

Advances in Plant Epigenome Editing Research and Its Application in Plants

 ,
, 
{kind=link}
Abstract
:1. Introduction
2. Genome Editing Tools
2.1. ZFN and TALEN
2.2. CRISPR/Cas
3. Types of Epigenetic Modifications of Genomes
3.1. DNA Methylation
3.2. Histone Modifications
3.3. Non-Coding RNA Regulation
4. Application of CRISPR in Plant Epigenetic Regulation
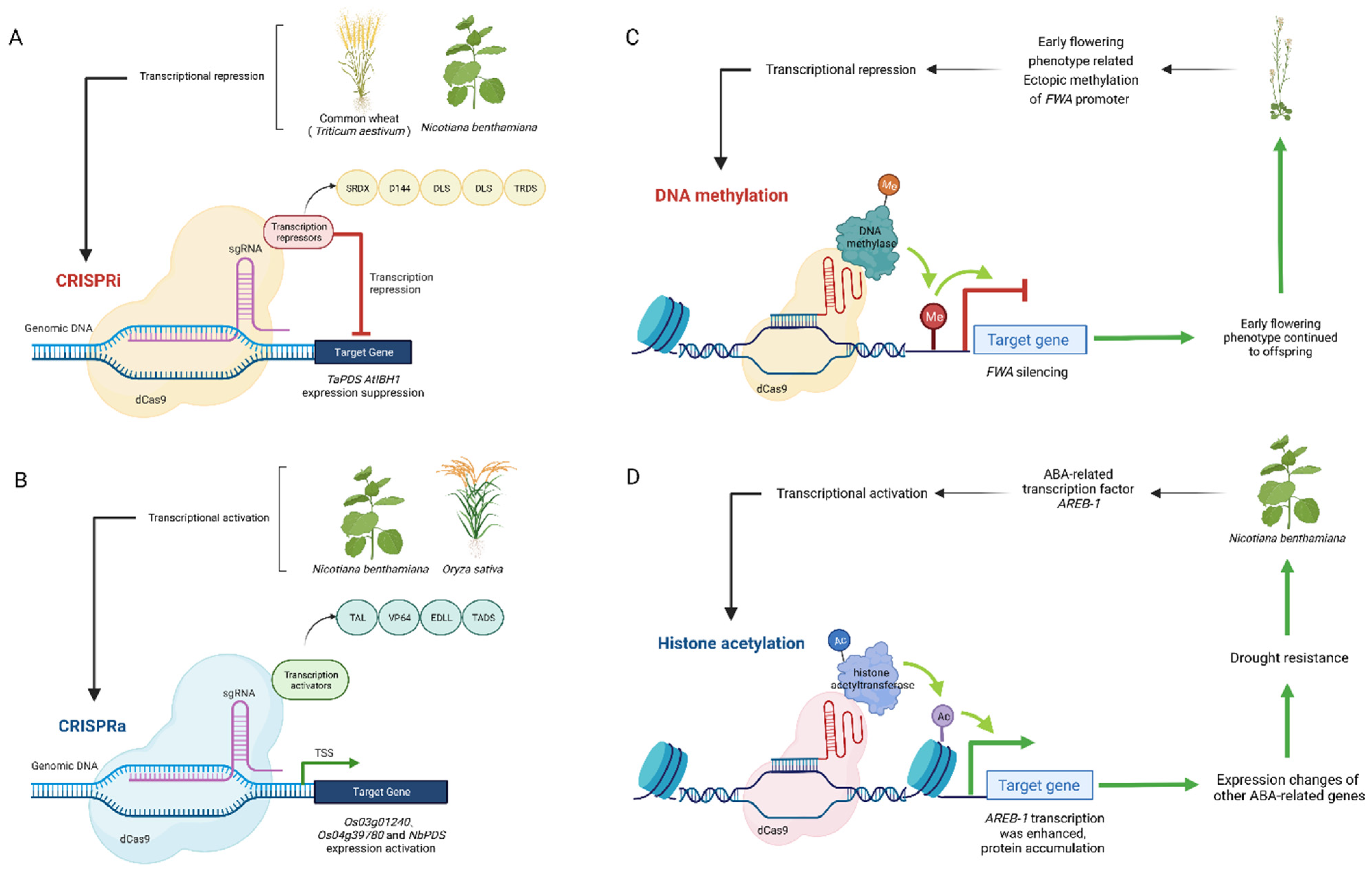
4.1. Transcriptional Regulation
4.2. DNA Methylated and Demethylated
4.3. Histone Acetylation
4.4. Non-Coding RNA
4.5. Summary and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Samanta, M.K.; Dey, A.; Gayen, S. CRISPR/Cas9: An advanced tool for editing plant genomes. Transgenic Res. 2016, 25, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Molla, K.A.; Sretenovic, S.; Bansal, K.C.; Qi, Y. Precise plant genome editing using base editors and prime editors. Nat. Plants 2021, 7, 1166–1187. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Niazian, M. Application of genetics and biotechnology for improving medicinal plants. Planta 2019, 249, 953–973. [Google Scholar] [CrossRef]
- Wright, D.A.; Li, T.; Yang, B.; Spalding, M.H. TALEN-mediated genome editing: Prospects and perspectives. Biochem. J. 2014, 462, 15–24. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Wyman, C.; Kanaar, R. DNA double-strand break repair: All’s well that ends well. Annu. Rev. Genet. 2006, 40, 363–383. [Google Scholar] [CrossRef]
- Sorek, R.; Lawrence, C.M.; Wiedenheft, B. CRISPR-mediated adaptive immune systems in bacteria and archaea. Annu. Rev. Biochem. 2013, 82, 237–266. [Google Scholar] [CrossRef]
- Hille, F.; Richter, H.; Wong, S.P.; Bratovič, M.; Ressel, S.; Charpentier, E. The Biology of CRISPR-Cas: Backward and Forward. Cell 2018, 172, 1239–1259. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef]
- Ghoshal, B.; Picard, C.L.; Vong, B.; Feng, S.; Jacobsen, S.E. CRISPR-based targeting of DNA methylation in Arabidopsis thaliana by a bacterial CG-specific DNA methyltransferase. Proc. Natl. Acad. Sci. USA 2021, 118, e2125016118. [Google Scholar] [CrossRef]
- Tirnaz, S.; Batley, J. DNA Methylation: Toward Crop Disease Resistance Improvement. Trends Plant Sci. 2019, 24, 1137–1150. [Google Scholar] [CrossRef]
- Wang, X.; Li, Z.; Zhang, Q.; Li, B.; Lu, C.; Li, W.; Cheng, T.; Xia, Q.; Zhao, P. DNA methylation on N6-adenine in lepidopteran Bombyx mori. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 815–825. [Google Scholar] [CrossRef]
- Ritter, E.J.; Niederhuth, C.E. Intertwined evolution of plant epigenomes and genomes. Curr. Opin. Plant Biol. 2021, 61, 101990. [Google Scholar] [CrossRef]
- Liu, R.; Lang, Z. The mechanism and function of active DNA demethylation in plants. J. Integr. Plant Biol. 2020, 62, 148–159. [Google Scholar] [CrossRef]
- Zhang, H.; Lang, Z.; Zhu, J.K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef]
- Lei, M.; Zhang, H.; Julian, R.; Tang, K.; Xie, S.; Zhu, J.K. Regulatory link between DNA methylation and active demethylation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2015, 112, 3553–3557. [Google Scholar] [CrossRef]
- Qüesta, J.I.; Song, J.; Geraldo, N.; An, H.; Dean, C. Arabidopsis transcriptional repressor VAL1 triggers Polycomb silencing at FLC during vernalization. Science 2016, 353, 485–488. [Google Scholar] [CrossRef]
- Zhang, S.F.; Li, X.R.; Sun, C.B.; He, Y.K. Epigenetics of plant vernalization regulated by non-coding RNAs. Yi Chuan Hereditas 2012, 34, 829–834. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, L.; Xia, H.; Wei, H.; Lou, Q.; Li, M.; Li, T.; Luo, L. Transgenerational epimutations induced by multi-generation drought imposition mediate rice plant’s adaptation to drought condition. Sci. Rep. 2017, 7, 39843. [Google Scholar] [CrossRef]
- Mao, D.; Tao, S.; Li, X.; Gao, D.; Tang, M.; Liu, C.; Wu, D.; Bai, L.; He, Z.; Wang, X.; et al. The Harbinger transposon-derived gene PANDA epigenetically coordinates panicle number and grain size in rice. Plant Biotechnol. J. 2022, 20, 1154–1166. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.L.; Laniel, M.A. Histones and histone modifications. Curr. Biol. 2004, 14, R546–R551. [Google Scholar] [CrossRef] [PubMed]
- Ninova, M.; Fejes Tóth, K.; Aravin, A.A. The control of gene expression and cell identity by H3K9 trimethylation. Development 2019, 146, dev181180. [Google Scholar] [CrossRef] [PubMed]
- Jambhekar, A.; Dhall, A.; Shi, Y. Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell Biol. 2019, 20, 625–641. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Du, J. Structure and mechanism of histone methylation dynamics in Arabidopsis. Curr. Opin. Plant Biol. 2022, 67, 102211. [Google Scholar] [CrossRef]
- Zhang, X.; Bernatavichute, Y.V.; Cokus, S.; Pellegrini, M.; Jacobsen, S.E. Genome-wide analysis of mono-, di- and trimethylation of histone H3 lysine 4 in Arabidopsis thaliana. Genome Biol. 2009, 10, R62. [Google Scholar] [CrossRef]
- Bai, L.; Chen, Q.; Jiang, L.; Lin, Y.; Ye, Y.; Liu, P.; Wang, X.; Tang, H. Comparative transcriptome analysis uncovers the regulatory functions of long noncoding RNAs in fruit development and color changes of Fragaria pentaphylla. Hortic Res. 2019, 6, 42. [Google Scholar] [CrossRef]
- Zhang, P.; Fan, Y.; Sun, X.; Chen, L.; Terzaghi, W.; Bucher, E.; Li, L.; Dai, M. A large-scale circular RNA profiling reveals universal molecular mechanisms responsive to drought stress in maize and Arabidopsis. Plant J. Cell Mol. Biol. 2019, 98, 697–713. [Google Scholar] [CrossRef]
- Chen, Y.; Cheng, C.; Feng, X.; Lai, R.; Gao, M.; Chen, W.; Wu, R. Integrated analysis of lncRNA and mRNA transcriptomes reveals the potential regulatory role of lncRNA in kiwifruit ripening and softening. Sci. Rep. 2021, 11, 1671. [Google Scholar] [CrossRef]
- Panni, S.; Lovering, R.C.; Porras, P.; Orchard, S. Non-coding RNA regulatory networks. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194417. [Google Scholar] [CrossRef]
- Quek, X.C.; Thomson, D.W.; Maag, J.L.; Bartonicek, N.; Signal, B.; Clark, M.B.; Gloss, B.S.; Dinger, M.E. lncRNAdb v2.0: Expanding the reference database for functional long noncoding RNAs. Nucleic Acids Res. 2015, 43, D168–D173. [Google Scholar] [CrossRef]
- Piatek, A.; Ali, Z.; Baazim, H.; Li, L.; Abulfaraj, A.; Al-Shareef, S.; Aouida, M.; Mahfouz, M.M. RNA-guided transcriptional regulation in planta via synthetic dCas9-based transcription factors. Plant Biotechnol. J. 2015, 13, 578–589. [Google Scholar] [CrossRef]
- Xu, L.; Sun, B.; Liu, S.; Gao, X.; Zhou, H.; Li, F.; Li, Y. The evaluation of active transcriptional repressor domain for CRISPRi in plants. Gene 2023, 851, 146967. [Google Scholar] [CrossRef]
- Lowder, L.G.; Zhou, J.; Zhang, Y.; Malzahn, A.; Zhong, Z.; Hsieh, T.-F.; Voytas, D.F.; Zhang, Y.; Qi, Y. Robust Transcriptional Activation in Plants Using Multiplexed CRISPR-Act2.0 and mTALE-Act Systems. Mol. Plant 2018, 11, 245–256. [Google Scholar] [CrossRef]
- Papikian, A.; Liu, W.; Gallego-Bartolomé, J.; Jacobsen, S.E. Site-specific manipulation of Arabidopsis loci using CRISPR-Cas9 SunTag systems. Nat. Commun. 2019, 10, 729. [Google Scholar] [CrossRef]
- De Melo, B.P.; Lourenço-Tessutti, I.T.; Paixão, J.F.R.; Noriega, D.D.; Silva, M.C.M.; de Almeida-Engler, J.; Fontes, E.P.B.; Grossi-de-Sa, M.F. Transcriptional modulation of AREB-1 by CRISPRa improves plant physiological performance under severe water deficit. Sci. Rep. 2020, 10, 16231. [Google Scholar] [CrossRef]
- Larson, M.H.; Gilbert, L.A.; Wang, X.; Lim, W.A.; Weissman, J.S.; Qi, L.S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013, 8, 2180–2196. [Google Scholar] [CrossRef]
- Zhou, H.; Xu, L.; Li, F.; Li, Y. Transcriptional regulation by CRISPR/dCas9 in common wheat. Gene 2022, 807, 145919. [Google Scholar] [CrossRef]
- Karlson, C.K.; Mohd Noor, S.N.; Khalid, N.; Tan, B.C. CRISPRi-Mediated Down-Regulation of the Cinnamate-4-Hydroxylase (C4H) Gene Enhances the Flavonoid Biosynthesis in Nicotiana tabacum. Biology 2022, 11, 1127. [Google Scholar] [CrossRef]
- Ding, X.; Yu, L.; Chen, L.; Li, Y.; Zhang, J.; Sheng, H.; Ren, Z.; Li, Y.; Yu, X.; Jin, S.; et al. Recent Progress and Future Prospect of CRISPR/Cas-Derived Transcription Activation (CRISPRa) System in Plants. Cells 2022, 11, 3045. [Google Scholar] [CrossRef]
- Bikard, D.; Jiang, W.; Samai, P.; Hochschild, A.; Zhang, F.; Marraffini, L.A. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013, 41, 7429–7437. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.W.; Wang, H.; Yang, H.; Shi, L.; Katz, Y.; Theunissen, T.W.; Rangarajan, S.; Shivalila, C.S.; Dadon, D.B.; Jaenisch, R. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013, 23, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Tsang, J.C.H.; Gaba, F.; Wu, D.; Lu, L.; Liu, P. Comparison of TALE designer transcription factors and the CRISPR/dCas9 in regulation of gene expression by targeting enhancers. Nucleic Acids Res. 2014, 42, e155. [Google Scholar] [CrossRef] [PubMed]
- Lowder, L.G.; Zhang, D.; Baltes, N.J.; Paul, J.W., III; Tang, X.; Zheng, X.; Voytas, D.F.; Hsieh, T.-F.; Zhang, Y.; Qi, Y. A CRISPR/Cas9 Toolbox for Multiplexed Plant Genome Editing and Transcriptional Regulation. Plant Physiol. 2015, 169, 971–985. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, D.; Xiong, X.; Yan, B.; Xie, W.; Sheen, J.; Li, J.F. A potent Cas9-derived gene activator for plant and mammalian cells. Nat. Plants 2017, 3, 930–936. [Google Scholar] [CrossRef]
- DeNizio, J.E.; Schutsky, E.K.; Berrios, K.N.; Liu, M.Y.; Kohli, R.M. Harnessing natural DNA modifying activities for editing of the genome and epigenome. Curr. Opin. Chem. Biol. 2018, 45, 10–17. [Google Scholar] [CrossRef]
- Song, J.; Teplova, M.; Ishibe-Murakami, S.; Patel, D.J. Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science 2012, 335, 709–712. [Google Scholar] [CrossRef]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Vojta, A.; Dobrinić, P.; Tadić, V.; Bočkor, L.; Korać, P.; Julg, B.; Klasić, M.; Zoldoš, V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef]
- Gallego-Bartolomé, J.; Gardiner, J.; Liu, W.; Papikian, A.; Ghoshal, B.; Kuo, H.Y.; Zhao, J.M.-C.; Segal, D.J.; Jacobsen, S.E. Targeted DNA demethylation of the Arabidopsis genome using the human TET1 catalytic domain. Proc. Natl. Acad. Sci. USA 2018, 115, E2125–E2134. [Google Scholar] [CrossRef]
- Zhong, X.; Du, J.; Hale, C.J.; Gallego-Bartolome, J.; Feng, S.; Vashisht, A.A.; Chory, J.; Wohlschlegel, J.A.; Patel, D.J.; Jacobsen, S.E. Molecular mechanism of action of plant DRM de novo DNA methyltransferases. Cell 2014, 157, 1050–1060. [Google Scholar] [CrossRef]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef]
- Lee, J.E.; Neumann, M.; Duro, D.I.; Schmid, M. CRISPR-based tools for targeted transcriptional and epigenetic regulation in plants. PLoS ONE 2019, 14, e0222778. [Google Scholar] [CrossRef]
- Lee, J.H.; Mazarei, M.; Pfotenhauer, A.C.; Dorrough, A.B.; Poindexter, M.R.; Hewezi, T.; Lenaghan, S.C.; Graham, D.E.; Stewart, C.N., Jr. Epigenetic Footprints of CRISPR/Cas9-Mediated Genome Editing in Plants. Front. Plant Sci. 2019, 10, 1720. [Google Scholar] [CrossRef]
- Zibitt, M.S.; Hartford, C.C.R.; Lal, A. Interrogating lncRNA functions via CRISPR/Cas systems. RNA Biol. 2021, 18, 2097–2106. [Google Scholar] [CrossRef]
- Joung, J.; Engreitz, J.M.; Konermann, S.; Abudayyeh, O.O.; Verdine, V.K.; Aguet, F.; Gootenberg, J.S.; Sanjana, N.E.; Wright, J.B.; Fulco, C.P.; et al. Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature 2017, 548, 343–346. [Google Scholar] [CrossRef]
- Chen, B.; Deng, S.; Ge, T.; Ye, M.; Yu, J.; Lin, S.; Ma, W.; Songyang, Z. Live cell imaging and proteomic profiling of endogenous NEAT1 lncRNA by CRISPR/Cas9-mediated knock-in. Protein Cell 2020, 11, 641–660. [Google Scholar] [CrossRef]
- Konermann, S.; Lotfy, P.; Brideau, N.J.; Oki, J.; Shokhirev, M.N.; Hsu, P.D. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 2018, 173, 665–676.e614. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, Q.; Hu, B.; Jiang, W.; Wang, Y.; Yan, J.; Ma, F.; Guan, Q.; Xu, J. Advances in Plant Epigenome Editing Research and Its Application in Plants. Int. J. Mol. Sci. 2023, 24, 3442. https://doi.org/10.3390/ijms24043442
Qi Q, Hu B, Jiang W, Wang Y, Yan J, Ma F, Guan Q, Xu J. Advances in Plant Epigenome Editing Research and Its Application in Plants. International Journal of Molecular Sciences. 2023; 24(4):3442. https://doi.org/10.3390/ijms24043442
Chicago/Turabian StyleQi, Qiaoyun, Bichun Hu, Weiyu Jiang, Yixiong Wang, Jinjiao Yan, Fengwang Ma, Qingmei Guan, and Jidi Xu. 2023. "Advances in Plant Epigenome Editing Research and Its Application in Plants" International Journal of Molecular Sciences 24, no. 4: 3442. https://doi.org/10.3390/ijms24043442
APA StyleQi, Q., Hu, B., Jiang, W., Wang, Y., Yan, J., Ma, F., Guan, Q., & Xu, J. (2023). Advances in Plant Epigenome Editing Research and Its Application in Plants. International Journal of Molecular Sciences, 24(4), 3442. https://doi.org/10.3390/ijms24043442