
ß-Adrenoreceptors in Human Cancers



Abstract
:1. Introduction
2. Review of the Literature
2.1. ß-Adrenoreceptors and Breast Cancer
2.2. ß-Adrenoreceptors and Lung Cancer
2.3. ß-Adrenoreceptors and Melanoma
2.4. ß-Adrenoreceptors and Gliomas
2.5. ß-Adrenergic Signaling Regulating Immune Components in the TME of Different Tumors
2.6. ß-Adrenoreceptors and Potential Treatment Options
3. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Smith, M.; Lutgendorf, S.K.; Sood, A.K.; Wong, K.Y.; Yu, L.; Chim, C.S.; Hardy, T.M.; O Tollefsbol, T.; Ubaldi, M.; Ricciardelli, E.; et al. Impact of stress on cancer metastasis. Futur. Oncol. 2010, 6, 1863–1881. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.; Soares-Silva, C.; Brandão, D.; Marino, F.; Cosentino, M.; Ribeiro, L. β-Adrenergic modulation of cancer cell proliferation: Available evidence and clinical perspectives. J. Cancer Res. Clin. Oncol. 2017, 143, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Ahles, A.; Engelhardt, S. Polymorphic variants of adrenoceptors: Pharmacology, physiology, and role in disease. Pharmacol. Rev. 2014, 66, 598–637. [Google Scholar] [CrossRef]
- Altosaar, K.; Balaji, P.; Bond, R.A.; Bylund, D.B.; Cotecchia, S.; Devost, D.; Zylbergold, P. Adrenoceptors in GtoPdb v. 2021.3. IUPHAR/BPS Guide Pharmacol. CITE 2021, 2021. [Google Scholar] [CrossRef]
- Velmurugan, B.K.; Baskaran, R.; Huang, C.Y. Detailed insight on β-adrenoceptors as therapeutic targets. Biomed. Pharmacother. 2019, 117, 109039. [Google Scholar] [CrossRef]
- Cole, S.W.; Sood, A.K. Molecular pathways: Beta-adrenergic signaling in cancer. Clin. Cancer Res. 2012, 18, 1201–1206. [Google Scholar] [CrossRef]
- Ciccarelli, M.; Sorriento, D.; Coscioni, E.; Iaccarino, G.; Santulli, G. Adrenergic receptors. In Endocrinology of the Heart in Health and Disease; Academic Press: Cambridge, MA, USA, 2017; pp. 285–315. [Google Scholar] [CrossRef]
- Hayward, L.F.; Mueller, P.J.; Hasser, E.M. Adrenergic receptors. In Encyclopedia of Endocrine Diseases; Elsevier: Amsterdam, The Netherlands, 2004; pp. 112–115. [Google Scholar]
- Ordovas-Montanes, J.; Rakoff-Nahoum, S.; Huang, S.; Riol-Blanco, L.; Barreiro, O.; von Andrian, U.H. The regulation of immunological processes by peripheral neurons in homeostasis and disease. Trends Immunol. 2015, 36, 578–604. [Google Scholar] [CrossRef]
- Erin, N.; Shurin, G.V.; Baraldi, J.H.; Shurin, M.R. Regulation of Carcinogenesis by Sensory Neurons and Neuromediators. Cancers 2022, 14, 2333. [Google Scholar] [CrossRef]
- Zhang, S.H.; Shurin, G.V.; Khosravi, H.; Kazi, R.; Kruglov, O.; Shurin, M.R.; Bunimovich, Y.L. Immunomodulation by Schwann cells in disease. Cancer Immunol. Immunother. 2020, 69, 245–253. [Google Scholar] [CrossRef]
- Restaino, A.C.; Vermeer, P.D. Neural regulations of the tumor microenvironment. FASEB BioAdvances 2022, 4, 29–42. [Google Scholar] [CrossRef]
- Wang, W.; Li, L.; Chen, N.; Niu, C.; Li, Z.; Hu, J.; Cui, J. Nerves in the tumor microenvironment: Origin and effects. Front. Cell Dev. Biol. 2020, 8, 601738. [Google Scholar] [CrossRef]
- Marino, F.; Cosentino, M. Adrenergic modulation of immune cells: An update. Amino Acids 2013, 45, 55–71. [Google Scholar] [CrossRef]
- Tang, J.; Li, Z.; Lu, L.; Cho, C.H. β-Adrenergic system, a backstage manipulator regulating tumour progression and drug target in cancer therapy. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2013; Volume 23, pp. 533–542. [Google Scholar] [CrossRef]
- Mravec, B.; Horvathova, L.; Hunakova, L. Neurobiology of cancer: The role of β-adrenergic receptor signaling in various tumor environments. Int. J. Mol. Sci. 2020, 21, 7958. [Google Scholar] [CrossRef] [PubMed]
- Kraboth, Z.; Galik, B.; Tompa, M.; Kajtar, B.; Urban, P.; Gyenesei, A.; Kalman, B. DNA CpG methylation in sequential glioblastoma specimens. J. Cancer Res. Clin. Oncol. 2020, 146, 2885–2896. [Google Scholar] [CrossRef]
- Kraboth, Z.; Kajtár, B.; Gálik, B.; Gyenesei, A.; Miseta, A.; Kalman, B. Involvement of the Catecholamine Pathway in Glioblastoma Development. Cells 2021, 10, 549. [Google Scholar] [CrossRef] [PubMed]
- Qiao, G.; Chen, M.; Bucsek, M.J.; Repasky, E.A.; Hylander, B.L. Adrenergic signaling: A targetable checkpoint limiting development of the antitumor immune response. Front. Immunol. 2018, 9, 164. [Google Scholar] [CrossRef] [PubMed]
- Maestroni, G.J. Adrenergic modulation of hematopoiesis. J. Neuroimmune Pharmacol. 2020, 15, 82–92. [Google Scholar] [CrossRef]
- Granot, Z. Neutrophils as a therapeutic target in cancer. Front. Immunol. 2019, 10, 1710. [Google Scholar] [CrossRef]
- Gysler, S.M.; Drapkin, R. Tumor innervation: Peripheral nerves take control of the tumor microenvironment. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Oleskin, A.V.; Sorokina, E.V.; Shilovsky, G.A. Interaction of Catecholamines with Microorganisms, Neurons, and Immune Cells. Biol. Bull. Rev. 2021, 11, 358–367. [Google Scholar] [CrossRef]
- Cosentino, M.; Marino, F.; Kustrimovic, N. Endogenous catecholamines in immune cells: Discovery, functions and clinical potential as therapeutic targets. In BrainImmune: Trends in Neuroendocrine Immunology; 5 October 2013; Available online: https://brainimmune.com/endogenous-catecholamines-in-immune-cells-discovery-functions-and-clinical-potential-as-pharmacotherapeutic-targets-3/ (accessed on 3 February 2023).
- Chhatar, S.; Lal, G. Role of adrenergic receptor signalling in neuroimmune communication. Curr. Res. Immunol. 2021, 2, 202–217. [Google Scholar] [CrossRef]
- Available online: https://www.cancer.net/cancer-types/breast-cancer/statistics (accessed on 18 December 2022).
- Lourenço, C.; Conceição, F.; Jerónimo, C.; Lamghari, M.; Sousa, D.M. Stress in Metastatic Breast Cancer: To the Bone and Beyond. Cancers 2022, 14, 1881. [Google Scholar] [CrossRef]
- Armaiz-Pena, G.N.; Cole, S.W.; Lutgendorf, S.K.; Sood, A.K. Neuroendocrine influences on cancer progression. Brain Behav. Immun. 2013, 30, S19–S25. [Google Scholar] [CrossRef]
- Thaker, P.H.; Sood, A.K.; Ramondetta, L.M. Importance of adrenergic pathways in women’s cancers. Cancer Biomark. 2013, 13, 145–154. [Google Scholar] [CrossRef]
- Gillis, R.D.; Botteri, E.; Chang, A.; Ziegler, A.I.; Chung, N.C.; Pon, C.K.; Sloan, E.K. Carvedilol blocks neural regulation of breast cancer progression in vivo and is associated with reduced breast cancer mortality in patients. Eur. J. Cancer 2021, 147, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Gruet, M.; Cotton, D.; Coveney, C.; Boocock, D.J.; Wagner, S.; Komorowski, L.; Powe, D.G. β2-adrenergic signalling promotes cell migration by upregulating expression of the metastasis-associated molecule LYPD3. Biology 2020, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Repasky, E.A.; Eng, J.; Hylander, B.L. Stress, metabolism and cancer: Integrated pathways contributing to immune suppression. Cancer J. 2015, 21, 97. [Google Scholar] [CrossRef]
- Silva, D.; Quintas, C.; Gonçalves, J.; Fresco, P. Contribution of adrenergic mechanisms for the stress-induced breast cancer carcinogenesis. J. Cell. Physiol. 2022, 237, 2107–2127. [Google Scholar] [CrossRef]
- Pon, C.K.; Lane, J.R.; Sloan, E.K.; Halls, M.L. The β2-adrenoceptor activates a positive cAMP-calcium feedforward loop to drive breast cancer cell invasion. FASEB J. 2016, 30, 1144–1154. [Google Scholar] [CrossRef]
- Craene, B.D.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Li, L.; Zhang, J.; Lei, Y.; Wang, L.; Liu, D.X.; Lu, J. CDK5 is essential for TGF-β1-induced epithelial-mesenchymal transition and breast cancer progression. Sci. Rep. 2013, 3, 2932. [Google Scholar] [CrossRef]
- Cui, B.; Luo, Y.; Tian, P.; Peng, F.; Lu, J.; Yang, Y.; Liu, Q. Stress-induced epinephrine enhances lactate dehydrogenase A and promotes breast cancer stem-like cells. J. Clin. Investig. 2019, 129, 1030–1046. [Google Scholar] [CrossRef]
- Du, P.; Zeng, H.; Xiao, Y.; Zhao, Y.; Zheng, B.; Deng, Y.; Ma, X. Chronic stress promotes EMT-mediated metastasis through activation of STAT3 signaling pathway by miR-337-3p in breast cancer. Cell Death Dis. 2020, 11, 761. [Google Scholar] [CrossRef]
- Ouyang, X.; Zhu, Z.; Yang, C.; Wang, L.; Ding, G.; Jiang, F. Epinephrine increases malignancy of breast cancer through p38 MAPK signaling pathway in depressive disorders. Int. J. Clin. Exp. Pathol. 2019, 12, 1932. ISSN 1936–2625/IJCEP0093897. [Google Scholar]
- Shi, M.; Liu, D.; Duan, H.; Qian, L.; Wang, L.; Niu, L.; Guo, N. The β2-adrenergic receptor and Her2 comprise a positive feedback loop in human breast cancer cells. Breast Cancer Res. Treat. 2011, 125, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Amaro, F.; Silva, D.; Reguengo, H.; Oliveira, J.C.; Quintas, C.; Vale, N.; Fresco, P. β-adrenoceptor activation in breast mcf-10a cells induces a pattern of catecholamine production similar to that of tumorigenic mcf-7 cells. Int. J. Mol. Sci. 2020, 21, 7968. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Cardoso, F. Breast cancer (Primer). Nat. Rev. Dis. Primers 2019, 66. [Google Scholar] [CrossRef]
- Chen, H.; Liu, D.; Guo, L.; Cheng, X.; Guo, N.; Shi, M. Chronic psychological stress promotes lung metastatic colonization of circulating breast cancer cells by decorating a pre-metastatic niche through activating β-adrenergic signaling. J. Pathol. 2018, 244, 49–60. [Google Scholar] [CrossRef]
- Denton, A.E.; Roberts, E.W.; Fearon, D.T. Stromal cells in the tumor microenvironment. Stromal Immunol. 2018, 99–114. [Google Scholar] [CrossRef]
- Cammarota, F.; Laukkanen, M.O. Mesenchymal stem/stromal cells in stromal evolution and cancer progression. Stem Cells Int. 2016, 2016, 4824573. [Google Scholar] [CrossRef]
- Campbell, J.P.; Karolak, M.R.; Ma, Y.; Perrien, D.S.; Masood-Campbell, S.K.; Penner, N.L.; Elefteriou, F. Stimulation of host bone marrow stromal cells by sympathetic nerves promotes breast cancer bone metastasis in mice. PLoS Biol. 2012, 10, e1001363. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Nilsson, M.B.; Sun, H.; Diao, L.; Tong, P.; Liu, D.; Li, L.; Heymach, J.V. Stress hormones promote EGFR inhibitor resistance in NSCLC: Implications for combinations with β-blockers. Sci. Transl. Med. 2017, 9, eaao4307. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.J.; Boo, H.J.; Lee, H.J.; Min, H.Y.; Lee, H.Y. Chronic stress facilitates lung tumorigenesis by promoting exocytosis of IGF2 in lung epithelial cells. Cancer Res. 2016, 76, 6607–6619. [Google Scholar] [CrossRef] [PubMed]
- Kaira, K.; Kamiyoshihara, M.; Kawashima, O.; Endoh, H.; Imaizumi, K.; Sugano, M.; Mogi, A. Prognostic impact of β2 adrenergic receptor expression in surgically resected pulmonary pleomorphic carcinoma. Anticancer. Res. 2019, 39, 395–403. [Google Scholar] [CrossRef] [PubMed]
- De Giorgi, V.; Geppetti, P.; Lupi, C.; Benemei, S. The role of β-blockers in melanoma. J. Neuroimmune Pharmacol. 2020, 15, 17–26. [Google Scholar] [CrossRef]
- Rains, S.L.; Amaya, C.N.; Bryan, B.A. Beta-adrenergic receptors are expressed across diverse cancers. Oncoscience 2017, 4, 95. [Google Scholar] [CrossRef]
- Batalla-Covello, J.; Ali, S.; Xie, T.; Amit, M. β-Adrenergic signaling in skin cancer. FASEB BioAdvances 2022, 4, 225–234. [Google Scholar] [CrossRef]
- Dal Monte, M.; Fornaciari, I.; Nicchia, G.P.; Svelto, M.; Casini, G.; Bagnoli, P. β3-adrenergic receptor activity modulates melanoma cell proliferation and survival through nitric oxide signaling. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2014, 387, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Calvani, M.; Cavallini, L.; Tondo, A.; Spinelli, V.; Ricci, L.; Pasha, A.; Filippi, L. β3-Adrenoreceptors control mitochondrial dormancy in melanoma and embryonic stem cells. Oxidative Med. Cell. Longev. 2018, 2018, 6816508. [Google Scholar] [CrossRef]
- Calvani, M.; Pelon, F.; Comito, G.; Taddei, M.L.; Moretti, S.; Innocenti, S.; Chiarugi, P. Norepinephrine promotes tumor microenvironment reactivity through β3-adrenoreceptors during melanoma progression. Oncotarget 2015, 6, 4615. [Google Scholar] [CrossRef]
- Dal Monte, M.; Calvani, M.; Cammalleri, M.; Favre, C.; Filippi, L.; Bagnoli, P. β-Adrenoceptors as drug targets in melanoma: Novel preclinical evidence for a role of β3-adrenoceptors. Br. J. Pharmacol. 2019, 176, 2496–2508. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Perrin, S.L.; Samuel, M.S.; Koszyca, B.; Brown, M.P.; Ebert, L.M.; Oksdath, M.; Gomez, G.A. Glioblastoma heterogeneity and the tumour microenvironment: Implications for preclinical research and development of new treatments. Biochem. Soc. Trans. 2019, 47, 625–638. [Google Scholar] [CrossRef]
- Sprugnoli, G.; Rossi, S.; Rotenberg, A.; Pascual-Leone, A.; El-Fakhri, G.; Golby, A.J.; Santarnecchi, E. Personalised, image-guided, noninvasive brain stimulation in gliomas: Rationale, challenges and opportunities. EbioMedicine 2021, 70, 103514. [Google Scholar] [CrossRef] [PubMed]
- Alphandéry, E. Nano-therapies for glioblastoma treatment. Cancers 2020, 12, 242. [Google Scholar] [CrossRef]
- Janjua, T.I.; Rewatkar, P.; Ahmed-Cox, A.; Saeed, I.; Mansfeld, F.M.; Kulshreshtha, R.; Popat, A. Frontiers in the treatment of glioblastoma: Past, present and emerging. Adv. Drug Deliv. Rev. 2021, 171, 108–138. [Google Scholar] [CrossRef]
- Thaker, P.H.; Han, L.Y.; Kamat, A.A.; Arevalo, J.M.; Takahashi, R.; Lu, C.; Sood, A.K. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat. Med. 2006, 12, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kang, J.H.; Jeong, K.J.; Lee, J.; Han, J.W.; Choi, W.S.; Lee, H.Y. Retracted: Norepinephrine induces VEGF expression and angiogenesis by a hypoxia-inducible factor-1α protein-dependent mechanism. Int. J. Cancer 2011, 128, 2306–2316. [Google Scholar] [CrossRef]
- Park, M.H.; Lee, H.S.; Lee, C.S.; You, S.T.; Kim, D.J.; Park, B.H.; Kim, E.G. p21-Activated kinase 4 promotes prostate cancer progression through CREB. Oncogene 2013, 32, 2475–2482. [Google Scholar] [CrossRef] [PubMed]
- Armento, A.; Ehlers, J.; Schotterl, S.; Naumann, U. Glioblastoma; de Vleeschouwer, S., Ed.; Exon Publications: Brisbane, Australia, 2017. [Google Scholar] [CrossRef]
- Mao, H.; LeBrun, D.G.; Yang, J.; Zhu, V.F.; Li, M. Deregulated signaling pathways in glioblastoma multiforme: Molecular mechanisms and therapeutic targets. Cancer Investig. 2012, 30, 48–56. [Google Scholar] [CrossRef]
- He, J.J.; Zhang, W.H.; Liu, S.L.; Chen, Y.F.; Liao, C.X.; Shen, Q.Q.; Hu, P. Activation of β-adrenergic receptor promotes cellular proliferation in human glioblastoma. Oncol. Lett. 2017, 14, 3846–3852. [Google Scholar] [CrossRef]
- Kang, T.W.; Choi, S.W.; Yang, S.R.; Shin, T.H.; Kim, H.S.; Yu, K.R.; Kang, K.S. Growth arrest and forced differentiation of human primary glioblastoma multiforme by a novel small molecule. Sci. Rep. 2014, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.; Epstein, P.M. Cyclic nucleotide phosphodiesterases as targets for treatment of haematological malignancies. Biochem. J. 2006, 393, 21–41. [Google Scholar] [CrossRef]
- Safitri, D.; Harris, M.; Potter, H.; Yeung, H.Y.; Winfield, I.; Kopanitsa, L.; Ladds, G. Elevated intracellular cAMP concentration mediates growth suppression in glioma cells. Biochem. Pharmacol. 2020, 174, 113823. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Q.; Wang, X.; Xue, B.H.; Zhao, Y.; Xie, F.; Wang, S.D.; Qian, L.J. Chronic stress promotes glioma cell proliferation via the PI3K/Akt signaling pathway. Oncol. Rep. 2021, 46, 1–12. [Google Scholar] [CrossRef]
- Shafaroudi, M.M.; Rezaei, N.; Noori, A.; Zarei, H.; Akhtari, J.; Shafaroudi, A.M. Effects of Epinephrine on Gastric Adenocarcinoma and Brain Glioblastoma Cancer Cells; Research Square: Durham, NC, USA, 2021. [Google Scholar] [CrossRef]
- Kwon, Y.; Mehta, S.; Clark, M.; Walters, G.; Zhong, Y.; Lee, H.N.; Zhang, J. Non-canonical β-adrenergic activation of ERK at endosomes. Nature 2022, 611, 173–179. [Google Scholar] [CrossRef]
- Pavlova, O.; Shirokov, A.; Fomin, A.; Navolokin, N.; Terskov, A.; Khorovodov, A.; Semyachkina-Glushkovskaya, O. Optical in vivo and ex vivo imaging of glioma cells migration via the cerebral vessels: Prospective clinical application of the beta2-adrenoreceptors blockade for glioma treatment. J. Innov. Opt. Health Sci. 2018, 11, 1850025. [Google Scholar] [CrossRef]
- Pekarová, M.; Kralova, J.; Kubala, L.; Ciz, M.; Papezikova, I.; Macicková, T.; Lojek, A. Carvedilol and adrenergic agonists suppress the lipopolysaccharide-induced NO production in RAW 264.7 macrophages via the adrenergic receptors. Acta Physiol. Pol. 2009, 60, 143. [Google Scholar]
- Du, Y.; Yan, L.; Du, H.; Wang, L.; Ding, F.; Quan, L.; Liu, H. β1-adrenergic receptor autoantibodies from heart failure patients enhanced TNF-α secretion in RAW264. 7 macrophages in a largely PKA-dependent fashion. J. Cell. Biochem. 2012, 113, 3218–3228. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.F.; Jin, F.J.; Li, N.; Guan, H.T.; Lan, L.; Ni, H.; Wang, Y. Adrenergic receptor β2 activation by stress promotes breast cancer progression through macrophages M2 polarization in tumor microenvironment. BMB Rep. 2015, 48, 295. [Google Scholar] [CrossRef]
- Kurozumi, S.; Kaira, K.; Matsumoto, H.; Hirakata, T.; Yokobori, T.; Inoue, K.; Shirabe, K. β 2-Adrenergic receptor expression is associated with biomarkers of tumor immunity and predicts poor prognosis in estrogen receptor-negative breast cancer. Breast Cancer Res. Treat. 2019, 177, 603–610. [Google Scholar] [CrossRef]
- Xia, Y.; Wei, Y.; Li, Z.Y.; Cai, X.Y.; Zhang, L.L.; Dong, X.R.; Wu, G. Catecholamines contribute to the neovascularization of lung cancer via tumor-associated macrophages. Brain Behav. Immun. 2019, 81, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Forsyth, P.A.; Algazi, A.; Hamid, O.; Hodi, F.S.; Moschos, S.J.; Margolin, K. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N. Engl. J. Med. 2018, 379, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Kambayashi, Y.; Aiba, S. Crosstalk between regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) during melanoma growth. Oncoimmunology 2012, 1, 1433–1434. [Google Scholar] [CrossRef]
- Cole, S.W.; Nagaraja, A.S.; Lutgendorf, S.K.; Green, P.A.; Sood, A.K. Sympathetic nervous system regulation of the tumour microenvironment. Nat. Rev. Cancer 2015, 15, 563–572. [Google Scholar] [CrossRef]
- Kokolus, K.M.; Zhang, Y.; Sivik, J.M.; Schmeck, C.; Zhu, J.; Repasky, E.A.; Schell, T.D. β blocker use correlates with better overall survival in metastatic melanoma patients and improves the efficacy of immunotherapies in mice. Oncoimmunology 2017, 7, e1405205. [Google Scholar] [CrossRef] [PubMed]
- Laureys, G.; Gerlo, S.; Spooren, A.; Demol, F.; De Keyser, J.; Aerts, J.L. β2-adrenergic agonists modulate TNF-α induced astrocytic inflammatory gene expression and brain. J. Neuroinflammation 2014, 11, 21. [Google Scholar] [CrossRef]
- Global Cancer Observatory 2020—Globocan 2020. Available online: https://gco.iarc.fr/ (accessed on 14 January 2023).
- de Oliveira, C.; Pataky, R.; Bremner, K.E.; Rangrej, J.; Chan, K.K.; Cheung, W.Y.; Krahn, M.D. Phase-specific and lifetime costs of cancer care in Ontario, Canada. BMC Cancer 2016, 16, 809. [Google Scholar] [CrossRef]
- Argulian, E.; Bangalore, S.; Messerli, F.H. Misconceptions and facts about beta-blockers. Am. J. Med. 2019, 132, 816–819. [Google Scholar] [CrossRef]
- Phadke, S.; Clamon, G. Beta blockade as adjunctive breast cancer therapy: A review. Crit. Rev. Oncol. Hematol. 2019, 138, 173–177. [Google Scholar] [CrossRef]
- Kim, H.Y.; Jung, Y.J.; Lee, S.H.; Jung, H.J.; Pak, K. Is beta-blocker use beneficial in breast cancer? A meta-analysis. Oncology 2017, 92, 264–268. [Google Scholar] [CrossRef]
- Melhem-Bertrandt, A.; Chavez-MacGregor, M.; Lei, X.; Brown, E.N.; Lee, R.T.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M. Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2011, 29, 2645. [Google Scholar] [CrossRef] [PubMed]
- Choy, C.; Raytis, J.L.; Smith, D.D.; Duenas, M.; Neman, J.; Jandial, R.; Lew, M.W. Inhibition of β2-adrenergic receptor reduces triple-negative breast cancer brain metastases: The potential benefit of perioperative β-blockade. Oncol. Rep. 2016, 35, 3135–3142. [Google Scholar] [CrossRef] [PubMed]
- Montoya, A.; Varela-Ramirez, A.; Dickerson, E.; Pasquier, E.; Torabi, A.; Aguilera, R.; Bryan, B. The beta adrenergic receptor antagonist propranolol alters mitogenic and apoptotic signaling in late stage breast cancer. Biomed. J. 2019, 42, 155–165. [Google Scholar] [CrossRef]
- Montoya, A.; Amaya, C.N.; Belmont, A.; Diab, N.; Trevino, R.; Villanueva, G.; Nahleh, Z. Use of non-selective β-blockers is associated with decreased tumor proliferative indices in early stage breast cancer. Oncotarget 2017, 8, 6446. [Google Scholar] [CrossRef]
- Lin, C.S.; Lin, W.S.; Lin, C.L.; Kao, C.H. Carvedilol use is associated with reduced cancer risk: A nationwide population-based cohort study. Int. J. Cardiol. 2015, 184, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Deng, W.; Han, Y.; Shi, Y.; Xu, T.; Shi, J.; Jin, H. Analysis of the correlation among hypertension, the intake of β-blockers, and overall survival outcome in patients undergoing chemoradiotherapy with inoperable stage III non-small cell lung cancer. Am. J. Cancer Res. 2017, 7, 946. [Google Scholar]
- Musselman, R.P.; Bennett, S.; Li, W.; Mamdani, M.; Gomes, T.; van Walraven, C.; Auer, R.C. Association between perioperative beta blocker use and cancer survival following surgical resection. Eur. J. Surg. Oncol. 2018, 44, 1164–1169. [Google Scholar] [CrossRef] [PubMed]
- Udumyan, R.; Montgomery, S.; Fang, F.; Valdimarsdottir, U.; Hardardottir, H.; Ekbom, A.; Fall, K. Beta-Blocker Use and Lung Cancer Mortality in a Nationwide Cohort Study of Patients with Primary Non–Small Cell Lung Cancerβ-Blocker Use and Lung Cancer Mortality. Cancer Epidemiol. Biomark. Prev. 2020, 29, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Aydiner, A.; Ciftci, R.; Karabulut, S.; Kilic, L. Does beta-blocker therapy improve the survival of patients with metastatic non-small cell lung cancer? Asian Pac. J. Cancer Prev. 2013, 14, 6109–6114. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, K.R.; Yan, S.X.; Heilbroner, S.P.; Sonett, J.R.; Stoopler, M.B.; Shu, C.; Cheng, S.K. Effects of β-Adrenergic antagonists on chemoradiation therapy for locally advanced non-small cell lung cancer. J. Clin. Med. 2019, 8, 575. [Google Scholar] [CrossRef]
- De Giorgi, V.; Grazzini, M.; Gandini, S.; Benemei, S.; Lotti, T.; Marchionni, N.; Geppetti, P. Treatment with β-blockers and reduced disease progression in patients with thick melanoma. Arch. Intern. Med. 2011, 171, 779–781. [Google Scholar] [CrossRef]
- De Giorgi, V.; Grazzini, M.; Benemei, S.; Marchionni, N.; Geppetti, P.; Gandini, S. β-Blocker use and reduced disease progression in patients with thick melanoma: 8 years of follow-up. Melanoma Res. 2017, 27, 268–270. [Google Scholar] [CrossRef]
- De Giorgi, V.; Grazzini, M.; Benemei, S.; Marchionni, N.; Botteri, E.; Pennacchioli, E.; Gandini, S. Propranolol for off-label treatment of patients with melanoma: Results from a cohort study. JAMA Oncol. 2018, 4, e172908. [Google Scholar] [CrossRef]
- Johansen, M.D.; Urup, T.; Holst, C.B.; Christensen, I.J.; Grunnet, K.; Lassen, U.; Poulsen, H.S. Outcome of bevacizumab therapy in patients with recurrent glioblastoma treated with angiotensin system inhibitors. Cancer Investig. 2018, 36, 512–519. [Google Scholar] [CrossRef]
- T Andresen, B. A pharmacological primer of biased agonism. Endocr. Metab. Immune Disord. -Drug Targets 2011, 11, 92–98. [Google Scholar] [CrossRef]
- Kim, I.M.; Tilley, D.G.; Chen, J.; Salazar, N.C.; Whalen, E.J.; Violin, J.D.; Rockman, H.A. β-Blockers alprenolol and carvedilol stimulate β-arrestin-mediated EGFR transactivation. Proc. Natl. Acad. Sci. 2008, 105, 14555–14560. [Google Scholar] [CrossRef]
- Deshpande, D.A.; Penn, R.B. Targeting G protein-coupled receptor signaling in asthma. Cell. Signal. 2006, 18, 2105–2120. [Google Scholar] [CrossRef]
- Lamichhane, R.; Liu, J.J.; White, K.L.; Katritch, V.; Stevens, R.C.; Wüthrich, K.; Millar, D.P. Biased signaling of the G-protein-coupled receptor β2AR is governed by conformational exchange kinetics. Structure 2020, 28, 371–377. [Google Scholar] [CrossRef]
- Choi, M.; Staus, D.P.; Wingler, L.M.; Ahn, S.; Pani, B.; Capel, W.D.; Lefkowitz, R.J. G protein–coupled receptor kinases (GRKs) orchestrate biased agonism at the β2-adrenergic receptor. Sci. Signal. 2018, 11, eaar7084. [Google Scholar] [CrossRef] [PubMed]
- Ippolito, M.; Benovic, J.L. Biased agonism at β-adrenergic receptors. Cell. Signal. 2021, 80, 109905. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T. Is the quest for signaling bias worth the effort? Mol. Pharmacol. 2018, 93, 266–269. [Google Scholar] [CrossRef]
- Gomez, J.L.; Bonaventura, J.; Lesniak, W.; Mathews, W.B.; Sysa-Shah, P.; Rodriguez, L.A.; Michaelides, M. Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science 2017, 357, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.A.; Hutchings, C.; Djamgoz, M.B. Clinical Potential of Nerve Input to Tumors: A Bioelectricity Perspective. Bioelectricity 2021, 3, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Armbruster, B.N.; Li, X.; Pausch, M.H.; Herlitze, S.; Roth, B.L. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl. Acad. Sci. USA 2007, 104, 5163–5168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Gumpper, R.H.; Huang, X.P.; Liu, Y.; Krumm, B.E.; Cao, C.; Roth, B.L. Molecular basis for selective activation of DREADD-based chemogenetics. Nature 2022, 612, 354–362. [Google Scholar] [CrossRef]
- Ojima, K.; Kakegawa, W.; Yamasaki, T.; Miura, Y.; Itoh, M.; Michibata, Y.; Kiyonaka, S. Coordination chemogenetics for activation of GPCR-type glutamate receptors in brain tissue. Nat. Commun. 2022, 13, 3167. [Google Scholar] [CrossRef]
- Costa, P.A.; Silva, W.N.; Prazeres, P.H.; Picoli, C.C.; Guardia, G.D.; Costa, A.C.; Birbrair, A. Chemogenetic modulation of sensory neurons reveals their regulating role in melanoma progression. Acta Neuropathol. Commun. 2021, 9, 183. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shaanan, T.L.; Azulay-Debby, H.; Dubovik, T.; Starosvetsky, E.; Korin, B.; Schiller, M.; Rolls, A. Activation of the reward system boosts innate and adaptive immunity. Nat. Med. 2016, 22, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shaanan, T.L.; Schiller, M.; Azulay-Debby, H.; Korin, B.; Boshnak, N.; Koren, T.; Rolls, A. Modulation of anti-tumor immunity by the brain’s reward system. Nat. Commun. 2018, 9, 2723. [Google Scholar] [CrossRef] [PubMed]



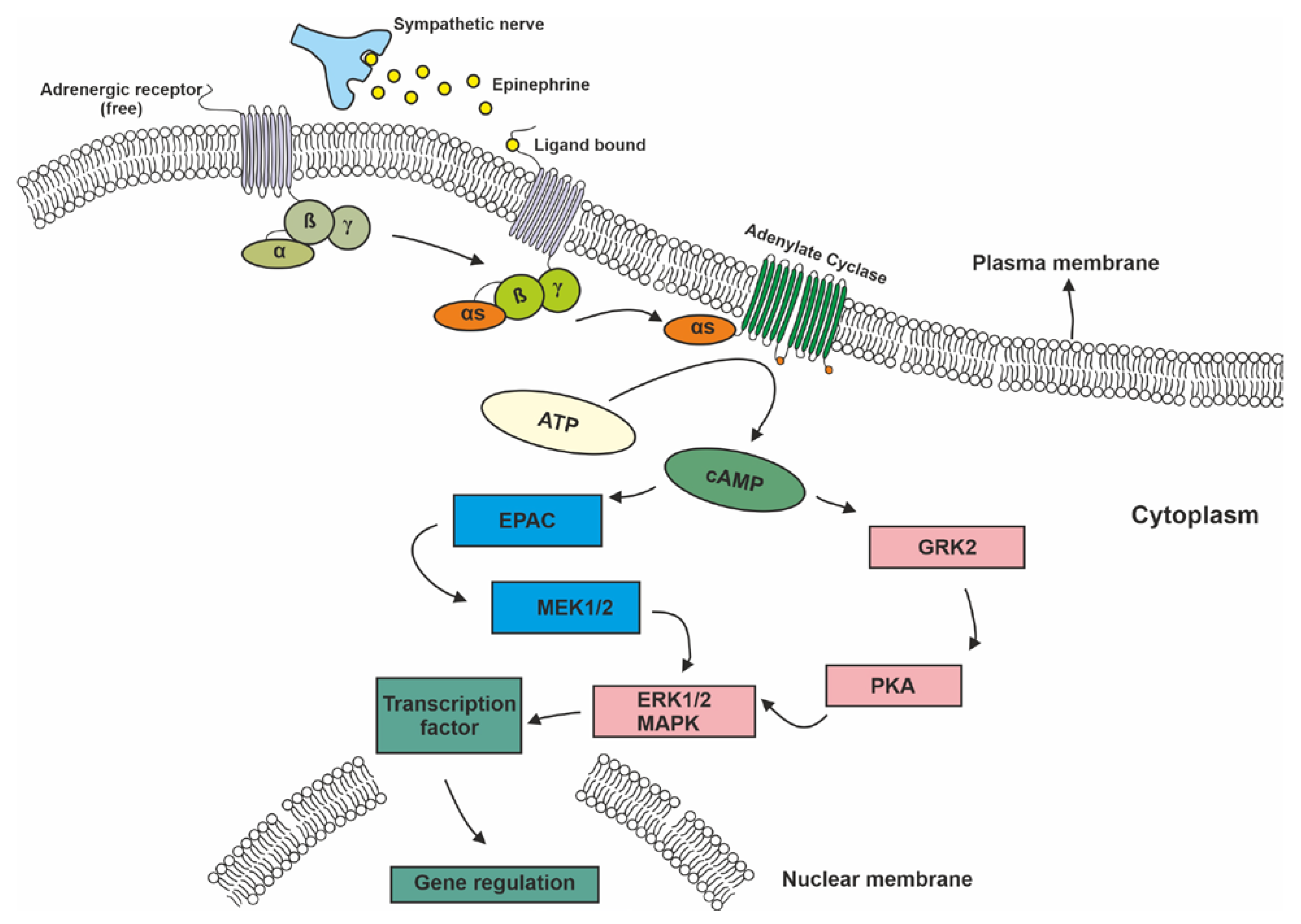{kind=link}
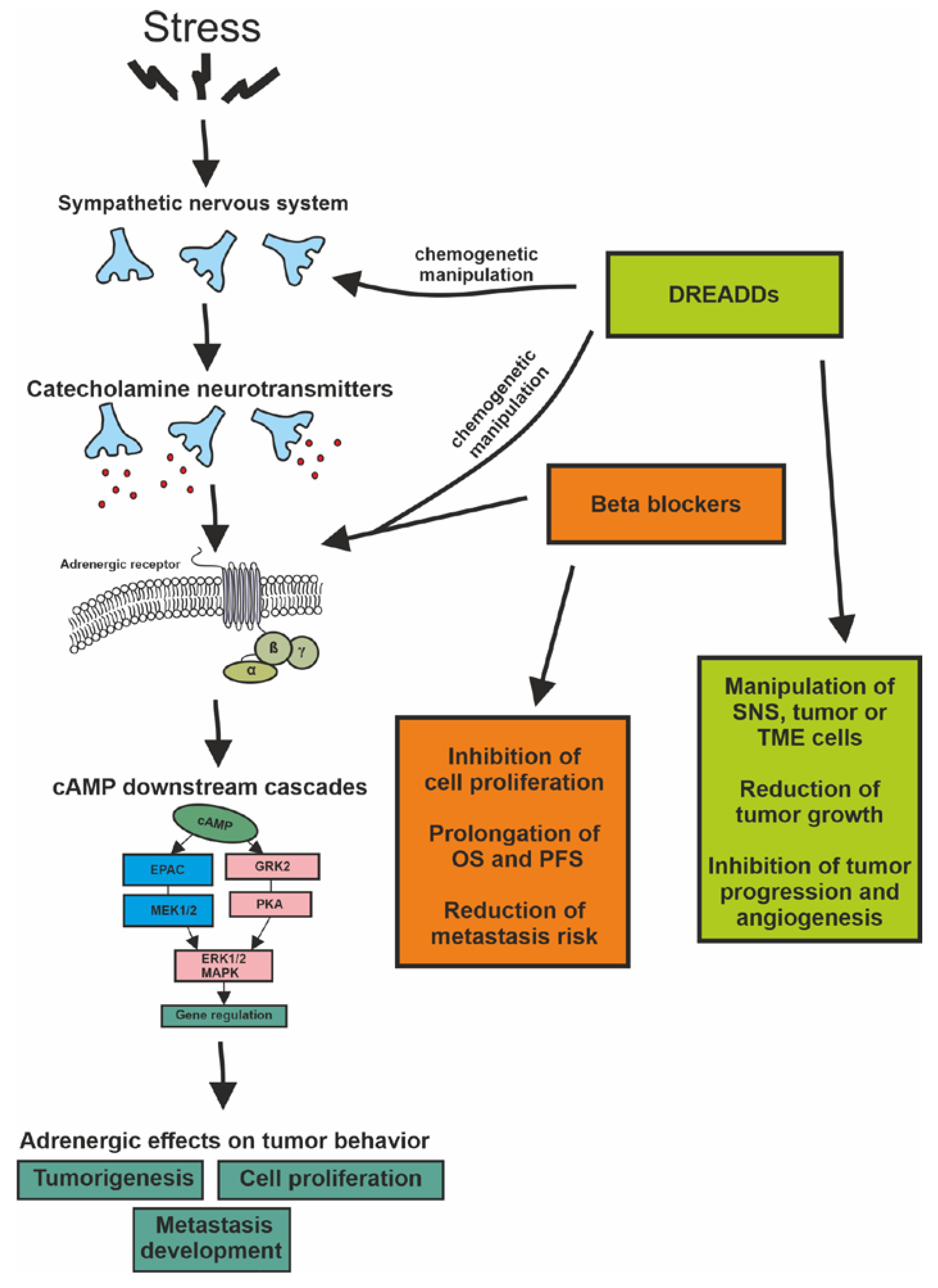{kind=link}
| Tumor Type | Receptor Subtype | Adaptor Protein | Second Messenger | Signaling Elements | Effect on Cell Physiology |
|---|---|---|---|---|---|
| Breast | α2, ß1, ß2 more significant | Gαs, Gi/Go | cAMP, Ca2+ | MAPK, HER2, ERK1/2, LDHA/USP28/MYC/SLUG, PI3K/AKT/mTOR, VEGF, STAT3 | Increased proliferation, metastatic rate, angiogenesis, and inflammation |
| Lung | ß1, ß2 more significant | Gαs | cAMP, Ca2+ | PKA, EPAC, ERK1/2, MAPK, IGF-1R | Increased proliferation, immune evasion, angiogenesis, migration, and invasion |
| Melanoma | ß1 most significant, ß2, ß3 | Gαs | cAMP, PKA, Ca2+ | VEGF, P38/MAPK, PI3K/AKT, STAT3,iNOS, FGF-2, IGF-1, Ang-2, Ras/ERK1/2 | Increased angiogenesis, proliferation, activation of stromal and inflammatory cells of TME, invasion, reduced apoptosis, and stem-cell trait induction |
| Glioma | ß1, ß2 | Gαs | cAMP, PKA | ERK1/2, MAPK, PI3K/AKT/mTOR, p53/MDM2/p21 | Increased proliferation rate, cell migration, angiogenesis, invasiveness, and metastasis formation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kraboth, Z.; Kalman, B. ß-Adrenoreceptors in Human Cancers. Int. J. Mol. Sci. 2023, 24, 3671. https://doi.org/10.3390/ijms24043671
Kraboth Z, Kalman B. ß-Adrenoreceptors in Human Cancers. International Journal of Molecular Sciences. 2023; 24(4):3671. https://doi.org/10.3390/ijms24043671
Chicago/Turabian StyleKraboth, Zoltan, and Bernadette Kalman. 2023. "ß-Adrenoreceptors in Human Cancers" International Journal of Molecular Sciences 24, no. 4: 3671. https://doi.org/10.3390/ijms24043671
APA StyleKraboth, Z., & Kalman, B. (2023). ß-Adrenoreceptors in Human Cancers. International Journal of Molecular Sciences, 24(4), 3671. https://doi.org/10.3390/ijms24043671