
Mitochondria-Targeted Antioxidants, an Innovative Class of Antioxidant Compounds for Neurodegenerative Diseases: Perspectives and Limitations
 ,
,  ,
,  and
and 
Abstract
:1. Introduction
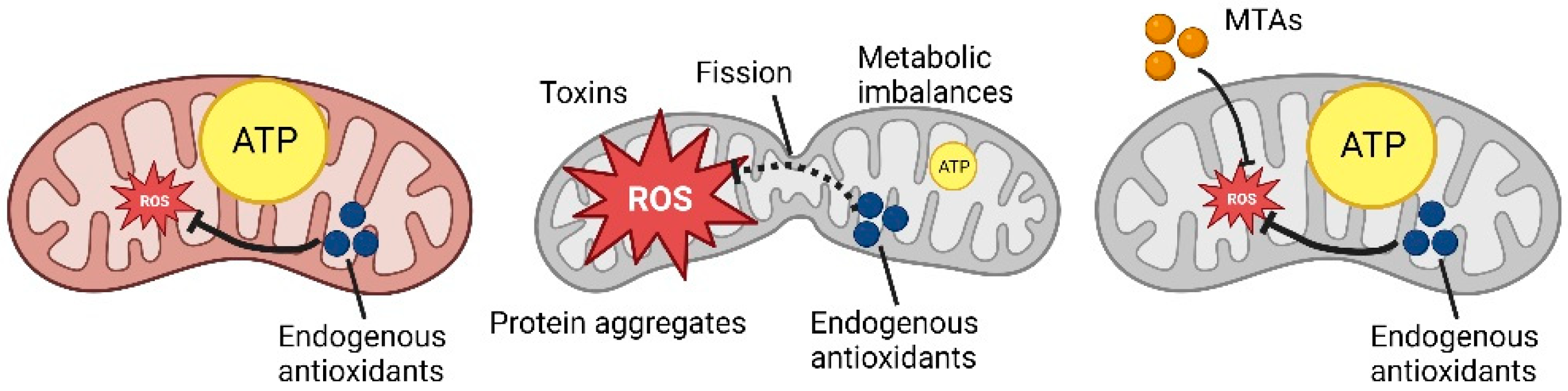
1.1. Mitochondrial Dynamics
1.2. Neurodegenerative Disorders Characterized by Mitochondrial Involvement
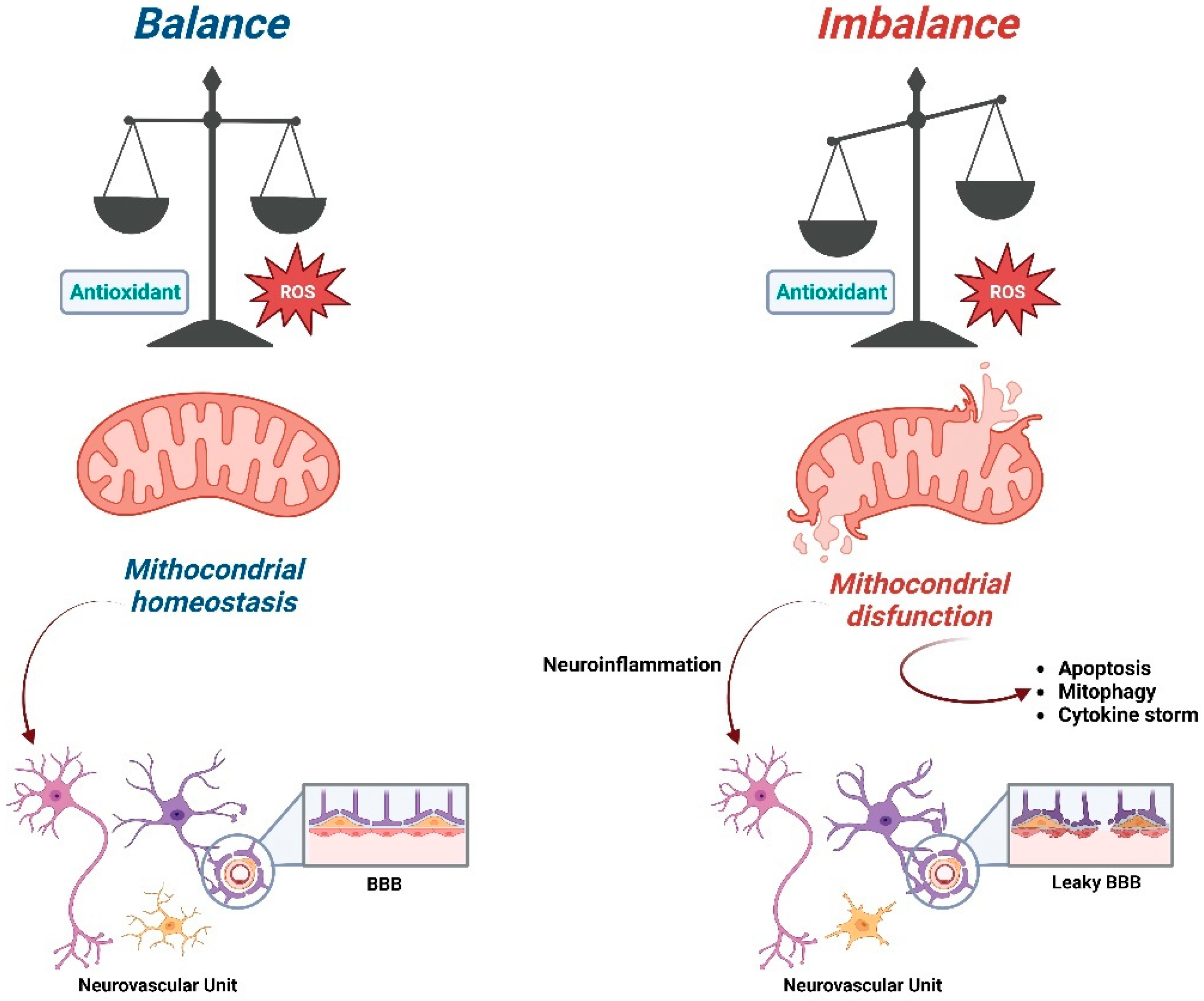
2. Role of Antioxidants on Mitochondrial Homeostasis
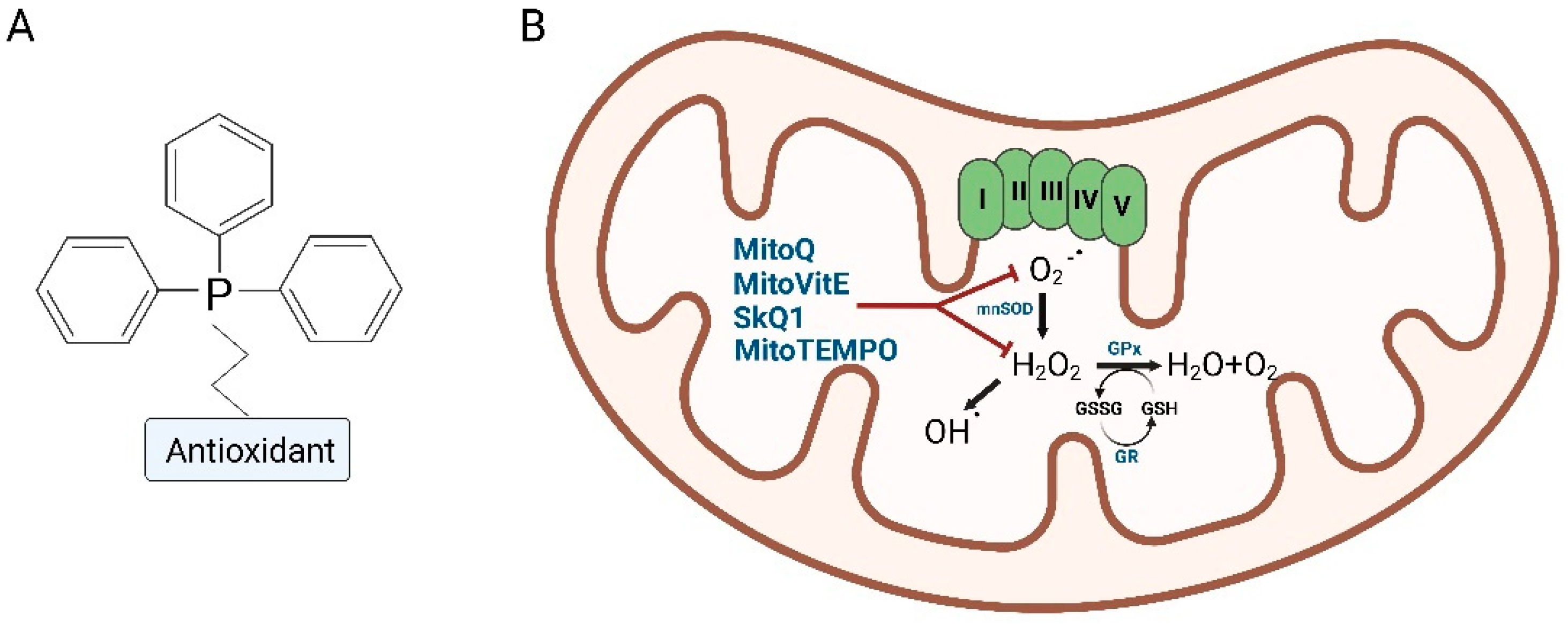
3. Mitochondria-Targeted Antioxidants (MTAs), Innovative Antioxidants Drugs
3.1. Mitoquinone (MitoQ)
3.2. SkQ1
3.3. MitoVitE
3.4. MitoTEMPO
4. ROS, Mitochondria and Neurodegenerative Diseases
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alimohammadi, M.; Makaremi, S.; Rahimi, A.; Asghariazar, V.; Taghadosi, M.; Safarzadeh, E. DNA methylation changes and inflammaging in aging-associated diseases. Epigenomics 2022, 14, 965–986. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dong, C.; Han, Y.; Gu, Z.; Sun, C. Immunosenescence, aging and successful aging. Front. Immunol. 2022, 13, 942796. [Google Scholar] [CrossRef] [PubMed]
- Shannon, O.M.; Gregory, S.; Siervo, M. Dietary nitrate, aging and brain health: The latest evidence. Curr. Opin. Clin. Nutr. Metab. Care 2022, 25, 393–400. [Google Scholar] [CrossRef]
- Stanga, S.; Caretto, A.; Boido, M.; Vercelli, A. Mitochondrial Dysfunctions: A Red Thread across Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 3719. [Google Scholar] [CrossRef] [PubMed]
- Millichap, L.E.; Damiani, E.; Tiano, L.; Hargreaves, I.P. Targetable Pathways for Alleviating Mitochondrial Dysfunction in Neurodegeneration of Metabolic and Non-Metabolic Diseases. Int. J. Mol. Sci. 2021, 22, 11444. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184. [Google Scholar] [CrossRef]
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Green, A.; Hossain, T.; Eckmann, D.M. Mitochondrial dynamics involves molecular and mechanical events in motility, fusion and fission. Front. Cell Dev. Biol. 2022, 10, 1010232. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar]
- Rambold, A.S.; Pearce, E.L. Mitochondrial Dynamics at the Interface of Immune Cell Metabolism and Function. Trends Immunol. 2018, 39, 6–18. [Google Scholar] [CrossRef]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef]
- Marchi, S.; Pinton, P. Mitochondria in the line of fire. Cell Death Differ. 2022, 29, 1301–1303. [Google Scholar] [CrossRef]
- Baechler, B.L.; Bloemberg, D.; Quadrilatero, J. Mitophagy regulates mitochondrial network signaling, oxidative stress, and apoptosis during myoblast differentiation. Autophagy 2019, 15, 1606–1619. [Google Scholar] [CrossRef]
- Iqbal, S.; Hood, D.A. Oxidative stress-induced mitochondrial fragmentation and movement in skeletal muscle myoblasts. Am. J. Physiol. Cell Physiol. 2014, 306, C1176–C1183. [Google Scholar] [CrossRef]
- Rambold, A.S.; Kostelecky, B.; Lippincott-Schwartz, J. Together we are stronger: Fusion protects mitochondria from autophagosomal degradation. Autophagy 2011, 7, 1568–1569. [Google Scholar] [CrossRef]
- Otera, H.; Ishihara, N.; Mihara, K. New insights into the function and regulation of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 1256–1268. [Google Scholar] [CrossRef]
- Kaur, S.; Sharma, N.; Kumar, V.; Sharma, D.; Devi, B.; Kapil, L.; Singh, C.; Singh, A. The Role of Mitophagy in Various Neurological Diseases as a Therapeutic Approach. Cell. Mol. Neurobiol. 2022; in press. [Google Scholar]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Kuroda, Y.; Mitsui, T.; Kunishige, M.; Shono, M.; Akaike, M.; Azuma, H.; Matsumoto, T. Parkin enhances mitochondrial biogenesis in proliferating cells. Hum. Mol. Genet. 2006, 15, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Chen, X.; Liao, J.; Yang, T.; Cheng, S.; Mei, Z.; Ge, J. Mitochondrial quality control in stroke: From the mechanisms to therapeutic potentials. J. Cell. Mol. Med. 2022, 26, 1000–1012. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Cen, X.; Chen, Y.; Xu, X.; Wu, R.; He, F.; Zhao, Q.; Sun, Q.; Yi, C.; Wu, J.; Najafov, A.; et al. Pharmacological targeting of MCL-1 promotes mitophagy and improves disease pathologies in an Alzheimer’s disease mouse model. Nat. Commun. 2020, 11, 5731. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Prescott, A.R.; Montava-Garriga, L.; Ball, G.; Singh, F.; Barini, E.; Muqit, M.M.; Brooks, S.P.; Ganley, I.G. Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metab. 2018, 27, 439–449.e5. [Google Scholar] [CrossRef]
- Bando, I.; Reus, M.I.; Andrés, D.; Cascales, M. Endogenous antioxidant defence system in rat liver following mercury chloride oral intoxication. J. Biochem. Mol. Toxicol. 2005, 19, 154–161. [Google Scholar] [CrossRef]
- Wang, Z.; Cai, L.; Li, H.; Liang, M.; Zhang, Y.; Wu, Q.; Yang, L. Rice protein stimulates endogenous antioxidant response attributed to methionine availability in growing rats. J. Food Biochem. 2020, 44, e13180. [Google Scholar] [CrossRef]
- Marchi, S.; Guilbaud, E.; Tait, S.W.G.; Yamazaki, T.; Galluzzi, L. Mitochondrial control of inflammation. Nat. Rev. Immunol. 2022; in press. [Google Scholar] [CrossRef]
- Tjahjono, E.; Kirienko, D.R.; Kirienko, N.V. The emergent role of mitochondrial surveillance in cellular health. Aging Cell. 2022, 21, e13710. [Google Scholar] [CrossRef]
- Trigo, D.; Avelar, C.; Fernandes, M.; Sá, J.; da Cruz ESilva, O. Mitochondria, energy, and metabolism in neuronal health and disease. FEBS Lett. 2022, 596, 1095–1110. [Google Scholar] [CrossRef]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: Can mitochondria be targeted therapeutically? Biochem. Soc. Trans. 2018, 46, 891–909. [Google Scholar] [CrossRef]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef]
- Monzio Compagnoni, G.; Di Fonzo, A.; Corti, S.; Comi, G.P.; Bresolin, N.; Masliah, E. The Role of Mitochondria in Neurodegenerative Diseases: The Lesson from Alzheimer’s Disease and Parkinson’s Disease. Mol. Neurobiol. 2020, 57, 2959–2980. [Google Scholar] [CrossRef]
- Rouaud, T.; Corbillé, A.G.; Leclair-Visonneau, L.; de Guilhem de Lataillade, A.; Lionnet, A.; Preterre, C.; Damier, P.; Derkinderen, P. Pathophysiology of Parkinson’s disease: Mitochondria, alpha-synuclein and much more. Rev. Neurol. 2021, 177, 260–271. [Google Scholar] [CrossRef]
- Nieto, M.; Gil-Bea, F.J.; Dalfó, E.; Cuadrado, M.; Cabodevilla, F.; Sánchez, B.; Catena, S.; Sesma, T.; Ribé, E.; Ferrer, I.; et al. Increased sensitivity to MPTP in human alpha-synuclein A30P transgenic mice. Neurobiol. Aging 2006, 27, 848–856. [Google Scholar] [CrossRef]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra78. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef]
- Vila, M.; Vukosavic, S.; Jackson-Lewis, V.; Neystat, M.; Jakowec, M.; Przedborski, S. Alpha-synuclein up-regulation in substantia nigra dopaminergic neurons following administration of the parkinsonian toxin MPTP. J. Neurochem. 2000, 74, 721–729. [Google Scholar] [CrossRef]
- Höglinger, G.U.; Féger, J.; Prigent, A.; Michel, P.P.; Parain, K.; Champy, P.; Ruberg, M.; Oertel, W.H.; Hirsch, E.C. Chronic systemic complex I inhibition induces a hypokinetic multisystem degeneration in rats. J. Neurochem. 2003, 84, 491–502. [Google Scholar] [CrossRef]
- Tieu, K. A guide to neurotoxic animal models of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2011, 1, a009316. [Google Scholar] [CrossRef]
- Nisticò, R.; Mehdawy, B.; Piccirilli, S.; Mercuri, N. Paraquat-and rotenone-induced models of Parkinson’s disease. Int. J. Immunopathol. Pharmacol. 2011, 24, 313–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Johri, A. Disentangling Mitochondria in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 11520. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Nahon-Crystal, E.; Shteinfer-Kuzmine, A.; Gupta, R. VDAC1, mitochondrial dysfunction, and Alzheimer’s disease. Pharmacol. Res. 2018, 131, 87–101. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Chen, J.; Bassot, A.; Giuliani, F.; Simmen, T. Amyotrophic Lateral Sclerosis (ALS): Stressed by Dysfunctional Mitochondria-Endoplasmic Reticulum Contacts (MERCs). Cells 2021, 10, 1789. [Google Scholar] [CrossRef]
- Manfredi, G.; Kawamata, H. Mitochondria and endoplasmic reticulum crosstalk in amyotrophic lateral sclerosis. Neurobiol. Dis. 2016, 90, 35–42. [Google Scholar] [CrossRef]
- Chen, W.; Guo, L.; Li, M.; Wei, C.; Li, S.; Xu, R. The pathogenesis of amyotrophic lateral sclerosis: Mitochondrial dysfunction, protein misfolding and epigenetics. Brain Res. 2022, 1786, 147904. [Google Scholar] [CrossRef]
- Patergnani, S.; Fossati, V.; Bonora, M.; Giorgi, C.; Marchi, S.; Missiroli, S.; Rusielewicz, T.; Wieckowski, M.R.; Pinton, P. Mitochondria in Multiple Sclerosis: Molecular Mechanisms of Pathogenesis. Int. Rev. Cell Mol. Biol. 2017, 328, 49–103. [Google Scholar]
- Ghafourifar, P.; Mousavizadeh, K.; Parihar, M.S.; Nazarewicz, R.R.; Parihar, A.; Zenebe, W.J. Mitochondria in multiple sclerosis. Front. Biosci. 2008, 13, 3116–3126. [Google Scholar] [CrossRef]
- Kozin, M.S.; Kulakova, O.G.; Favorova, O.O. Involvement of Mitochondria in Neurodegeneration in Multiple Sclerosis. Biochemistry 2018, 83, 813–830. [Google Scholar] [CrossRef]
- Franssen, H. Physiology of Myelinated Nerve Conduction and Pathophysiology of Demyelination. Adv. Exp. Med. Biol. 2019, 1190, 85–106. [Google Scholar]
- Mahad, D.; Lassmann, H.; Turnbull, D. Review: Mitochondria and disease progression in multiple sclerosis. Neuropathol. Appl. Neurobiol. 2008, 34, 577–589. [Google Scholar] [CrossRef]
- Fão, L.; Rego, A.C. Mitochondrial and Redox-Based Therapeutic Strategies in Huntington’s Disease. Antioxid. Redox Signal. 2021, 34, 650–673. [Google Scholar] [CrossRef]
- Pini, L.; Jacquemot, C.; Cagnin, A.; Meneghello, F.; Semenza, C.; Mantini, D.; Vallesi, A. Aberrant brain network connectivity in presymptomatic and manifest Huntington’s disease: A systematic review. Hum. Brain Mapp. 2020, 41, 256–269. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Beal, M.F. Mitochondrial approaches for neuroprotection. Ann. N. Y. Acad. Sci. 2008, 1147, 395–412. [Google Scholar] [CrossRef]
- Ellrichmann, G.; Petrasch-Parwez, E.; Lee, D.H.; Reick, C.; Arning, L.; Saft, C.; Gold, R.; Linker, R.A. Efficacy of fumaric acid esters in the R6/2 and YAC128 models of Huntington’s disease. PLoS ONE 2011, 6, e16172. [Google Scholar] [CrossRef]
- Brennan, W.A., Jr.; Bird, E.D.; Aprille, J.R. Regional mitochondrial respiratory activity in Huntington’s disease brain. J. Neurochem. 1985, 44, 1948–1950. [Google Scholar] [CrossRef]
- Gu, M.; Gash, M.T.; Mann, V.M.; Javoy-Agid, F.; Cooper, J.M.; Schapira, A.H. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann. Neurol. 1996, 39, 385–389. [Google Scholar] [CrossRef]
- Song, W.; Chen, J.; Petrilli, A.; Liot, G.; Klinglmayr, E.; Zhou, Y.; Poquiz, P.; Tjong, J.; Pouladi, M.A.; Hayden, M.R.; et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med. 2011, 17, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Shirendeb, U.P.; Calkins, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; McBride, J.L.; Mao, P.; Reddy, P.H. Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum. Mol. Genet. 2012, 21, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.N.; Gupta, G.; Sharma, P. A Comprehensive Review of Free Radicals, Antioxidants, and Their Relationship with Human Ailments. Crit. Rev. Eukaryot. Gene Expr. 2018, 28, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Poljsak, B. Strategies for reducing or preventing the generation of oxidative stress. Oxidative Med. Cell. Longev. 2011, 2011, 194586. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.P.; Sulaiman Rahman, H. Antioxidant and Oxidative Stress: A Mutual Interplay in Age-Related Diseases. Front. Pharmacol. 2018, 9, 1162. [Google Scholar] [CrossRef]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for commonn pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef]
- Błaszczyk, J.W. Energy Metabolism Decline in the Aging Brain-Pathogenesis of Neurodegenerative Disorders. Metabolites 2020, 10, 450. [Google Scholar] [CrossRef]
- Błaszczyk, J.W. Pathogenesis of Dementia. Int. J. Mol. Sci. 2022, 24, 543. [Google Scholar] [CrossRef]
- Jiang, Q.; Yin, J.; Chen, J.; Ma, X.; Wu, M.; Liu, G.; Yao, K.; Tan, B.; Yin, Y. Mitochondria- Targeted Antioxidants: A Step towards Disease Treatment. Oxidative Med. Cell. Longev. 2020, 2020, 8837893. [Google Scholar] [CrossRef]
- Apostolova, N.; Victor, V.M. Molecular strategies for targeting antioxidants to mitochondria: Therapeutic implications. Antioxid. Redox Signal. 2015, 22, 686–729. [Google Scholar] [CrossRef]
- Murphy, M.P. Antioxidants as therapies: Can we improve on nature? Free Radic. Biol. Med. 2014, 66, 20–23. [Google Scholar] [CrossRef]
- Cerimele, F.; Battle, T.; Lynch, R.; Frank, D.A.; Murad, E.; Cohen, C.; Macaron, N.; Sixbey, J.; Smith, K.; Watnick, R.S.; et al. Reactive oxygen signaling and MAPK activation distinguish Epstein-Barr Virus (EBV)-positive versus EBV-negative Burkitt’s lymphoma. Proc. Natl. Acad. Sci. USA 2005, 102, 175–179. [Google Scholar] [CrossRef]
- Oyewole, A.O.; Birch-Machin, M.A. Mitochondria-targeted antioxidants. FASEB J. 2015, 29, 4766–4771. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.A.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412. [Google Scholar] [CrossRef]
- Oliver, D.M.A.; Reddy, P.H. Small molecules as therapeutic drugs for Alzheimer’s disease. Mol. Cell. Neurosci. 2019, 96, 47–62. [Google Scholar] [CrossRef]
- Murphy, M.P.; Echtay, K.S.; Blaikie, F.H.; Asin-Cayuela, J.; Cochemé, H.M.; Green, K.; Buckingham, J.A.; Taylor, E.R.; Hurrell, F.; Hughes, G.; et al. Superoxide activates uncoupling proteins by generating carbon-centered radicals and initiating lipid peroxidation: Studies using a mitochondria-targeted spin trap derived from alpha-phenyl-N-tert-butylnitrone. J. Biol. Chem. 2003, 278, 48534–48545. [Google Scholar] [CrossRef]
- Filipovska, A.; Kelso, G.F.; Brown, S.E.; Beer, S.M.; Smith, R.A.; Murphy, M.P. Synthesis and characterization of a triphenylphosphonium-conjugated peroxidase mimetic. Insights into the interaction of ebselen with mitochondria. J. Biol. Chem. 2005, 280, 24113–24126. [Google Scholar] [CrossRef]
- Rokitskaya, T.I.; Klishin, S.S.; Severina, I.I.; Skulachev, V.P.; Antonenko, Y.N. Kinetic analysis of permeation of mitochondria-targeted antioxidants across bilayer lipid membranes. J. Membr. Biol. 2008, 224, 9–19. [Google Scholar] [CrossRef]
- Zinovkin, R.A.; Zamyatnin, A.A. Mitochondria-Targeted Drugs. Curr. Mol. Pharmacol. 2019, 12, 202–214. [Google Scholar] [CrossRef]
- Solesio, M.E.; Prime, T.A.; Logan, A.; Murphy, M.P.; Del Mar Arroyo-Jimenez, M.; Jordán, J.; Galindo, M.F. The mitochondria-targeted anti-oxidant MitoQ reduces aspects of mitochondrial fission in the 6-OHDA cell model of Parkinson’s disease. Biochim. Biophys. Acta 2013, 1832, 174–182. [Google Scholar] [CrossRef]
- Snow, B.J.; Rolfe, F.L.; Lockhart, M.M.; Frampton, C.M.; O’Sullivan, J.D.; Fung, V.; Smith, R.A.; Murphy, M.P.; Taylor, K.M. Protect Study Group. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord. 2010, 25, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Shinn, L.J.; Lagalwar, S. Treating Neurodegenerative Disease with Antioxidants: Efficacy of the Bioactive Phenol Resveratrol and Mitochondrial-Targeted MitoQ and SkQ. Antioxidants 2021, 10, 573. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lu, M.; Zhu, X.; Gu, X.; Zhang, T.; Xia, C.; Yang, L.; Xu, Y.; Zhou, M. The role of microglia immunometabolism in neurodegeneration: Focus on molecular determinants and metabolic intermediates of metabolic reprogramming. Biomed. Pharmacother. 2022, 153, 113412. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, Y.; Zhao, J. Inhibitory effect of mitoquinone against the α-synuclein fibrillation and relevant neurotoxicity: Possible role in inhibition of Parkinson’s disease. Biol. Chem. 2021, 403, 253–263. [Google Scholar] [CrossRef]
- Ünal, İ.; Çalışkan-Ak, E.; Üstündağ, Ü.V.; Ateş, P.S.; Alturfan, A.A.; Altinoz, M.A.; Elmaci, I.; Emekli-Alturfan, E. Neuroprotective effects of mitoquinone and oleandrin on Parkinson’s disease model in zebrafish. Int. J. Neurosci. 2020, 130, 574–582. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.N. Mitochondrial enhancement for neurodegenerative movement disorders: A systematic review of trials involving creatine, coenzyme Q10, idebenone and mitoquinone. CNS Drugs. 2014, 28, 63–68. [Google Scholar] [CrossRef]
- Saez-Atienzar, S.; Bonet-Ponce, L.; Blesa, J.R.; Romero, F.J.; Murphy, M.P.; Jordan, J.; Galindo, M.F. The LRRK2 inhibitor GSK2578215A induces protective autophagy in SH-SY5Y cells: Involvement of Drp-1-mediated mitochondrial fission and mitochondrial-derived ROS signaling. Cell Death Dis. 2014, 5, e1368. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, C.; Yang, L.; Ji, T.; Zhang, N.; Dong, X.; Chen, X.; Ma, J.; Gao, W.; Huang, S.; et al. NOX2-derived hydrogen peroxide impedes the AMPK/Akt-mTOR signaling pathway contributing to cell death in neuronal cells. Cell Signal. 2022, 94, 110330. [Google Scholar] [CrossRef]
- Kim, H.; Kim, S.H.; Cha, H.; Kim, S.R.; Lee, J.H.; Park, J.W. IDH2 deficiency promotes mitochondrial dysfunction and dopaminergic neurotoxicity: Implications for Parkinson’s disease. Free Radic. Res. 2016, 50, 853–860. [Google Scholar] [CrossRef]
- Almikhlafi, M.A.; Karami, M.M.; Jana, A.; Alqurashi, T.M.; Majrashi, M.; Alghamdi, B.S.; Ashraf, G.M. Mitochondrial Medicine: A Promising Therapeutic Option Against Various Neurodegenerative Disorders. Curr. Neuropharmacol. 2023. [Google Scholar] [CrossRef]
- Zhelev, Z.; Bakalova, R.; Aoki, I.; Lazarova, D.; Saga, T. Imaging of superoxide generation in the dopaminergic area of the brain in Parkinson’s disease, using mito-TEMPO. ACS Chem. Neurosci. 2013, 4, 1439–1445. [Google Scholar] [CrossRef]
- McManus, M.J.; Murphy, M.P.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 15703–15715. [Google Scholar] [CrossRef]
- Young, M.L.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ inhibits memory loss, neuropathology, and extends lifespan in aged 3xTg-AD mice. Mol. Cell. Neurosci. 2019, 101, 103409. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, A. A review on mitochondrial restorative mechanism of antioxidants in Alzheimer’s disease and other neurological conditions. Front. Pharmacol. 2015, 6, 206. [Google Scholar] [CrossRef]
- Aborode, A.T.; Pustake, M.; Awuah, W.A.; Alwerdani, M.; Shah, P.; Yarlagadda, R.; Ahmad, S.; Silva Correia, I.F.; Chandra, A.; Nansubuga, E.P.; et al. Targeting Oxidative Stress Mechanisms to Treat Alzheimer’s and Parkinson’s Disease: A Critical Review. Oxidative Med. Cell. Longev. 2022, 2022, 7934442. [Google Scholar] [CrossRef]
- Ng, L.F.; Gruber, J.; Cheah, I.K.; Goo, C.K.; Cheong, W.F.; Shui, G.; Sit, K.P.; Wenk, M.R.; Halliwell, B. The mitochondria-targeted antioxidant MitoQ extends lifespan and improves healthspan of a transgenic Caenorhabditis elegans model of Alzheimer disease. Free Radic. Biol. Med. 2014, 71, 390–401. [Google Scholar] [CrossRef]
- Du, F.; Yu, Q.; Kanaan, N.M.; Yan, S.S. Mitochondrial oxidative stress contributes to the pathological aggregation and accumulation of tau oligomers in Alzheimer’s disease. Hum. Mol. Genet. 2022, 31, 2498–2507. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Matera, R.; Minarini, A.; Rosini, M.; Melchiorre, C. Alzheimer’s disease: New approaches to drug discovery. Curr. Opin. Chem. Biol. 2009, 13, 303–308. [Google Scholar] [CrossRef]
- Manabe, T.; Matsumura, A.; Yokokawa, K.; Saito, T.; Fujikura, M.; Iwahara, N.; Matsushita, T.; Suzuki, S.; Hisahara, S.; Kawamata, J.; et al. Evaluation of Mitochondrial Oxidative Stress in the Brain of a Transgenic Mouse Model of Alzheimer’s Disease by in vitro Electron Paramagnetic Resonance Spectroscopy. J. Alzheimer’s Dis. 2019, 67, 1079–1087. [Google Scholar] [CrossRef]
- Yin, X.; Manczak, M.; Reddy, P.H. Mitochondria-targeted molecules MitoQ and SS31 reduce mutant huntingtin-induced mitochondrial toxicity and synaptic damage in Huntington’s disease. Hum. Mol. Genet. 2016, 25, 1739–1753. [Google Scholar] [CrossRef]
- Miquel, E.; Cassina, A.; Martínez-Palma, L.; Souza, J.M.; Bolatto, C.; Rodríguez-Bottero, S.; Logan, A.; Smith, R.A.; Murphy, M.P.; Barbeito, L.; et al. Neuroprotective effects of the mitochondria-targeted antioxidant MitoQ in a model of inherited amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2014, 70, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Junghans, M.; John, F.; Cihankaya, H.; Schliebs, D.; Winklhofer, K.F.; Bader, V.; Matschke, J.; Theiss, C.; Matschke, V. ROS scavengers decrease γH2ax spots in motor neuronal nuclei of ALS model mice in vitro. Front. Cell Neurosci. 2022, 16, 963169. [Google Scholar] [CrossRef] [PubMed]
- Brisset, M.; Nicolas, G. Peripheral neuropathies and aging. Neuropathies périphériques et vieillissement. Geriatr. Psychol. Neuropsychiatr. Vieil. 2018, 16, 409–413. [Google Scholar] [PubMed]
- Mao, P.; Manczak, M.; Shirendeb, U.P.; Reddy, P.H. MitoQ, a mitochondria-targeted antioxidant, delays disease progression and alleviates pathogenesis in an experimental autoimmune encephalomyelitis mouse model of multiple sclerosis. Biochim. Biophys. Acta 2013, 1832, 2322–2331. [Google Scholar] [CrossRef] [PubMed]
- López-Pedrera, C.; Villalba, J.M.; Patiño-Trives, A.M.; Luque-Tévar, M.; Barbarroja, N.; Aguirre, M.Á.; Escudero-Contreras, A.; Pérez-Sánchez, C. Therapeutic Potential and Immunomodulatory Role of Coenzyme Q10 and Its Analogues in Systemic Autoimmune Diseases. Antioxidants 2021, 10, 600. [Google Scholar] [CrossRef]
- Fetisova, E.K.; Muntyan, M.S.; Lyamzaev, K.G.; Chernyak, B.V. Therapeutic Effect of the Mitochondria-Targeted Antioxidant SkQ1 on the Culture Model of Multiple Sclerosis. Oxidative Med. Cell. Longev. 2019, 2019, 2082561. [Google Scholar] [CrossRef]
- Magalon, K.; Le Grand, M.; El Waly, B.; Moulis, M.; Pruss, R.; Bordet, T.; Cayre, M.; Belenguer, P.; Carré, M.; Durbec, P. Olesoxime favors oligodendrocyte differentiation through a functional interplay between mitochondria and microtubules. Neuropharmacology 2016, 111, 293–303. [Google Scholar] [CrossRef]
- Piscianz, E.; Tesser, A.; Rimondi, E.; Melloni, E.; Celeghini, C.; Marcuzzi, A. MitoQ Is Able to Modulate Apoptosis and Inflammation. Int. J. Mol. Sci. 2021, 22, 4753. [Google Scholar] [CrossRef]
- Shill, D.D.; Southern, W.M.; Willingham, T.B.; Lansford, K.A.; McCully, K.K.; Jenkins, N.T. Mitochondria-specific antioxidant supplementation does not influence endurance exercise training-induced adaptations in circulating angiogenic cells, skeletal muscle oxidative capacity or maximal oxygen uptake. J. Physiol. 2016, 594, 7005–7014. [Google Scholar] [CrossRef]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.; Murphy, M.P.; Sammut, I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005, 19, 1088–1095. [Google Scholar] [CrossRef]
- Xi, Y.; Feng, D.; Tao, K.; Wang, R.; Shi, Y.; Qin, H.; Murphy, M.P.; Yang, Q.; Zhao, G. MitoQ protects dopaminergic neurons in a 6-OHDA induced PD model by enhancing Mfn2-dependent mitochondrial fusion via activation of PGC-1α. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2859–2870. [Google Scholar] [CrossRef]
- Ghosh, A.; Chandran, K.; Kalivendi, S.V.; Joseph, J.; Antholine, W.E.; Hillard, C.J.; Kanthasamy, A.; Kanthasamy, A.; Kalyanaraman, B. Neuroprotection by a mitochondria-targeted drug in a Parkinson’s disease model. Free Radic. Biol. Med. 2010, 49, 1674–1684. [Google Scholar] [CrossRef]
- Manczak, M.; Mao, P.; Calkins, M.J.; Cornea, A.; Reddy, A.P.; Murphy, M.P.; Szeto, H.H.; Park, B.; Reddy, P.H. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer’s disease neurons. J. Alzheimer’s Dis. 2010, 20 (Suppl. 2), S609–S631. [Google Scholar] [CrossRef] [Green Version]
- Dupuis, L.; Gonzalez de Aguilar, J.L.; Oudart, H.; de Tapia, M.; Barbeito, L.; Loeffler, J.P. Mitochondria in amyotrophic lateral sclerosis: A trigger and a target. Neurodegener. Dis. 2004, 1, 245–254. [Google Scholar] [CrossRef]
- Tsitkanou, S.; Della Gatta, P.A.; Russell, A.P. Skeletal Muscle Satellite Cells, Mitochondria, and MicroRNAs: Their Involvement in the Pathogenesis of ALS. Front. Physiol. 2016, 7, 403. [Google Scholar] [CrossRef]
- Pinho, B.R.; Duarte, A.I.; Canas, P.M.; Moreira, P.I.; Murphy, M.P.; Oliveira, J.M.A. The interplay between redox signalling and proteostasis in neurodegeneration: In vivo effects of a mitochondria-targeted antioxidant in Huntington’s disease mice. Free Radic. Biol. Med. 2020, 146, 372–382. [Google Scholar] [CrossRef]
- Shabalina, I.G.; Vyssokikh, M.Y.; Gibanova, N.; Csikasz, R.I.; Edgar, D.; Hallden-Waldemarson, A.; Rozhdestvenskaya, Z.; Bakeeva, L.E.; Vays, V.B.; Pustovidko, A.V.; et al. Improved health-span and lifespan in mtDNA mutator mice treated with the mitochondrially targeted antioxidant SkQ1. Aging 2017, 9, 315–339. [Google Scholar] [CrossRef]
- Petrov, A.; Perekhvatova, N.; Skulachev, M.; Stein, L.; Ousler, G. SkQ1 Ophthalmic Solution for Dry Eye Treatment: Results of a Phase 2 Safety and Efficacy Clinical Study in the Environment and During Challenge in the Controlled Adverse Environment Model. Adv. Ther. 2016, 33, 96–115. [Google Scholar] [CrossRef]
- Sacks, B.; Onal, H.; Martorana, R.; Sehgal, A.; Harvey, A.; Wastella, C.; Ahmad, H.; Ross, E.; Pjetergjoka, A.; Prasad, S.; et al. Mitochondrial targeted antioxidants, mitoquinone and SKQ1, not vitamin C, mitigate doxorubicin-induced damage in H9c2 myoblast: Pretreatment vs. co-treatment. BMC Pharmacol. Toxicol. 2021, 22, 49. [Google Scholar] [CrossRef]
- Pavshintsev, V.V.; Podshivalova, L.S.; Frolova, O.Y.; Belopolskaya, M.V.; Averina, O.A.; Kushnir, E.A.; Marmiy, N.V.; Lovat, M.L. Effects of Mitochondrial Antioxidant SkQ1 on Biochemical and Behavioral Parameters in a Parkinsonism Model in Mice. Biochemistry 2017, 82, 1513–1520. [Google Scholar] [CrossRef]
- Stefanova, N.A.; Muraleva, N.A.; Skulachev, V.P.; Kolosova, N.G. Alzheimer’s disease-like pathology in senescence-accelerated OXYS rats can be partially retarded with mitochondria-targeted antioxidant SkQ1. J. Alzheimer’s Dis. 2014, 38, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, N.A.; Muraleva, N.A.; Maksimova, K.Y.; Rudnitskaya, E.A.; Kiseleva, E.; Telegina, D.V.; Kolosova, N.G. An antioxidant specifically targeting mitochondria delays progression of Alzheimer’s disease-like pathology. Aging 2016, 8, 2713–2733. [Google Scholar] [CrossRef] [PubMed]
- Tucker, J.M.; Townsend, D.M. Alpha-tocopherol: Roles in prevention and therapy of human disease. Biomed. Pharmacother. 2005, 59, 380–387. [Google Scholar] [CrossRef]
- Smith, R.A.; Porteous, C.M.; Coulter, C.V.; Murphy, M.P. Selective targeting of an antioxidant to mitochondria. Eur. J. Biochem. 1999, 263, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, A.; Kotamraju, S.; Kalivendi, S.V.; Matsunaga, T.; Shang, T.; Keszler, A.; Joseph, J.; Kalyanaraman, B. Supplementation of endothelial cells with mitochondria-targeted antioxidants inhibit peroxide-induced mitochondrial iron uptake, oxidative damage, and apoptosis. J. Biol. Chem. 2004, 279, 37575–37587. [Google Scholar] [CrossRef]
- Jauslin, M.L.; Meier, T.; Smith, R.A.; Murphy, M.P. Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J. 2003, 17, 1972–1974. [Google Scholar] [CrossRef]
- Hu, H.; Li, M. Mitochondria-targeted antioxidant mitotempo protects mitochondrial function against amyloid beta toxicity in primary cultured mouse neurons. Biochem. Biophys. Res. Commun. 2016, 478, 174–180. [Google Scholar] [CrossRef]
- Mitchell, J.B.; DeGraff, W.; Kaufman, D.; Krishna, M.C.; Samuni, A.; Finkelstein, E.; Ahn, M.S.; Hahn, S.M.; Gamson, J.; Russo, A. Inhibition of oxygen-dependent radiation-induced damage by the nitroxide superoxide dismutase mimic, tempol. Arch. Biochem. Biophys. 1991, 289, 62–70. [Google Scholar] [CrossRef]
- Trnka, J.; Blaikie, F.H.; Smith, R.A.; Murphy, M.P. A mitochondria-targeted nitroxide is reduced to its hydroxylamine by ubiquinol in mitochondria. Free Radic. Biol. Med. 2008, 44, 1406–1419. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Flint Beal, M. Mitochondrial diseases of the brain. Free Radic. Biol. Med. 2013, 63, 1–29. [Google Scholar] [CrossRef]
- Petrozzi, L.; Ricci, G.; Giglioli, N.J.; Siciliano, G.; Mancuso, M. Mitochondria and neurodegeneration. Biosci. Rep. 2007, 27, 87–104. [Google Scholar] [CrossRef]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxidative Med. Cell. Longev. 2019, 2019, 2105607. [Google Scholar] [CrossRef]
- Marcuzzi, A.; Loganes, C.; Valencic, E.; Piscianz, E.; Monasta, L.; Bilel, S.; Bortul, R.; Celeghini, C.; Zweyer, M.; Tommasini, A. Neuronal Dysfunction Associated with Cholesterol Deregulation. Int. J. Mol. Sci. 2018, 19, 1523. [Google Scholar] [CrossRef] [Green Version]
- Marcuzzi, A.; Piscianz, E.; Zweyer, M.; Bortul, R.; Loganes, C.; Girardelli, M.; Baj, G.; Monasta, L.; Celeghini, C. Geranylgeraniol and Neurological Impairment: Involvement of Apoptosis and Mitochondrial Morphology. Int. J. Mol. Sci. 2016, 17, 365. [Google Scholar] [CrossRef]
- Di Matteo, V.; Esposito, E. Biochemical and therapeutic effects of antioxidants in the treatment of Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Curr. Drug Targets CNS Neurol. Disord. 2003, 2, 95–107. [Google Scholar] [CrossRef]
- Kalincik, T. Multiple Sclerosis Relapses: Epidemiology, Outcomes and Management. A Systematic Review. Neuroepidemiology 2015, 44, 199–214. [Google Scholar] [CrossRef]
- Mehanna, R.; Jankovic, J. Movement disorders in multiple sclerosis and other demyelinating diseases. J. Neurol. Sci. 2013, 328, 1–8. [Google Scholar] [CrossRef]
- Katsara, M.; Apostolopoulos, V. Editorial: Multiple Sclerosis: Pathogenesis and Therapeutics. Med. Chem. 2018, 14, 104–105. [Google Scholar] [CrossRef]
- Garg, N.; Smith, T.W. An update on immunopathogenesis, diagnosis, and treatment of multiple sclerosis. Brain Behav. 2015, 5, e00362. [Google Scholar] [CrossRef]
- Lemus, H.N.; Warrington, A.E.; Rodriguez, M. Multiple Sclerosis: Mechanisms of Disease and Strategies for Myelin and Axonal Repair. Neurol. Clin. 2018, 36, 1–11. [Google Scholar] [CrossRef]
- Nicholas, R.; Rashid, W. Multiple sclerosis. Am. Fam. Physician 2013, 87, 712–714. [Google Scholar] [PubMed]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent Advancements in Pathogenesis, Diagnostics and Treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Oboudiyat, C.; Glazer, H.; Seifan, A.; Greer, C.; Isaacson, R.S. Alzheimer’s disease. Semin. Neurol. 2013, 33, 313–329. [Google Scholar] [CrossRef]
- Jeremic, D.; Jiménez-Díaz, L.; Navarro-López, J.D. Past, present and future of therapeutic strategies against amyloid-β peptides in Alzheimer’s disease: A systematic review. Ageing Res. Rev. 2021, 72, 101496. [Google Scholar] [CrossRef]
- Kurz, C.; Walker, L.; Rauchmann, B.S.; Perneczky, R. Dysfunction of the blood-brain barrier in Alzheimer’s disease: Evidence from human studies. Neuropathol. Appl. Neurobiol. 2022, 48, e12782. [Google Scholar] [CrossRef]
- Logan, S.; Pharaoh, G.A.; Marlin, M.C.; Masser, D.R.; Matsuzaki, S.; Wronowski, B.; Yeganeh, A.; Parks, E.E.; Premkumar, P.; Farley, J.A.; et al. Insulin-like growth factor receptor signaling regulates working memory, mitochondrial metabolism, and amyloid-β uptake in astrocytes. Mol. Metab. 2018, 9, 141–155. [Google Scholar] [CrossRef]
- Thenganatt, M.A.; Jankovic, J. Parkinson disease subtypes. JAMA Neurol. 2014, 71, 499–504. [Google Scholar] [CrossRef]
- Roheger, M.; Kalbe, E.; Liepelt-Scarfone, I. Progression of Cognitive Decline in Parkinson’s Disease. J. Parkinsons Dis. 2018, 8, 183–193. [Google Scholar] [CrossRef]
- Reich, S.G.; Savitt, J.M. Parkinson’s Disease. Med. Clin. N. Am. 2019, 103, 337–350. [Google Scholar] [CrossRef]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Samii, A.; Nutt, J.G.; Ransom, B.R. Parkinson’s disease. Lancet 2004, 363, 1783–1793. [Google Scholar] [CrossRef] [Green Version]
- Ganley, I.G. Strengthening the link between mitophagy and Parkinson’s disease. Brain 2022, 145, 4154–4156. [Google Scholar] [CrossRef]
- Blagov, A.V.; Grechko, A.V.; Nikiforov, N.G.; Borisov, E.E.; Sadykhov, N.K.; Orekhov, A.N. Role of Impaired Mitochondrial Dynamics Processes in the Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 6954. [Google Scholar] [CrossRef]
- Coelho, P.; Fão, L.; Mota, S.; Rego, A.C. Mitochondrial function and dynamics in neural stem cells and neurogenesis: Implications for neurodegenerative diseases. Ageing Res. Rev. 2022, 80, 101667. [Google Scholar] [CrossRef]
- Rauf, A.; Badoni, H.; Abu-Izneid, T.; Olatunde, A.; Rahman, M.M.; Painuli, S.; Semwal, P.; Wilairatana, P.; Mubarak, M.S. Neuroinflammatory Markers: Key Indicators in the Pathology of Neurodegenerative Diseases. Molecules 2022, 27, 3194. [Google Scholar] [CrossRef]
- Bazzani, V.; Equisoain Redin, M.; McHale, J.; Perrone, L.; Vascotto, C. Mitochondrial DNA Repair in Neurodegenerative Diseases and Ageing. Int. J. Mol. Sci. 2022, 23, 11391. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Nikiforov, N.G.; Zhuravlev, A.D.; Orekhov, N.A.; Grechko, A.V.; Orekhov, A.N. Role of the mtDNA Mutations and Mitophagy in Inflammaging. Int. J. Mol. Sci. 2022, 23, 1323. [Google Scholar] [CrossRef]
- Jose, S.; Groves, N.J.; Roper, K.E.; Gordon, R. Mechanisms of NLRP3 activation and pathology during neurodegeneration. Int. J. Biochem. Cell Biol. 2022, 151, 106273. [Google Scholar] [CrossRef]
- Litwiniuk, A.; Baranowska-Bik, A.; Domańska, A.; Kalisz, M.; Bik, W. Contribution of Mitochondrial Dysfunction Combined with NLRP3 Inflammasome Activation in Selected Neurodegenerative Diseases. Pharmaceuticals 2021, 14, 1221. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Ji, C.; Shao, A. Neurovascular Unit Dysfunction and Neurodegenerative Disorders. Front. Neurosci. 2020, 14, 334. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yuan, Z.; Yang, S.; Zhu, Y.; Xue, M.; Zhang, J.; Leng, L. Brain Energy Metabolism: Astrocytes in Neurodegenerative Diseases. CNS Neurosci. Ther. 2022; in press. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, E.Y.; Zorov, D.B. Pros and Cons of Use of Mitochondria-Targeted Antioxidants. Antioxidants 2019, 8, 316. [Google Scholar] [CrossRef]
- Kulkarni, C.A.; Fink, B.D.; Gibbs, B.E.; Chheda, P.R.; Wu, M.; Sivitz, W.I.; Kerns, R.J. A Novel Triphenylphosphonium Carrier to Target Mitochondria without Uncoupling Oxidative Phosphorylation. J. Med. Chem. 2021, 64, 662–676. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Neurodegenerative Diseases | Mechanisms Involved | References |
|---|---|---|
| Parkinson’s disease (PD) |
| [32,33,34,35,36,37,38,39,40,41,42] |
| Alzheimer’s disease (AD) |
| [32,34,43,44,45,46] |
| Amyotrophic lateral sclerosis (ALS) |
| [47,48,49,50] |
| Multiple Sclerosis (MS) |
| [51,52,53,54,55] |
| Huntington’s disease (HD) |
| [19,56,57,58,59,60,61,62,63] |
| Neurodegenerative Diseases | MitoQ | MitoTEMPO | MitoVitE | SKQ1 |
|---|---|---|---|---|
| Parkinson’s disease (PD) | [81,82,83,84,85,86,87,88] | [89,90,91,92] | [88,92] | [83,92] |
| Alzheimer’s disease (AD) | [76,88,93,94,95,96,97] | [76,92,95,98,99] | [76,88,92,95] | [92,100] |
| Huntington’s disease (HD) | [56,88,101] | [76] | [88] | |
| Amyotrophic lateral sclerosis (ALS) | [102,88] | [103] | [88] | |
| Multiple Sclerosis | [104,105] | [106] | [107,108] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fields, M.; Marcuzzi, A.; Gonelli, A.; Celeghini, C.; Maximova, N.; Rimondi, E. Mitochondria-Targeted Antioxidants, an Innovative Class of Antioxidant Compounds for Neurodegenerative Diseases: Perspectives and Limitations. Int. J. Mol. Sci. 2023, 24, 3739. https://doi.org/10.3390/ijms24043739
Fields M, Marcuzzi A, Gonelli A, Celeghini C, Maximova N, Rimondi E. Mitochondria-Targeted Antioxidants, an Innovative Class of Antioxidant Compounds for Neurodegenerative Diseases: Perspectives and Limitations. International Journal of Molecular Sciences. 2023; 24(4):3739. https://doi.org/10.3390/ijms24043739
Chicago/Turabian StyleFields, Matteo, Annalisa Marcuzzi, Arianna Gonelli, Claudio Celeghini, Natalia Maximova, and Erika Rimondi. 2023. "Mitochondria-Targeted Antioxidants, an Innovative Class of Antioxidant Compounds for Neurodegenerative Diseases: Perspectives and Limitations" International Journal of Molecular Sciences 24, no. 4: 3739. https://doi.org/10.3390/ijms24043739
APA StyleFields, M., Marcuzzi, A., Gonelli, A., Celeghini, C., Maximova, N., & Rimondi, E. (2023). Mitochondria-Targeted Antioxidants, an Innovative Class of Antioxidant Compounds for Neurodegenerative Diseases: Perspectives and Limitations. International Journal of Molecular Sciences, 24(4), 3739. https://doi.org/10.3390/ijms24043739