
Contribution of Glucosylsphingosine (Lyso-Gb1) to Treatment Decisions in Patients with Gaucher Disease

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Study Cohort
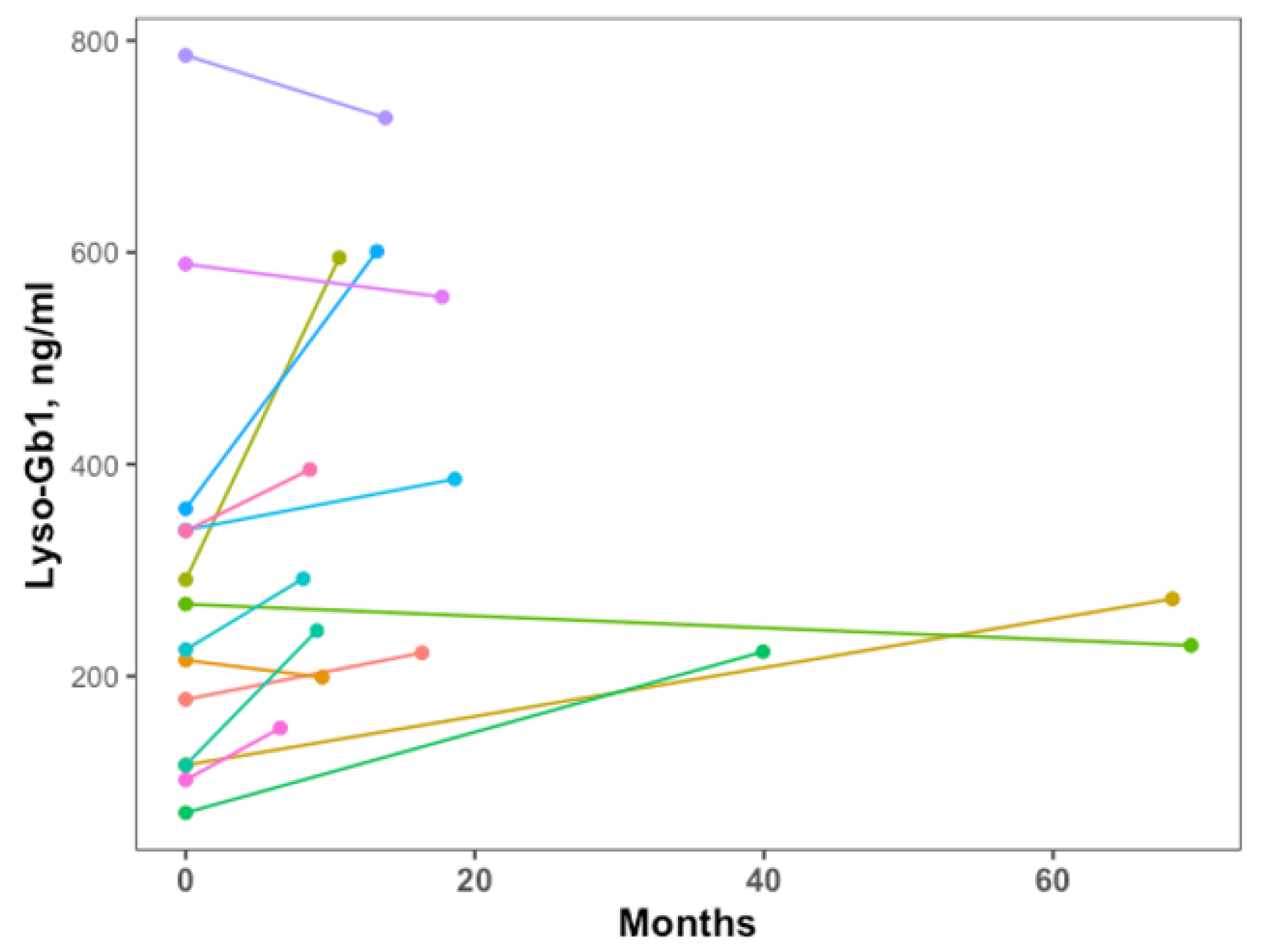
2.2. Lyso-Gb1 Levels
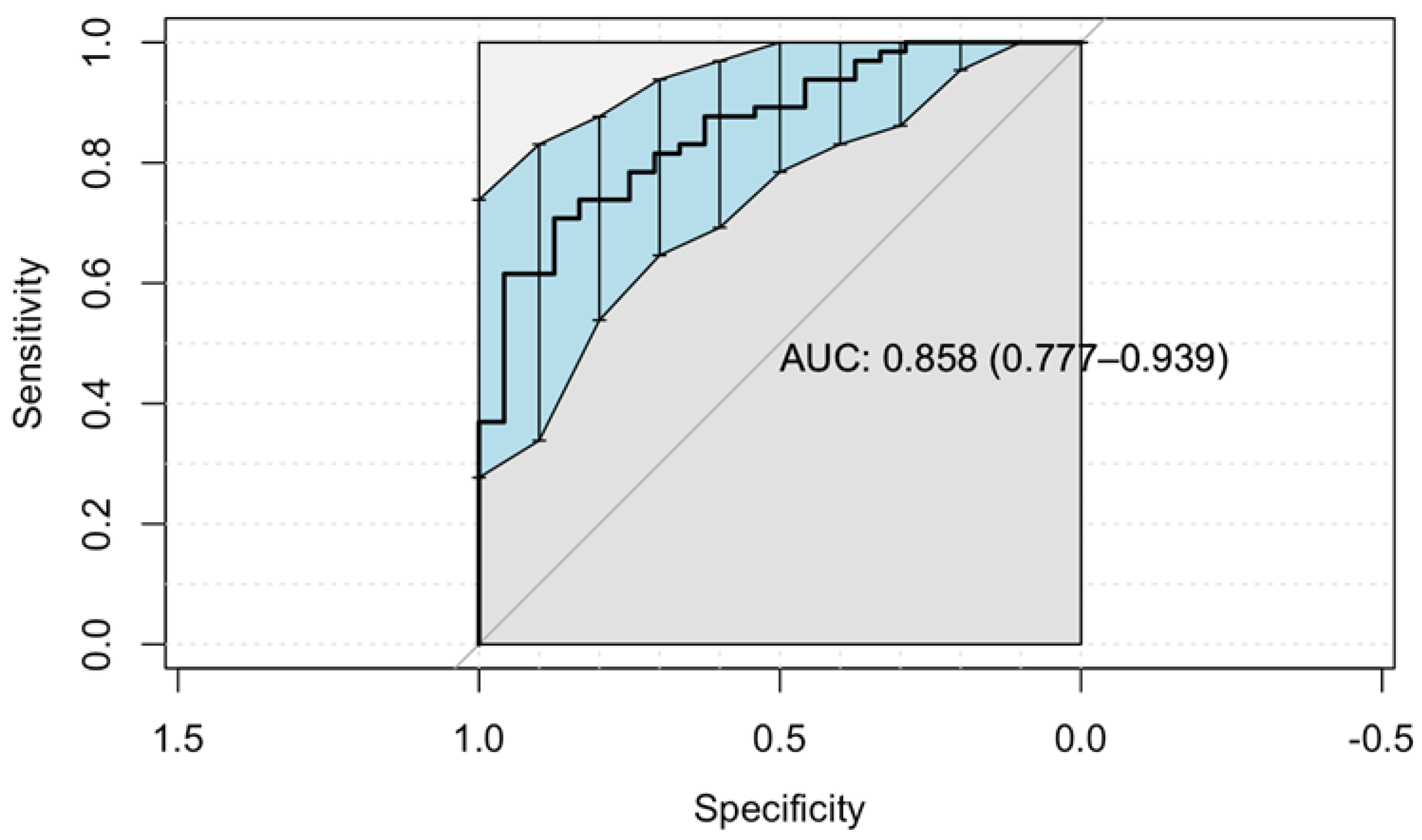
2.3. Predictors for Starting Therapy
3. Discussion
4. Materials and Methods
4.1. Study Cohort
4.2. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Revel-Vilk, S.; Szer, J.; Zimran, A. Gaucher disease and related lysosomal storage diseases. In Williams Hematology, 10th ed.; Kaushansky, K., Lichtman, M., Prchal, J., Levi, M., Press, O., Burns, L., Caligiuri, M., Eds.; McGraw-Hill: New York, NY, USA, 2021; pp. 1189–1202. [Google Scholar]
- Gary, S.E.; Ryan, E.; Steward, A.M.; Sidransky, E. Recent advances in the diagnosis and management of Gaucher disease. Expert Rev. Endocrinol. Metab. 2018, 13, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Drelichman, G.; Castaneda-Hernandez, G.; Cem Ar, M.; Dragosky, M.; Garcia, R.; Lee, H.; Moiseev, S.; Naderi, M.; Rosenbaum, H.; Znidar, I.; et al. The road to biosimilars in rare diseases—ongoing lessons from Gaucher disease. Am. J. Hematol. 2020, 95, 233–237. [Google Scholar] [CrossRef] [Green Version]
- Revel-Vilk, S.; Szer, J.; Mehta, A.; Zimran, A. How we manage Gaucher Disease in the era of choices. Br. J. Haematol. 2018, 182, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Dekker, N.; van Dussen, L.; Hollak, C.E.; Overkleeft, H.; Scheij, S.; Ghauharali, K.; van Breemen, M.J.; Ferraz, M.J.; Groener, J.E.; Maas, M.; et al. Elevated plasma glucosylsphingosine in Gaucher disease: Relation to phenotype, storage cell markers, and therapeutic response. Blood 2011, 118, e118–e127. [Google Scholar] [CrossRef] [Green Version]
- Rolfs, A.; Giese, A.K.; Grittner, U.; Mascher, D.; Elstein, D.; Zimran, A.; Bottcher, T.; Lukas, J.; Hubner, R.; Golnitz, U.; et al. Glucosylsphingosine is a highly sensitive and specific biomarker for primary diagnostic and follow-up monitoring in Gaucher disease in a non-Jewish, Caucasian cohort of Gaucher disease patients. PLoS ONE 2013, 8, e79732. [Google Scholar] [CrossRef] [PubMed]
- Arkadir, D.; Dinur, T.; Revel-Vilk, S.; Becker Cohen, M.; Cozma, C.; Hovakimyan, M.; Eichler, S.; Rolfs, A.; Zimran, A. Glucosylsphingosine is a reliable response biomarker in Gaucher disease. Am. J. Hematol. 2018, 93, E140–E142. [Google Scholar] [CrossRef] [Green Version]
- Saville, J.T.; McDermott, B.K.; Chin, S.J.; Fletcher, J.M.; Fuller, M. Expanding the clinical utility of glucosylsphingosine for Gaucher disease. J. Inherit. Metab. Dis. 2020, 43, 558–563. [Google Scholar] [CrossRef]
- Ida, H.; Watanabe, Y.; Sagara, R.; Inoue, Y.; Fernandez, J. An observational study to investigate the relationship between plasma glucosylsphingosine (lyso-Gb1) concentration and treatment outcomes of patients with Gaucher disease in Japan. Orphanet. J. Rare Dis. 2022, 17, 401. [Google Scholar] [CrossRef]
- Dinur, T.; Bauer, P.; Beetz, C.; Kramp, G.; Cozma, C.; Iurascu, M.I.; Becker-Cohen, M.; Istaiti, M.; Rolfs, A.; Zimran, A.; et al. Gaucher Disease Diagnosis Using Lyso-Gb1 on Dry Blood Spot Samples: Time to Change the Paradigm? Int. J. Mol. Sci. 2022, 23, 1627. [Google Scholar] [CrossRef]
- Nahm, F.S. Receiver operating characteristic curve: Overview and practical use for clinicians. Korean J. Anesthesiol. 2022, 75, 25–36. [Google Scholar] [CrossRef]
- Revel-Vilk, S.; Szer, J.; Zimran, A. Hematological manifestations and complications of Gaucher disease. Expert Rev. Hematol. 2021, 14, 347–354. [Google Scholar] [CrossRef]
- Revel-Vilk, S.; Naamad, M.; Frydman, D.; Freund, M.R.; Dinur, T.; Istaiti, M.; Becker-Cohen, M.; Falk, R.; Broide, E.; Michelson, A.D.; et al. Platelet Activation and Reactivity in a Large Cohort of Patients with Gaucher Disease. Thromb. Haemost. 2022, 122, 951–960. [Google Scholar] [CrossRef]
- Raskovalova, T.; Deegan, P.B.; Mistry, P.K.; Pavlova, E.; Yang, R.; Zimran, A.; Berger, J.; Bourgne, C.; Pereira, B.; Labarere, J.; et al. Accuracy of chitotriosidase activity and CCL18 concentration in assessing type I Gaucher disease severity. A systematic review with meta-analysis of individual participant data. Haematologica 2021, 106, 437–445. [Google Scholar] [CrossRef] [Green Version]
- Lukas, J.; Cozma, C.; Yang, F.; Kramp, G.; Meyer, A.; Nesslauer, A.M.; Eichler, S.; Bottcher, T.; Witt, M.; Brauer, A.U.; et al. Glucosylsphingosine Causes Hematological and Visceral Changes in Mice-Evidence for a Pathophysiological Role in Gaucher Disease. Int. J. Mol. Sci. 2017, 18, 2192. [Google Scholar] [CrossRef] [Green Version]
- Cozma, C.; Cullufi, P.; Kramp, G.; Hovakimyan, M.; Velmishi, V.; Gjikopulli, A.; Tomori, S.; Fischer, S.; Oppermann, S.; Grittner, U.; et al. Treatment Efficiency in Gaucher Patients Can Reliably Be Monitored by Quantification of Lyso-Gb1 Concentrations in Dried Blood Spots. Int. J. Mol. Sci. 2020, 21, 4577. [Google Scholar] [CrossRef]
- Available online: https://www.ewggd.com/ewggd-working-groups/ (accessed on 10 February 2023).
- Dinur, T.; Zimran, A.; Becker-Cohen, M.; Arkadir, D.; Cozma, C.; Hovakimyan, M.; Oppermann, S.; Demuth, L.; Rolfs, A.; Revel-Vilk, S. Long Term Follow-Up of 103 Untreated Adult Patients with Type 1 Gaucher Disease. J. Clin. Med. 2019, 8, 1662. [Google Scholar] [CrossRef] [Green Version]
- Weinreb, N.J.; Goker-Alpan, O.; Kishnani, P.S.; Longo, N.; Burrow, T.A.; Bernat, J.A.; Gupta, P.; Henderson, N.; Pedro, H.; Prada, C.E.; et al. The diagnosis and management of Gaucher disease in pediatric patients: Where do we go from here? Mol. Genet Metab. 2022, 136, 4–21. [Google Scholar] [CrossRef]
- Hurvitz, N.; Dinur, T.; Becker-Cohen, M.; Cozma, C.; Hovakimyan, M.; Oppermann, S.; Demuth, L.; Rolfs, A.; Abramov, A.; Zimran, A.; et al. Glucosylsphingosine (lyso-Gb1) as a Biomarker for Monitoring Treated and Untreated Children with Gaucher Disease. Int. J. Mol. Sci. 2019, 20, 3033. [Google Scholar] [CrossRef] [Green Version]
- Revel-Vilk, S.; Fuller, M.; Zimran, A. Value of Glucosylsphingosine (Lyso-Gb1) as a Biomarker in Gaucher Disease: A Systematic Literature Review. Int. J. Mol. Sci. 2020, 21, 7159. [Google Scholar] [CrossRef]
- Matern, D.; Gavrilov, D.; Oglesbee, D.; Raymond, K.; Rinaldo, P.; Tortorelli, S. Newborn screening for lysosomal storage disorders. Semin. Perinatol. 2015, 39, 206–216. [Google Scholar] [CrossRef]
- Burlina, A.B.; Polo, G.; Salviati, L.; Duro, G.; Zizzo, C.; Dardis, A.; Bembi, B.; Cazzorla, C.; Rubert, L.; Zordan, R.; et al. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J. Inherit. Metab. Dis. 2018, 41, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Bender, F.; Burin, M.G.; Tirelli, K.M.; Medeiros, F.; Bitencourt, F.H.; Civallero, G.; Kubaski, F.; Bravo, H.; Daher, A.; Carnier, V.; et al. Newborn screening for lysosomal disorders in Brazil: A pilot study using customized fluorimetric assays. Genet. Mol. Biol. 2020, 43, e20180334. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Zhan, X.; Gu, X.; Zhang, H. Successful newborn screening for Gaucher disease using fluorometric assay in China. J. Hum. Genet. 2017, 62, 763–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisi, E.C.; McCandless, S.E. Newborn Screening for Lysosomal Storage Disorders: Views of Genetic Healthcare Providers. J. Genet. Couns. 2016, 25, 373–384. [Google Scholar] [CrossRef]
- Cohen, Y.; Frydman, D.; Rotem, R.; Kofman, R.; Zimran, A.; Revel-Vilk, S.; Grisaru-Granovsky, S. Risk of postpartum hemorrhage in multiparous women with Gaucher disease: A call for reconsidering enzyme replacement therapy in all pregnant patients. J. Inherit. Metab. Dis. 2021, 44, 1165–1173. [Google Scholar] [CrossRef]
- Elstein, D.; Abrahamov, A.; Hadas-Halpern, I.; Meyer, A.; Zimran, A. Low-dose low-frequency imiglucerase as a starting regimen of enzyme replacement therapy for patients with type I Gaucher disease. QJM 1998, 91, 483–488. [Google Scholar] [CrossRef] [Green Version]
- Harel, R.; Gavish, I.; Aviv, A.; Greenman Maravi, N.; Trougouboff, P.; Zimran, A.; Revel-Vilk, S. Enzyme replacement therapy leading to improvement in myeloma indices in a patient with concomitant Gaucher disease. Intern. Med. J. 2022, 52, 872–875. [Google Scholar] [CrossRef]
- Nguyen, Y.; Stirnemann, J.; Lautredoux, F.; Cador, B.; Bengherbia, M.; Yousfi, K.; Hamroun, D.; Astudillo, L.; Billette de Villemeur, T.; Brassier, A.; et al. Immunoglobulin Abnormalities in Gaucher Disease: An Analysis of 278 Patients Included in the French Gaucher Disease Registry. Int. J. Mol. Sci. 2020, 21, 1247. [Google Scholar] [CrossRef] [Green Version]
- Grosbois, B.; Rose, C.; Noel, E.; Serratrice Cde, R.; Dobbelaere, D.; Gressin, V.; Cherin, P.; Hartmann, A.; Javier, R.M.; Clerson, P.; et al. Gaucher disease and monoclonal gammopathy: A report of 17 cases and impact of therapy. Blood Cells Mol. Dis. 2009, 43, 138–139. [Google Scholar] [CrossRef]
- Horowitz, M.; Braunstein, H.; Zimran, A.; Revel-Vilk, S.; Goker-Alpan, O. Lysosomal functions and dysfunctions: Molecular and cellular mechanisms underlying Gaucher disease and its association with Parkinson disease. Adv. Drug Deliv. Rev. 2022, 187, 114402. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Type 1- Mild GBA1 Genotype(s) | Type 1- Severe GBA1 Genotype(s) | Neuronopathic (Type 2/3/3c) | p | |
|---|---|---|---|---|
| n | 58 | 29 | 10 | |
| Male, n (%) | 23, 39.7% | 13, 44.8% | 3, 30% | 0.7 |
| Age, years: median (range) | 27.5 (2–78) | 11 (1–42) | 2.5 (1–25) | <0.001 |
| Children, % | 13, 22.4% | 19, 65.5% | 30, 46.2% | <0.001 |
| Platelet count, ×109/L: median (range) | 114 (48–297) | 125 (56–245) | 98 (41–257) | 0.7 |
| Hemoglobin level, g/dL: median (range) | 12.15 (9.4–15.8) | 11.4 (7.1–15.5) | 8.95 (6.3–12.7) | <0.001 |
| Lyso-Gb1, ng/mL: median (range) | 220 (9–786) | 447 (102–1340) | 290 (136–1270) | <0.001 |
| GD-Specific Treatment | |||
|---|---|---|---|
| No | Yes | p | |
| n | 24 * | 65 | |
| Male, n (%) | 11, 45.8% | 24, 38% | 0.68 |
| Age, years: median (range) | 26.5 (1–78) | 21 (1–71) | 0.29 |
| Children, % | 9, 37.5% | 30, 46.2% | 0.62 |
| Type of Gaucher disease, % | 0.005 | ||
| Type 1- mild GBA1 genotype(s) | 21, 87.5% | 32, 49.2% | |
| Type 1- severe GBA1 genotype(s) | 2, 8.3% | 24, 36.9% | |
| Neuronopathic (Type 2/3/3c) | 1, 4.2% | 9, 13.8% | |
| Platelet count, ×109/L: median (range) | 165 (53–297) | 98 (41–257) | <0.001 |
| Hemoglobin level, g/dL: median (range) | 13.2 (10.6–15.8) | 11.6 (6.3–15.6) | <0.001 |
| Lyso-Gb1, ng/mL: median (range) | 153.5 (9–442) | 337 (60–1340) | <0.001 |
| Variables | GD-Specific Therapy | Odd Ratio (OR) (95% CI) | p | |
|---|---|---|---|---|
| No | Yes | |||
| Platelet count, ×109/L | ||||
| ≥ 100 | 22 (92%) | 31 (48%) | 1 | |
| < 100 | 2 (8%) | 34 (52%) | 16.5 (3.4–143) | 0.002 |
| Hemoglobin level, mg/dL | ||||
| ≥ 11.5 | 22 (92%) | 36 (55.5%) | 1 | |
| < 11.5 | 2 (8%) | 29 (44.5%) | 7.5 (1.5–58) | 0.02 |
| Lyso-Gb1 level, ng/mL | ||||
| <100 | 8 (46%) | 3 (5%) | 1 | |
| 100–250 | 12 (50%) | 17 (26%) | 3.2 (0.5–29) | 0.2 |
| >250 | 4 (17%) | 45 (69%) | 22.8 (3.4–239) | 0.003 |
| Israeli Ministry of Health Criteria for Imiglucerase, 1998 [28] | Suggested Updated Criteria for ERT/SRT |
|---|---|
| When symptomatic or high lyso-Gb1 * |
| Age is not a criterion by itself |
| Stays as is |
| Gaucher-related significant, symptomatic cytopenia and/or bleeding disorder, irrespective of lyso-Gb1 levels |
| Redundant |
| With high lyso-Gb1 |
| Bone pain or evidence of significant bone involvement in MRI/ DEXA or any imaging abnormalities and the presence of high lyso-Gb1 |
| Stay as is |
| Short stature after exclusion of other causes or with the presence of high lyso-Gb1 |
| Molecular diagnosis of a ‘severe’ genotype ** and high lyso-Gb1 |
| - | Any patient who was diagnosed with malignancy requiring myelosuppressive therapy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dinur, T.; Bauer, P.; Beetz, C.; Cozma, C.; Becker-Cohen, M.; Istaiti, M.; Rolfs, A.; Skrahina, V.; Zimran, A.; Revel-Vilk, S. Contribution of Glucosylsphingosine (Lyso-Gb1) to Treatment Decisions in Patients with Gaucher Disease. Int. J. Mol. Sci. 2023, 24, 3945. https://doi.org/10.3390/ijms24043945
Dinur T, Bauer P, Beetz C, Cozma C, Becker-Cohen M, Istaiti M, Rolfs A, Skrahina V, Zimran A, Revel-Vilk S. Contribution of Glucosylsphingosine (Lyso-Gb1) to Treatment Decisions in Patients with Gaucher Disease. International Journal of Molecular Sciences. 2023; 24(4):3945. https://doi.org/10.3390/ijms24043945
Chicago/Turabian StyleDinur, Tama, Peter Bauer, Christian Beetz, Claudia Cozma, Michal Becker-Cohen, Majdolen Istaiti, Arndt Rolfs, Volha Skrahina, Ari Zimran, and Shoshana Revel-Vilk. 2023. "Contribution of Glucosylsphingosine (Lyso-Gb1) to Treatment Decisions in Patients with Gaucher Disease" International Journal of Molecular Sciences 24, no. 4: 3945. https://doi.org/10.3390/ijms24043945
APA StyleDinur, T., Bauer, P., Beetz, C., Cozma, C., Becker-Cohen, M., Istaiti, M., Rolfs, A., Skrahina, V., Zimran, A., & Revel-Vilk, S. (2023). Contribution of Glucosylsphingosine (Lyso-Gb1) to Treatment Decisions in Patients with Gaucher Disease. International Journal of Molecular Sciences, 24(4), 3945. https://doi.org/10.3390/ijms24043945