renal cell carcinoma. Anticancer. Res. 2007, 27, 4345–4350. [Google Scholar]
- Saito, S.; Orikasa, S.; Satoh, M.; Ohyama, C.; Ito, A.; Takahashi, T. Expression of globo-series gangliosides in human renal cell carcinoma. Jpn. J. Cancer Res. 1997, 88, 652–659. [Google Scholar] [CrossRef]
- Satoh, M.; Nejad, F.M.; Nakano, O.; Ito, A.; Kawamura, S.; Ohyama, C.; Saito, S.; Orikasa, S. Four new human renal cell carcinoma cell lines expressing globo-series gangliosides. Tohoku J. Exp. Med. 1999, 189, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Tringali, C.; Lupo, B.; Silvestri, I.; Papini, N.; Anastasia, L.; Tettamanti, G.; Venerando, B. The plasma membrane sialidase NEU3 regulates the malignancy of renal carcinoma cells by controlling β1 integrin internalization and recycling. J. Biol. Chem. 2012, 287, 42835–42845. [Google Scholar] [CrossRef]
- Saito, S.; Nojiri, H.; Satoh, M.; Ito, A.; Ohyama, C.; Orikasa, S. Inverse relationship of expression between GM3 and globo-series ganglioside in human renal cell carcinoma. Tohoku J. Exp. Med. 2000, 190, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Woutersen, R.A.; Appel, M.J.; van Garderen-Hoetmer, A.; Wijnands, M.V. Dietary fat and carcinogenesis. Mutat. Res. 1999, 443, 111–127. [Google Scholar] [CrossRef]
- Bojkova, B.; Winklewski, P.J.; Wszedybyl-Winklewska, M. Dietary Fat and Cancer-Which Is Good, Which Is Bad, and the Body of Evidence. Int. J. Mol. Sci. 2020, 21, 4114. [Google Scholar] [CrossRef]
- Gerber, M. Omega-3 fatty acids and cancers: A systematic update review of epidemiological studies. Br. J. Nutr. 2012, 107 (Suppl. S2), S228–S239. [Google Scholar] [CrossRef] [PubMed]
- Liotti, A.; Cosimato, V.; Mirra, P.; Cali, G.; Conza, D.; Secondo, A.; Luongo, G.; Terracciano, D.; Formisano, P.; Beguinot, F.; et al. Oleic acid promotes prostate cancer malignant phenotype via the G protein-coupled receptor FFA1/GPR40. J. Cell. Physiol. 2018, 233, 7367–7378. [Google Scholar] [CrossRef] [PubMed]
- Brasky, T.M.; Darke, A.K.; Song, X.; Tangen, C.M.; Goodman, P.J.; Thompson, I.M.; Meyskens, F.L., Jr.; Goodman, G.E.; Minasian, L.M.; Parnes, H.L.; et al. Plasma phospholipid fatty acids and prostate cancer risk in the SELECT trial. J. Natl. Cancer Inst. 2013, 105, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xiao, Y.; Yuan, Y.; Zhang, X.; Qin, C.; Xie, J.; Hao, Y.; Xu, T.; Wang, X. Effects of oleic acid on cell proliferation through an integrin-linked kinase signaling pathway in 786-O renal cell carcinoma cells. Oncol. Lett. 2013, 5, 1395–1399. [Google Scholar] [CrossRef]
- Xiang, F.; Wu, K.; Liu, Y.; Shi, L.; Wang, D.; Li, G.; Tao, K.; Wang, G. Omental adipocytes enhance the invasiveness of gastric cancer cells by oleic acid-induced activation of the PI3K-Akt signaling pathway. Int. J. Biochem. Cell Biol. 2017, 84, 14–21. [Google Scholar] [CrossRef]
- Soto-Guzman, A.; Navarro-Tito, N.; Castro-Sanchez, L.; Martinez-Orozco, R.; Salazar, E.P. Oleic acid promotes MMP-9 secretion and invasion in breast cancer cells. Clin. Exp. Metastasis 2010, 27, 505–515. [Google Scholar] [CrossRef]
- McCabe, A.J.; Wallace, J.M.; Gilmore, W.S.; McGlynn, H.; Strain, S.J. Docosahexaenoic acid reduces in vitro invasion of renal cell carcinoma by elevated levels of tissue inhibitor of metalloproteinase-1. J. Nutr. Biochem. 2005, 16, 17–22. [Google Scholar] [CrossRef]
- Bidoli, E.; Talamini, R.; Zucchetto, A.; Polesel, J.; Bosetti, C.; Negri, E.; Maruzzi, D.; Montella, M.; Franceschi, S.; La Vecchia, C. Macronutrients, fatty acids, cholesterol and renal cell cancer risk. Int. J. Cancer 2008, 122, 2586–2589. [Google Scholar] [CrossRef] [PubMed]
- Brock, K.E.; Gridley, G.; Chiu, B.C.; Ershow, A.G.; Lynch, C.F.; Cantor, K.P. Dietary fat and risk of renal cell carcinoma in the USA: A case-control study. Br. J. Nutr. 2009, 101, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Spiegelman, D.; Hunter, D.J.; Albanes, D.; Bernstein, L.; van den Brandt, P.A.; Buring, J.E.; Cho, E.; English, D.R.; Freudenheim, J.L.; et al. Fat, protein, and meat consumption and renal cell cancer risk: A pooled analysis of 13 prospective studies. J. Natl. Cancer Inst. 2008, 100, 1695–1706. [Google Scholar] [CrossRef]
- Schwenk, R.W.; Holloway, G.P.; Luiken, J.J.; Bonen, A.; Glatz, J.F. Fatty acid transport across the cell membrane: Regulation by fatty acid transporters. Prostaglandins Leukot Essent Fat. Acids 2010, 82, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, J.E.; Lodish, H.F. Expression cloning and characterization of a novel adipocyte long chain fatty acid transport protein. Cell 1994, 79, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Kimura, I.; Inoue, D.; Ichimura, A.; Hirasawa, A. Free fatty acid receptors and their role in regulation of energy metabolism. Rev. Physiol. Biochem. Pharmacol. 2013, 164, 77–116. [Google Scholar]
- Hopkins, M.M.; Meier, K.E. Free Fatty Acid Receptors and Cancer: From Nutrition to Pharmacology. Handb. Exp. Pharmacol. 2017, 236, 233–251. [Google Scholar] [PubMed]
- Hopkins, M.M.; Meier, K.E. Free fatty acid receptor (FFAR) agonists inhibit proliferation of human ovarian cancer cells. Prostaglandins Leukot Essent Fat. Acids 2017, 122, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, M.M.; Zhang, Z.; Liu, Z.; Meier, K.E. Eicosopentaneoic Acid and Other Free Fatty Acid Receptor Agonists Inhibit Lysophosphatidic Acid- and Epidermal Growth Factor-Induced Proliferation of Human Breast Cancer Cells. J. Clin. Med. 2016, 5, 16. [Google Scholar] [CrossRef]
- Wang, J.; Hong, Y.; Shao, S.; Zhang, K.; Hong, W. FFAR1-and FFAR4-dependent activation of Hippo pathway mediates DHA-induced apoptosis of androgen-independent prostate cancer cells. Biochem. Biophys. Res. Commun. 2018, 506, 590–596. [Google Scholar] [CrossRef]
- Takahashi, K.; Fukushima, K.; Onishi, Y.; Minami, K.; Otagaki, S.; Ishimoto, K.; Fukushima, N.; Honoki, K.; Tsujiuchi, T. Involvement of FFA1 and FFA4 in the regulation of cellular functions during tumor progression in colon cancer cells. Exp. Cell Res. 2018, 369, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Monaco, M.E. Fatty acid metabolism in breast cancer subtypes. Oncotarget 2017, 8, 29487–29500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, S.K.; Hougen, H.Y.; Merchan, J.R.; Gonzalgo, M.L.; Welford, S.M. Fatty acid metabolism reprogramming in ccRCC: Mechanisms and potential targets. Nat. Rev. Urol. 2022, 20, 48–60. [Google Scholar] [CrossRef]
- Alo, P.L.; Visca, P.; Marci, A.; Mangoni, A.; Botti, C.; Di Tondo, U. Expression of fatty acid synthase (FAS) as a predictor of recurrence in stage I breast carcinoma patients. Cancer 1996, 77, 474–482. [Google Scholar] [CrossRef]
- Epstein, J.I.; Carmichael, M.; Partin, A.W. OA-519 (fatty acid synthase) as an independent predictor of pathologic state in adenocarcinoma of the prostate. Urology 1995, 45, 81–86. [Google Scholar] [CrossRef]
- Rashid, A.; Pizer, E.S.; Moga, M.; Milgraum, L.Z.; Zahurak, M.; Pasternack, G.R.; Kuhajda, F.P.; Hamilton, S.R. Elevated expression of fatty acid synthase and fatty acid synthetic activity in colorectal neoplasia. Am. J. Pathol. 1997, 150, 201–208. [Google Scholar] [PubMed]
- Nguyen, P.L.; Ma, J.; Chavarro, J.E.; Freedman, M.L.; Lis, R.; Fedele, G.; Fiore, C.; Qiu, W.; Fiorentino, M.; Finn, S.; et al. Fatty acid synthase polymorphisms, tumor expression, body mass index, prostate cancer risk, and survival. J. Clin. Oncol. 2010, 28, 3958–3964. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, A.; Asano, T.; Asano, T.; Ito, K.; Sumitomo, M.; Hayakawa, M. Pharmacological inhibitor of fatty acid synthase suppresses growth and invasiveness of renal cancer cells. J. Urol. 2008, 180, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Hu, X.; Anwaier, A.; Wang, J.; Liu, W.; Tian, X.; Zhu, W.; Ma, C.; Wan, F.; Shi, G.; et al. Fatty Acid Synthase Correlates With Prognosis-Related Abdominal Adipose Distribution and Metabolic Disorders of Clear Cell Renal Cell Carcinoma. Front. Mol. Biosci. 2020, 7, 610229. [Google Scholar] [CrossRef]
- Yuan, Y.; Yang, X.; Li, Y.; Liu, Q.; Wu, F.; Qu, H.; Gao, H.; Ge, J.; Xu, Y.; Wang, H.; et al. Expression and prognostic significance of fatty acid synthase in clear cell renal cell carcinoma. Pathol. Res. Pract. 2020, 216, 153227. [Google Scholar] [CrossRef]
- Che, L.; Paliogiannis, P.; Cigliano, A.; Pilo, M.G.; Chen, X.; Calvisi, D.F. Pathogenetic, Prognostic, and Therapeutic Role of Fatty Acid Synthase in Human Hepatocellular Carcinoma. Front. Oncol. 2019, 9, 1412. [Google Scholar] [CrossRef]
- Lu, T.; Sun, L.; Wang, Z.; Zhang, Y.; He, Z.; Xu, C. Fatty acid synthase enhances colorectal cancer cell proliferation and metastasis via regulating AMPK/mTOR pathway. Onco Targets Ther. 2019, 12, 3339–3347. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Fang, S.; Chen, Y.; Yang, Z.; Yuan, Y.; Zhang, J.; Ye, L.; Gu, W. Inhibition of FASN suppresses the malignant biological behavior of non-small cell lung cancer cells via deregulating glucose metabolism and AKT/ERK pathway. Lipids Health Dis. 2019, 18, 118. [Google Scholar] [CrossRef]
- Wettersten, H.I.; Hakimi, A.A.; Morin, D.; Bianchi, C.; Johnstone, M.E.; Donohoe, D.R.; Trott, J.F.; Aboud, O.A.; Stirdivant, S.; Neri, B.; et al. Grade-Dependent Metabolic Reprogramming in Kidney Cancer Revealed by Combined Proteomics and Metabolomics Analysis. Cancer Res. 2015, 75, 2541–2552. [Google Scholar] [CrossRef]
- Albiges, L.; Hakimi, A.A.; Xie, W.; McKay, R.R.; Simantov, R.; Lin, X.; Lee, J.L.; Rini, B.I.; Srinivas, S.; Bjarnason, G.A.; et al. Body Mass Index and Metastatic Renal Cell Carcinoma: Clinical and Biological Correlations. J. Clin. Oncol. 2016, 34, 3655–3663. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Huang, C.; Sun, J.; Qiu, W.; Zhang, J.; Li, H.; Jiang, T.; Huang, K.; Cao, J. RNA interference-mediated signal transducers and activators of transcription 3 gene silencing inhibits invasion and metastasis of human pancreatic cancer cells. Cancer Sci. 2007, 98, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Slade, R.F.; Hunt, D.A.; Pochet, M.M.; Venema, V.J.; Hennigar, R.A. Characterization and inhibition of fatty acid synthase in pediatric tumor cell lines. Anticancer. Res. 2003, 23, 1235–1243. [Google Scholar] [PubMed]
- Wang, X.; Du, G.; Wu, Y.; Zhang, Y.; Guo, F.; Liu, W.; Wu, R. Association between different levels of lipid metabolismrelated enzymes and fatty acid synthase in Wilms’ tumor. Int. J. Oncol. 2020, 56, 568–580. [Google Scholar]
- Camassei, F.D.; Jenkner, A.; Rava, L.; Bosman, C.; Francalanci, P.; Donfrancesco, A.; Alo, P.L.; Boldrini, R. Expression of the lipogenic enzyme fatty acid synthase (FAS) as a predictor of poor outcome in nephroblastoma: An interinstitutional study. Med. Pediatr. Oncol. 2003, 40, 302–308. [Google Scholar] [CrossRef]
- Quan, J.; Bode, A.M.; Luo, X. ACSL family: The regulatory mechanisms and therapeutic implications in cancer. Eur. J. Pharmacol. 2021, 909, 174397. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Hu, H.L.; Huang, R.Z.; Huang, G.M.; Xi, X.Q. ACSL3 is a potential prognostic biomarker for immune infiltration in clear cell renal cell carcinoma. Front. Surg. 2022, 9, 909854. [Google Scholar] [CrossRef]
- Klasson, T.D.; LaGory, E.L.; Zhao, H.; Huynh, S.K.; Papandreou, I.; Moon, E.J.; Giaccia, A.J. ACSL3 regulates lipid droplet biogenesis and ferroptosis sensitivity in clear cell renal cell carcinoma. Cancer Metab. 2022, 10, 14. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, H.; Zhang, J.; Lv, J.; Huang, Y. Positive feedback loop and synergistic effects between hypoxia-inducible factor-2alpha and stearoyl-CoA desaturase-1 promote tumorigenesis in clear cell renal cell carcinoma. Cancer Sci. 2013, 104, 416–422. [Google Scholar] [CrossRef]
- von Roemeling, C.A.; Marlow, L.A.; Wei, J.J.; Cooper, S.J.; Caulfield, T.R.; Wu, K.; Tan, W.W.; Tun, H.W.; Copland, J.A. Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target for clear cell renal cell carcinoma. Clin. Cancer Res. 2013, 19, 2368–2380. [Google Scholar] [CrossRef]
- Tracz-Gaszewska, Z.; Dobrzyn, P. Stearoyl-CoA Desaturase 1 as a Therapeutic Target for the Treatment of Cancer. Cancers 2019, 11, 948. [Google Scholar] [CrossRef] [PubMed]
- Budhu, A.; Roessler, S.; Zhao, X.; Yu, Z.; Forgues, M.; Ji, J.; Karoly, E.; Qin, L.X.; Ye, Q.H.; Jia, H.L.; et al. Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology 2013, 144, 1066–1075.e1. [Google Scholar] [CrossRef]
- Chavarro, J.E.; Kenfield, S.A.; Stampfer, M.J.; Loda, M.; Campos, H.; Sesso, H.D.; Ma, J. Blood levels of saturated and monounsaturated fatty acids as markers of de novo lipogenesis and risk of prostate cancer. Am. J. Epidemiol. 2013, 178, 1246–1255. [Google Scholar] [CrossRef] [PubMed]
- Raeisi, M.; Hassanbeigi, L.; Khalili, F.; Kharrati-Shishavan, H.; Yousefi, M.; Mehdizadeh, A. Stearoyl-CoA desaturase 1 as a therapeutic target for cancer: A focus on hepatocellular carcinoma. Mol. Biol. Rep. 2022, 49, 8871–8882. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Eicosanoids. Essays Biochem. 2020, 64, 423–441. [Google Scholar]
- Gomes, R.N.; Felipe da Costa, S.; Colquhoun, A. Eicosanoids and cancer. Clinics 2018, 73, e530s. [Google Scholar] [CrossRef]
- Wang, D.; DuBois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef]
- Wang, Q.; Morris, R.J.; Bode, A.M.; Zhang, T. Prostaglandin pathways: Opportunities for cancer prevention and therapy. Cancer Res. 2022, 82, 949. [Google Scholar] [CrossRef] [PubMed]
- De Keijzer, S.; Meddens, M.B.; Torensma, R.; Cambi, A. The multiple faces of prostaglandin E2 G-protein coupled receptor signaling during the dendritic cell life cycle. Int. J. Mol. Sci. 2013, 14, 6542–6555. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.C. COX-2 and the kidney. J. Cardiovasc. Pharmacol. 2006, 47, S37–S42. [Google Scholar] [CrossRef]
- Tuna, B.; Yorukoglu, K.; Gurel, D.; Mungan, U.; Kirkali, Z. Significance of COX-2 expression in human renal cell carcinoma. Urology 2004, 64, 1116–1120. [Google Scholar] [CrossRef]
- Osman, W.M.; Youssef, N.S. Combined use of COX-1 and VEGF immunohistochemistry refines the histopathologic prognosis of renal cell carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 8165. [Google Scholar]
- Miyata, Y.; Koga, S.; Kanda, S.; Nishikido, M.; Hayashi, T.; Kanetake, H. Expression of cyclooxygenase-2 in renal cell carcinoma: Correlation with tumor cell proliferation, apoptosis, angiogenesis, expression of matrix metalloproteinase-2, and survival. Clin. Cancer Res. 2003, 9, 1741–1749. [Google Scholar] [PubMed]
- Cho, D.S.; Joo, H.J.; Oh, D.K.; Kang, J.H.; Kim, Y.S.; Lee, K.B.; Kim, S.J. Cyclooxygenase-2 and p53 expression as prognostic indicators in conventional renal cell carcinoma. Yonsei Med. J. 2005, 46, 133–140. [Google Scholar] [CrossRef]
- Kankuri-Tammilehto, M.K.; Söderström, K.-O.; Pelliniemi, T.-T.; Vahlberg, T.; Pyrhönen, S.O.; Salminen, E.K. Prognostic evaluation of COX-2 expression in renal cell carcinoma. Anticancer. Res. 2010, 30, 3023–3030. [Google Scholar] [PubMed]
- Hashemi Goradel, N.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in cancer: A review. J. Cell. Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef] [PubMed]
- Park, G.; Song, N.-Y.; Kim, D.-H.; Lee, S.-J.; Chun, K.-S. Thymoquinone suppresses migration of human renal carcinoma caki-1 cells through inhibition of the PGE2-mediated activation of the EP2 receptor pathway. Biomol. Ther. 2021, 29, 64. [Google Scholar] [CrossRef]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. 2015, 15, 1–6. [Google Scholar] [CrossRef]
- Li, J.; Feng, G.; Liu, J.; Rong, R.; Luo, F.; Guo, L.; Zhu, T.; Wang, G.; Chu, Y. Renal cell carcinoma may evade the immune system by converting CD4+ Foxp3-T cells into CD4+ CD25+ Foxp3+ regulatory T cells: Role of tumor COX-2-derived PGE2. Mol. Med. Rep. 2010, 3, 959–963. [Google Scholar]
- Li, J.F.; Chu, Y.W.; Wang, G.M.; Zhu, T.Y.; Rong, R.M.; Hou, J.; Xu, M. The prognostic value of peritumoral regulatory T cells and its correlation with intratumoral cyclooxygenase-2 expression in clear cell renal cell carcinoma. BJU Int. 2009, 103, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.P.; Stapleton, P.P.; Barden, C.B.; Mestre, J.R.; Freeman, T.A.; Duff, M.D.; Maddali, S.; Yan, Z.; Daly, J.M. Renal cell carcinoma induces prostaglandin E2 and T-helper type 2 cytokine production in peripheral blood mononuclear cells. Ann. Surg. Oncol. 2003, 10, 455–462. [Google Scholar] [CrossRef]
- Menetrier-Caux, C.; Bain, C.; Favrot, M.; Duc, A.; Blay, J. Renal cell carcinoma induces interleukin 10 and prostaglandin E2 production by monocytes. Br. J. Cancer 1999, 79, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Hamieh, L.; Moreira, R.B.; Lin, X.; Simantov, R.; Choueiri, T.K.; McKay, R.R. Impact of Aspirin and Non-Aspirin Nonsteroidal Anti-Inflammatory Drugs on Outcomes in Patients with Metastatic Renal Cell Carcinoma. Kidney Cancer 2018, 2, 37–46. [Google Scholar] [CrossRef]
- Ohba, K.; Miyata, Y.; Sakai, H. Expression and function of E prostanoid receptors in urological cancer. Hinyokika Kiyo. Acta Urol. Jpn. 2013, 59, 83–89. [Google Scholar]
- Sato, N.; Mizutani, Y.; Li, Y.N.; Fujiwara, J.; Ishida, H.; Toiyama, D.; Abe, K.; Hayashi, I.; Nakanishi, H.; Kawauchi, A. Enhancement of the sensitivity of renal cell carcinoma cells to fas-mediated cytotoxicity and apoptosis by the selective cyclooxygenase-2 inhibitor JTE-522. Urol. Int. 2010, 84, 362–368. [Google Scholar] [CrossRef]
- Yoshimura, R.; Matsuyama, M.; Kawahito, Y.; Takemoto, Y.; Tsuchida, K.; Kuratsukuri, K.; Segawa, Y.; Shinnka, T.; Sano, H.; Nakatani, T. The effects of cyclooxygenase-2 inhibitors on urological cancer cells. Int. J. Mol. Med. 2004, 13, 789–793. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, L.; O’neill, A.; Bahamon, B.; Alsop, D.C.; Mier, J.W.; Goldberg, S.N.; Signoretti, S.; Atkins, M.; Wood, C. Cox-2 inhibition enhances the activity of sunitinib in human renal cell carcinoma xenografts. Br. J. Cancer 2013, 108, 319–326. [Google Scholar] [CrossRef]
- Shinohara, N.; Kumagai, A.; Kanagawa, K.; Maruyama, S.; Abe, T.; Sazawa, A.; Nonomura, K. Multicenter phase II trial of combination therapy with meloxicam, a COX-2 inhibitor, and natural interferon-α for metastatic renal cell carcinoma. Jpn. J. Clin. Oncol. 2009, 39, 720–726. [Google Scholar] [CrossRef]
- Rini, B.I.; Weinberg, V.; Dunlap, S.; Elchinoff, A.; Yu, N.; Bok, R.; Simko, J.; Small, E.J. Maximal COX-2 immunostaining and clinical response to celecoxib and interferon alpha therapy in metastatic renal cell carcinoma. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2006, 106, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Schwandt, A.; Garcia, J.A.; Elson, P.; Wyckhouse, J.; Finke, J.H.; Ireland, J.; Triozzi, P.; Zhou, M.; Dreicer, R.; Rini, B.I. Clinical and immunomodulatory effects of celecoxib plus interferon-alpha in metastatic renal cell carcinoma patients with COX-2 tumor immunostaining. J. Clin. Immunol. 2011, 31, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Tatokoro, M.; Fujii, Y.; Kawakami, S.; Saito, K.; Koga, F.; Matsuoka, Y.; Iimura, Y.; Masuda, H.; Kihara, K. Phase-II trial of combination treatment of interferon-α, cimetidine, cyclooxygenase-2 inhibitor and renin-angiotensin-system inhibitor (I-CCA therapy) for advanced renal cell carcinoma. Cancer Sci. 2011, 102, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Liang, Y.; Ding, X.; Ma, X.; Zhang, G.; Sun, L. Significance of cyclooxygenase-2, prostaglandin E2 and CD133 levels in sunitinib-resistant renal cell carcinoma. Oncol. Lett. 2019, 18, 1442–1450. [Google Scholar] [CrossRef] [Green Version]
- Ohba, K.; Miyata, Y.; Watanabe, S.-I.; Hayashi, T.; Kanetake, H.; Kanda, S.; Sakai, H. Clinical significance and predictive value of prostaglandin E2 receptors (EPR) 1–4 in patients with renal cell carcinoma. Anticancer. Res. 2011, 31, 597–605. [Google Scholar]
- Wu, J.; Zhang, Y.; Frilot, N.; Kim, J.I.; Kim, W.-J.; Daaka, Y. Prostaglandin E2 regulates renal cell carcinoma invasion through the EP4 receptor-Rap GTPase signal transduction pathway. J. Biol. Chem. 2011, 286, 33954–33962. [Google Scholar] [CrossRef]
- Zhang, Y.; Purayil, H.T.; Black, J.B.; Fetto, F.; Lynch, L.D.; Masannat, J.N.; Daaka, Y. Prostaglandin E2 receptor 4 mediates renal cell carcinoma intravasation and metastasis. Cancer Lett. 2017, 391, 50–58. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.; Kim, W.; Daaka, Y. PGE2 promotes renal carcinoma cell invasion through activated RalA. Oncogene 2013, 32, 1408–1415. [Google Scholar] [CrossRef]
- Chen, Q.; Shinohara, N.; Abe, T.; Harabayashi, T.; Nonomura, K. Impact of cyclooxygenase-2 gene expression on tumor invasiveness in a human renal cell carcinoma cell line. J. Urol. 2004, 172 6 Part 1, 2153–2157. [Google Scholar] [CrossRef]
- Hong, D.S.; Parikh, A.; Shapiro, G.I.; Varga, A.; Naing, A.; Meric-Bernstam, F.; Ataman, Ö.; Reyderman, L.; Binder, T.A.; Ren, M. First-in-human phase I study of immunomodulatory E7046, an antagonist of PGE2-receptor E-type 4 (EP4), in patients with advanced cancers. J. Immunother. Cancer 2020, 8, e000222. [Google Scholar] [CrossRef]
- Take, Y.; Koizumi, S.; Nagahisa, A. Prostaglandin E receptor 4 antagonist in cancer immunotherapy: Mechanisms of action. Front. Immunol. 2020, 11, 324. [Google Scholar] [CrossRef] [PubMed]
- Remuzzi, G.; FitzGerald, G.A.; Patrono, C. Thromboxane synthesis and action within the kidney. Kidney Int. 1992, 41, 1483–1493. [Google Scholar] [CrossRef]
- Ashton, A.W.; Zhang, Y.; Cazzolli, R.; Honn, K.V. The Role and Regulation of Thromboxane A(2) Signaling in Cancer-Trojan Horses and Misdirection. Molecules 2022, 27, 6234. [Google Scholar] [CrossRef]
- Moussa, O.; Ashton, A.W.; Fraig, M.; Garrett-Mayer, E.; Ghoneim, M.A.; Halushka, P.V.; Watson, D.K. Novel role of thromboxane receptors β isoform in bladder cancer pathogenesis. Cancer Res. 2008, 68, 4097–4104. [Google Scholar] [CrossRef]
- Faronato, M.; Muzzonigro, G.; Milanese, G.; Menna, C.; Bonfigli, A.; Catalano, A.; Procopio, A. Increased expression of 5-lipoxygenase is common in clear cell renal cell carcinoma. Histol. Histopathol. 2007, 22, 1109–1118. [Google Scholar]
- Matsuyama, M.; Yoshimura, R.; Mitsuhashi, M.; Tsuchida, K.; Takemoto, Y.; Kawahito, Y.; Sano, H.; Nakatani, T. 5-Lipoxygenase inhibitors attenuate growth of human renal cell carcinoma and induce apoptosis through arachidonic acid pathway. Oncol. Rep. 2005, 14, 73–79. [Google Scholar] [PubMed]
- Matsuyama, M.; Yoshimura, R. Relationship between arachidonic acid pathway and human renal cell carcinoma. OncoTargets Ther. 2008, 1, 41. [Google Scholar] [CrossRef]
- Selka, A.; Doiron, J.A.; Lyons, P.; Dastous, S.; Chiasson, A.; Cormier, M.; Turcotte, S.; Surette, M.E.; Touaibia, M. Discovery of a novel 2, 5-dihydroxycinnamic acid-based 5-lipoxygenase inhibitor that induces apoptosis and may impair autophagic flux in RCC4 renal cancer cells. Eur. J. Med. Chem. 2019, 179, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Thalanayar Muthukrishnan, P.; Nouraie, M.; Parikh, A.; Holguin, F. Zileuton use and phenotypic features in asthma. Pulm. Pharmacol. Ther. 2020, 60, 101872. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, H.; Yan, X.; Ma, J.; Chen, Z. LTB4R Promotes the Occurrence and Progression of Clear Cell Renal Cell Carcinoma (ccRCC) by Regulating the AKT/mTOR Signaling Pathway. Cells 2022, 11, 3606. [Google Scholar] [CrossRef]
- Wu, H.-H.; Yan, X.; Chen, Z.; Du, G.-W.; Bai, X.-J.; Tuoheti, K.; Liu, T.-Z. GNRH1 and LTB4R might be novel immune-related prognostic biomarkers in clear cell renal cell carcinoma (ccRCC). Cancer Cell Int. 2021, 21, 1–14. [Google Scholar] [CrossRef]
- Yuan, X.; He, Y.; Luo, C.; Wang, W. Leukotriene B4 receptor 2 correlates with prognosis and immune infiltration in clear cell renal cell carcinoma. Investig. New Drugs 2022, 40, 232–244. [Google Scholar] [CrossRef]
- Funao, K.; Matsuyama, M.; Naganuma, T.; Kawahito, Y.; Sano, H.; Nakatani, T.; Yoshimura, R. The cysteinylLT1 receptor in human renal cell carcinoma. Mol. Med. Rep. 2008, 1, 185–189. [Google Scholar] [PubMed]
- Matsuyama, M.; Yoshimura, R. Cysteinyl-leukotriene1 receptor is a potent target for the prevention and treatment of human urological cancer. Mol. Med. Rep. 2010, 3, 245–251. [Google Scholar] [PubMed]
- Tsai, M.-J.; Wu, P.-H.; Sheu, C.-C.; Hsu, Y.-L.; Chang, W.-A.; Hung, J.-Y.; Yang, C.-J.; Yang, Y.-H.; Kuo, P.-L.; Huang, M.-S. Cysteinyl leukotriene receptor antagonists decrease cancer risk in asthma patients. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wolf, C.; Smith, S.; van Wijk, S.J. Zafirlukast Induces VHL-and HIF-2α-Dependent Oxidative Cell Death in 786-O Clear Cell Renal Carcinoma Cells. Int. J. Mol. Sci. 2022, 23, 3567. [Google Scholar] [CrossRef]
- Yoshimura, R.; Inoue, K.; Kawahito, Y.; Mitsuhashi, M.; Tsuchida, K.; Matsuyama, M.; Sano, H.; Nakatani, T. Expression of 12-lipoxygenase in human renal cell carcinoma and growth prevention by its inhibitor. Int. J. Mol. Med. 2004, 13, 41–46. [Google Scholar] [CrossRef]
- Daurkin, I.; Eruslanov, E.; Stoffs, T.; Perrin, G.Q.; Algood, C.; Gilbert, S.M.; Rosser, C.J.; Su, L.-M.; Vieweg, J.; Kusmartsev, S. Tumor-Associated Macrophages Mediate Immunosuppression in the Renal Cancer Microenvironment by Activating the 15-Lipoxygenase-2 PathwayTumor-Associated Macrophages in Human Kidney Cancer. Cancer Res. 2011, 71, 6400–6409. [Google Scholar] [CrossRef]
- Kusmartsev, S. Enhanced 15-lipoxygenase activity and elevated eicosanoid production in kidney tumor microenvironment contribute to the inflammation and immune suppression. Oncoimmunology 2012, 1, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Gohara, A.; Eltaki, N.; Sabry, D.; Murtagh, D.; Jankun, J.; Selman, S.H.; Skrzypczak-Jankun, E. Human 5-, 12-and 15-lipoxygenase-1 coexist in kidney but show opposite trends and their balance changes in cancer. Oncol. Rep. 2012, 28, 1275–1282. [Google Scholar] [CrossRef]
- Alexanian, A.; Sorokin, A. Targeting 20-HETE producing enzymes in cancer–rationale, pharmacology, and clinical potential. OncoTargets Ther. 2013, 6, 243. [Google Scholar]
- Alexanian, A.; Rufanova, V.A.; Miller, B.; Flasch, A.; Roman, R.J.; Sorokin, A. Down-regulation of 20-HETE synthesis and signaling inhibits renal adenocarcinoma cell proliferation and tumor growth. Anticancer. Res. 2009, 29, 3819–3824. [Google Scholar]
- Liu, J.; Wang, L.; Harvey-White, J.; Osei-Hyiaman, D.; Razdan, R.; Gong, Q.; Chan, A.C.; Zhou, Z.; Huang, B.X.; Kim, H.Y.; et al. A biosynthetic pathway for anandamide. Proc. Natl. Acad. Sci. USA 2006, 103, 13345–13350. [Google Scholar] [CrossRef] [PubMed]
- Simon, G.M.; Cravatt, B.F. Anandamide biosynthesis catalyzed by the phosphodiesterase GDE1 and detection of glycerophospho-N-acyl ethanolamine precursors in mouse brain. J. Biol. Chem. 2008, 283, 9341–9349. [Google Scholar] [CrossRef]
- Maccarrone, M. Need for Methods to Investigate Endocannabinoid Signaling. Methods Mol. Biol. 2016, 1412, 1–8. [Google Scholar] [PubMed]
- Pisanti, S.; Bifulco, M. Endocannabinoid system modulation in cancer biology and therapy. Pharmacol. Res. 2009, 60, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Galve-Roperh, I.; Canova, C.; Brachet, P.; Guzman, M. Delta9-tetrahydrocannabinol induces apoptosis in C6 glioma cells. FEBS Lett. 1998, 436, 6–10. [Google Scholar] [CrossRef]
- Elbaz, M.; Ahirwar, D.; Ravi, J.; Nasser, M.W.; Ganju, R.K. Novel role of cannabinoid receptor 2 in inhibiting EGF/EGFR and IGF-I/IGF-IR pathways in breast cancer. Oncotarget 2017, 8, 29668–29678. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, L.; Miguel, A.; Diaz-Laviada, I. Delta9-tetrahydrocannabinol induces apoptosis in human prostate PC-3 cells via a receptor-independent mechanism. FEBS Lett. 1999, 458, 400–404. [Google Scholar] [CrossRef]
- Maccarrone, M.; Lorenzon, T.; Bari, M.; Melino, G.; Finazzi-Agro, A. Anandamide induces apoptosis in human cells via vanilloid receptors. Evidence for a protective role of cannabinoid receptors. J. Biol. Chem. 2000, 275, 31938–31945. [Google Scholar] [CrossRef]
- Petrosino, S.; Di Marzo, V. FAAH and MAGL inhibitors: Therapeutic opportunities from regulating endocannabinoid levels. Curr. Opin. Investig. Drugs 2010, 11, 51–62. [Google Scholar]
- Hu, W.R.; Lian, Y.F.; Peng, L.X.; Lei, J.J.; Deng, C.C.; Xu, M.; Feng, Q.S.; Chen, L.Z.; Bei, J.X.; Zeng, Y.X. Monoacylglycerol lipase promotes metastases in nasopharyngeal carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 3704–3713. [Google Scholar] [PubMed]
- Gong, X.; Zheng, X.; Huang, Y.; Song, W.; Chen, G.; Chen, T. Monoacylglycerol Lipase (MAGL) Inhibition Impedes the Osteosarcoma Progression by Regulating Epithelial Mesenchymal Transition. Tohoku J. Exp. Med. 2022, 256, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Endsley, M.P.; Thill, R.; Choudhry, I.; Williams, C.L.; Kajdacsy-Balla, A.; Campbell, W.B.; Nithipatikom, K. Expression and function of fatty acid amide hydrolase in prostate cancer. Int. J. Cancer 2008, 123, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Winkler, K.; Ramer, R.; Dithmer, S.; Ivanov, I.; Merkord, J.; Hinz, B. Fatty acid amide hydrolase inhibitors confer anti-invasive and antimetastatic effects on lung cancer cells. Oncotarget 2016, 7, 15047–15064. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.A.; Aviello, G.; Petrosino, S.; Orlando, P.; Marsicano, G.; Lutz, B.; Borrelli, F.; Capasso, R.; Nigam, S.; Capasso, F.; et al. Increased endocannabinoid levels reduce the development of precancerous lesions in the mouse colon. J. Mol. Med. 2008, 86, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Larrinaga, G.; Sanz, B.; Perez, I.; Blanco, L.; Candenas, M.L.; Pinto, F.M.; Gil, J.; Lopez, J.I. Cannabinoid CB(1) receptor is downregulated in clear cell renal cell carcinoma. J. Histochem. Cytochem. 2010, 58, 1129–1134. [Google Scholar] [CrossRef]
- Wang, J.; Xu, Y.; Zhu, L.; Zou, Y.; Kong, W.; Dong, B.; Huang, J.; Chen, Y.; Xue, W.; Huang, Y.; et al. Cannabinoid receptor 2 as a novel target for promotion of renal cell carcinoma prognosis and progression. J. Cancer Res. Clin. Oncol. 2018, 144, 39–52. [Google Scholar] [CrossRef]
- Khan, M.I.; Sobocinska, A.A.; Brodaczewska, K.K.; Zielniok, K.; Gajewska, M.; Kieda, C.; Czarnecka, A.M.; Szczylik, C. Involvement of the CB2 cannabinoid receptor in cell growth inhibition and G0/G1 cell cycle arrest via the cannabinoid agonist WIN 55,212-2 in renal cell carcinoma. BMC Cancer 2018, 18, 583. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.; Cao, X.; Zhang, K.; Li, Y.; Zheng, Q.Y.; Li, G.Q.; He, Q.H.; Li, S.J.; Xu, G.L.; Zhang, K.Q. Celastrol alleviates renal fibrosis by upregulating cannabinoid receptor 2 expression. Cell Death Dis. 2018, 9, 601. [Google Scholar] [CrossRef]
- Jiang, X.; Chen, S.; Zhang, Q.; Yi, C.; He, J.; Ye, X.; Liu, M.; Lu, W. Celastrol is a novel selective agonist of cannabinoid receptor 2 with anti-inflammatory and anti-fibrotic activity in a mouse model of systemic sclerosis. Phytomedicine 2020, 67, 153160. [Google Scholar] [CrossRef]
- Croxford, J.L.; Yamamura, T. Cannabinoids and the immune system: Potential for the treatment of inflammatory diseases? J. Neuroimmunol. 2005, 166, 3–18. [Google Scholar] [CrossRef]
- Katz-Talmor, D.; Katz, I.; Porat-Katz, B.S.; Shoenfeld, Y. Cannabinoids for the treatment of rheumatic diseases-where do we stand? Nat. Rev. Rheumatol. 2018, 14, 488–498. [Google Scholar] [CrossRef]
- Taha, T.; Meiri, D.; Talhamy, S.; Wollner, M.; Peer, A.; Bar-Sela, G. Cannabis Impacts Tumor Response Rate to Nivolumab in Patients with Advanced Malignancies. Oncologist 2019, 24, 549–554. [Google Scholar] [CrossRef]
- Samanic, C.; Chow, W.H.; Gridley, G.; Jarvholm, B.; Fraumeni, J.F., Jr. Relation of body mass index to cancer risk in 362,552 Swedish men. Cancer Causes Control 2006, 17, 901–909. [Google Scholar] [CrossRef]
- Adams, K.F.; Leitzmann, M.F.; Albanes, D.; Kipnis, V.; Moore, S.C.; Schatzkin, A.; Chow, W.H. Body size and renal cell cancer incidence in a large US cohort study. Am. J. Epidemiol. 2008, 168, 268–277. [Google Scholar] [CrossRef]
- Reeves, G.K.; Pirie, K.; Beral, V.; Green, J.; Spencer, E.; Bull, D.; Million Women Study, C. Cancer incidence and mortality in relation to body mass index in the Million Women Study: Cohort study. BMJ 2007, 335, 1134. [Google Scholar] [CrossRef]
- Gebhard, R.L.; Clayman, R.V.; Prigge, W.F.; Figenshau, R.; Staley, N.A.; Reesey, C.; Bear, A. Abnormal cholesterol metabolism in renal clear cell carcinoma. J. Lipid Res. 1987, 28, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Heravi, G.; Yazdanpanah, O.; Podgorski, I.; Matherly, L.H.; Liu, W. Lipid metabolism reprogramming in renal cell carcinoma. Cancer Metastasis Rev. 2022, 41, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Riscal, R.; Bull, C.J.; Mesaros, C.; Finan, J.M.; Carens, M.; Ho, E.S.; Xu, J.P.; Godfrey, J.; Brennan, P.; Johansson, M.; et al. Cholesterol Auxotrophy as a Targetable Vulnerability in Clear Cell Renal Cell Carcinoma. Cancer Discov. 2021, 11, 3106–3125. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Huang, D.; Zheng, H.; Cai, Q.; Guo, Z.; Wang, S. Preoperative serum total cholesterol is a predictor of prognosis in patients with renal cell carcinoma: A meta- analysis of observational studies. Int. Braz. J. Urol. 2020, 46, 158–168. [Google Scholar] [CrossRef]
- Santoni, M.; Monteiro, F.S.M.; Massari, F.; Abahssain, H.; Aurilio, G.; Molina-Cerrillo, J.; Myint, Z.W.; Zabalza, I.O.; Battelli, N.; Grande, E. Statins and renal cell carcinoma: Antitumor activity and influence on cancer risk and survival. Crit. Rev. Oncol. Hematol. 2022, 176, 103731. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 2012, 367, 1792–1802. [Google Scholar] [CrossRef]
- Luo, Y.; She, D.L.; Xiong, H.; Fu, S.J.; Yang, L. The Prognostic Effect of Statin Use on Urologic Cancers: An Updated Meta-Analysis of 35 Observational Studies. Medicine 2015, 94, e1523. [Google Scholar] [CrossRef]
- Wu, P.; Xiang, T.; Wang, J.; Lv, R.; Zhuang, Y.; Wu, G. Statin use and the overall survival of renal cell carcinoma: A meta-analysis. Clin. Investig. Med. 2020, 43, E17–E23. [Google Scholar] [CrossRef] [PubMed]
- Jonasch, E.; Donskov, F.; Iliopoulos, O.; Rathmell, W.K.; Narayan, V.K.; Maughan, B.L.; Oudard, S.; Else, T.; Maranchie, J.K.; Welsh, S.J.; et al. Belzutifan for Renal Cell Carcinoma in von Hippel-Lindau Disease. N. Engl. J. Med. 2021, 385, 2036–2046. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, J.; Claps, F.; Mir, M.C.; Ischia, J. Promising Biomarkers in Renal Cell Carcinoma. Soc. Int. Urol. J. 2021, 2, 43–52. [Google Scholar] [CrossRef]
- Claps, F.; Mir, M.C. Novel Expanding Renal Cell Carcinoma Biomarkers. Soc. Int. Urol. J. 2021, 2, 32–42. [Google Scholar] [CrossRef]
- Lucarelli, G.; Rutigliano, M.; Sallustio, F.; Ribatti, D.; Giglio, A.; Signorile, M.L.; Grossi, V.; Sanese, P.; Napoli, A.; Maiorano, E.; et al. Integrated multi-omics characterization reveals a distinctive metabolic signature and the role of NDUFA4L2 in promoting angiogenesis, chemoresistance, and mitochondrial dysfunction in clear cell renal cell carcinoma. Aging (Albany NY) 2018, 10, 3957–3985. [Google Scholar] [CrossRef]
- Ragone, R.; Sallustio, F.; Piccinonna, S.; Rutigliano, M.; Vanessa, G.; Palazzo, S.; Lucarelli, G.; Ditonno, P.; Battaglia, M.; Fanizzi, F.P.; et al. Renal Cell Carcinoma: A Study through NMR-Based Metabolomics Combined with Transcriptomics. Diseases 2016, 4, 7. [Google Scholar] [CrossRef] [Green Version]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stepanovska Tanturovska, B.; Manaila, R.; Fabbro, D.; Huwiler, A. Lipids as Targets for Renal Cell Carcinoma Therapy. Int. J. Mol. Sci. 2023, 24, 3272. https://doi.org/10.3390/ijms24043272
Stepanovska Tanturovska B, Manaila R, Fabbro D, Huwiler A. Lipids as Targets for Renal Cell Carcinoma Therapy. International Journal of Molecular Sciences. 2023; 24(4):3272. https://doi.org/10.3390/ijms24043272
Chicago/Turabian StyleStepanovska Tanturovska, Bisera, Roxana Manaila, Doriano Fabbro, and Andrea Huwiler. 2023. "Lipids as Targets for Renal Cell Carcinoma Therapy" International Journal of Molecular Sciences 24, no. 4: 3272. https://doi.org/10.3390/ijms24043272
APA StyleStepanovska Tanturovska, B., Manaila, R., Fabbro, D., & Huwiler, A. (2023). Lipids as Targets for Renal Cell Carcinoma Therapy. International Journal of Molecular Sciences, 24(4), 3272. https://doi.org/10.3390/ijms24043272